
Identification of the Microbial Transformation Products of Secoisolariciresinol Using an Untargeted Metabolomics Approach and Evaluation of the Osteogenic Activities of the Metabolites

 , ,
, ,  , ,
, ,
Abstract
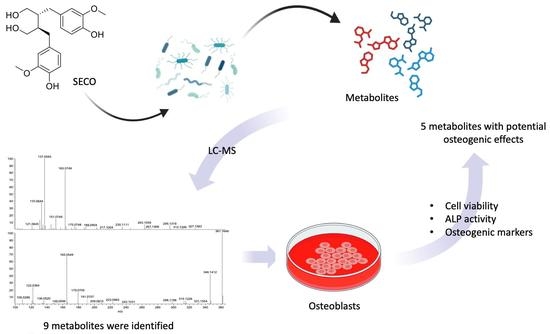
:
1. Introduction
2. Results
2.1. Multi-Variance Analysis
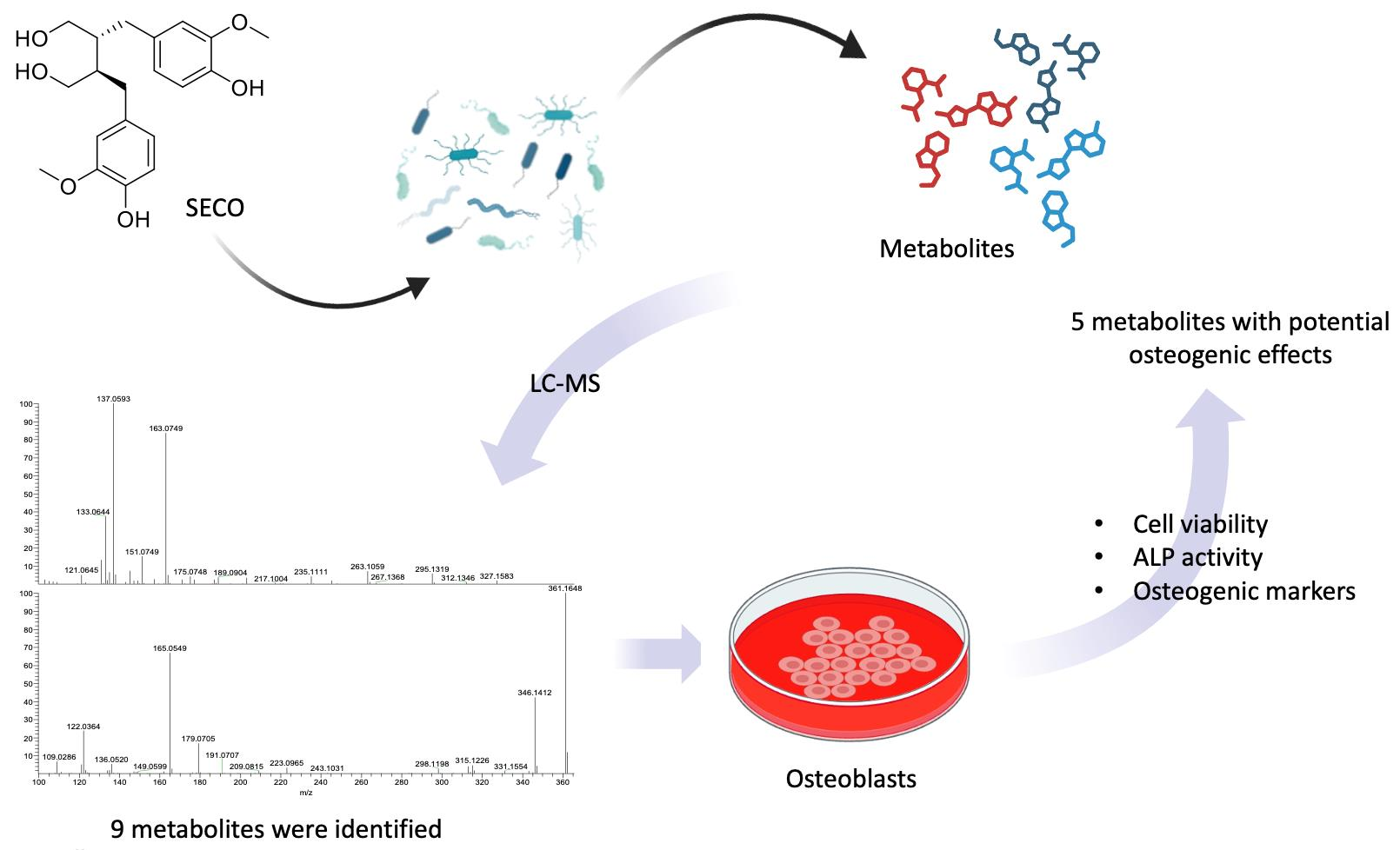
2.2. Metabolite Identification
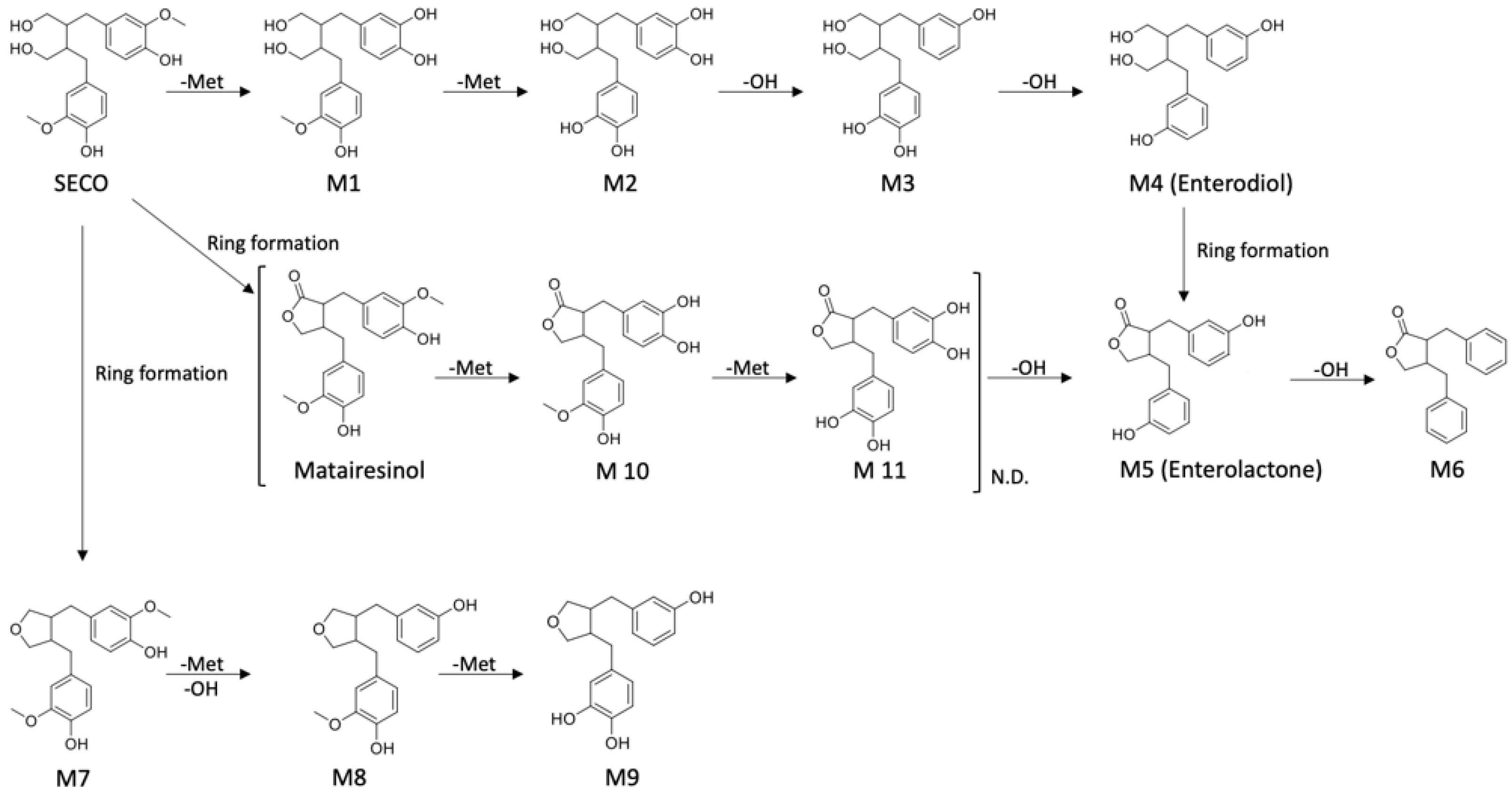
2.3. Proposed Metabolic Pathway
2.4. Biotransformation of SECO over Time
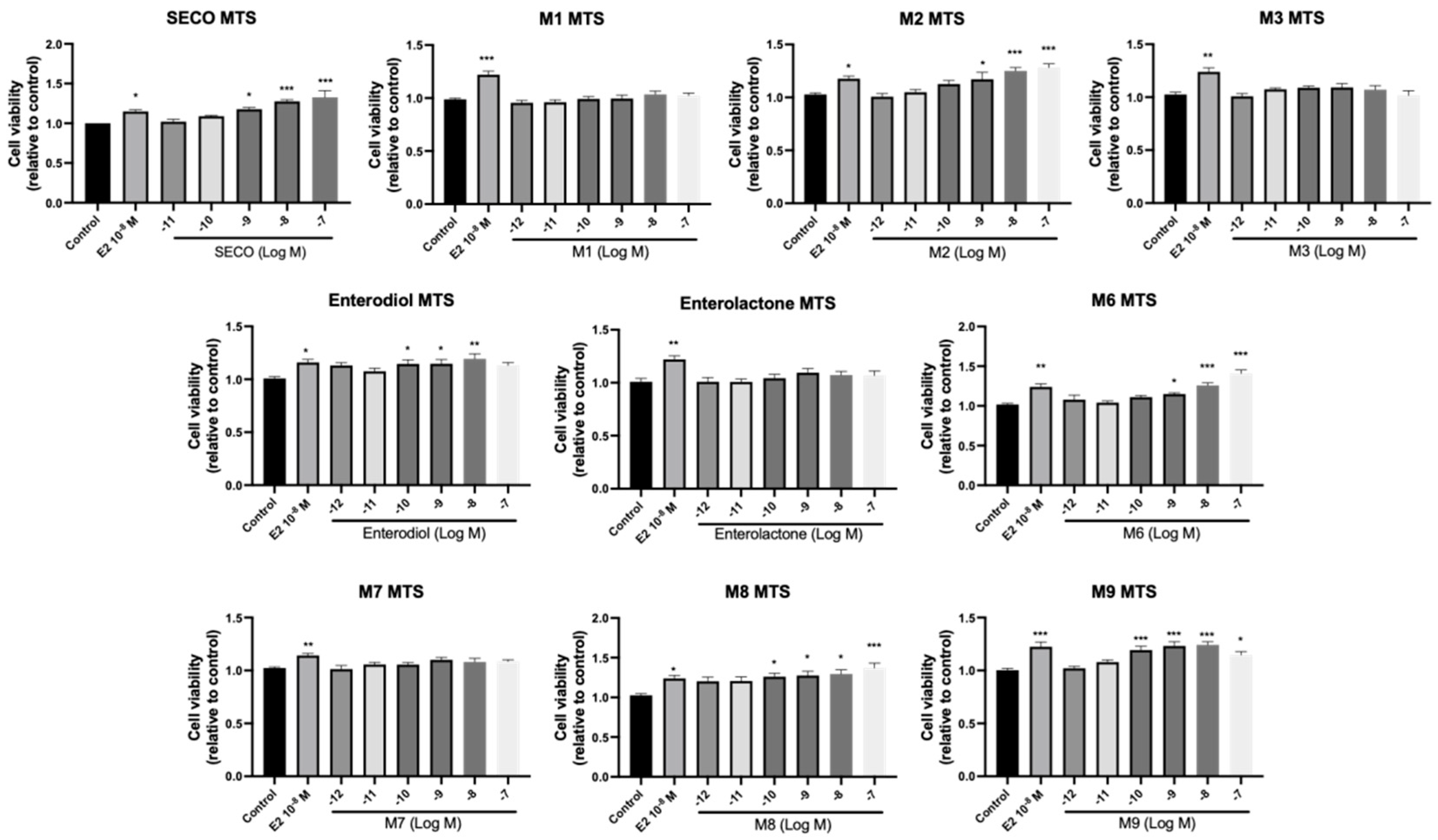
2.5. Osteogenic Effects of SECO and Microbial Transformation Products
2.6. Effects of Microbial Transformation Products on the Gene Expression of Bone Markers
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Human Fecal Sample Collection and Preparation
4.3. Microbial Transformation of SECO
4.4. Ultra-High-Performance Liquid Chromatography–Orbitrap–MS/MS Analysis
4.5. Data Processing and Metabolite Identification
4.6. Synthesis of Identified Metabolites
4.7. Culture of MC3T3-E1 and MG-63 Cells
4.8. Cell Availability Assay
4.9. Alkaline Phosphatase (ALP) Activity Assay
4.10. Real-Time Quantitative RT- PCR Analysis
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Lorentzon, M.; Johansson, H.; Harvey, N.; Liu, E.; Vandenput, L.; McCloskey, E.; Kanis, J. Osteoporosis and fractures in women: The burden of disease. Climacteric 2022, 25, 4–10. [Google Scholar] [PubMed]
- Bowring, C.; Francis, R. National Osteoporosis Society’s Position statement on hormone replacement therapy in the prevention and treatment of osteoporosis. Menopause Int. 2011, 17, 63–65. [Google Scholar] [CrossRef]
- Langer, R.; Hodis, H.; Lobo, R.; Allison, M. Hormone replacement therapy–where are we now? Climacteric 2021, 24, 3–10. [Google Scholar] [CrossRef]
- Cagnacci, A.; Venier, M. The controversial history of hormone replacement therapy. Medicina 2019, 55, 602. [Google Scholar] [CrossRef] [Green Version]
- Ranich, T.; Bhathena, S.J.; Velasquez, M.T. Protective effects of dietary phytoestrogens in chronic renal disease. J. Ren. Nutr. 2001, 11, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Adlercreutz, H.; Höckerstedt, K.; Bannwart, C.; Bloigu, S.; Hämäläinen, E.; Fotsis, T.; Ollus, A. Effect of dietary components, including lignans and phytoestrogens, on enterohepatic circulation and liver metabolism of estrogens and on sex hormone binding globulin (SHBG). J. Steroid Biochem. 1987, 27, 1135–1144. [Google Scholar]
- Landete Iranzo, J.M. Plant and mammalian lignans: A review of source, intake, metabolism, intestinal bacteria and health. Food Res. Int. 2012, 46, 410–424. [Google Scholar]
- Mazur, W. 11 Phytoestrogen content in foods. Baillière’s Clin. Endocrinol. Metab. 1998, 12, 729–742. [Google Scholar] [CrossRef]
- Kitts, D.; Yuan, Y.; Wijewickreme, A.; Thompson, L. Antioxidant activity of the flaxseed lignan secoisolariciresinol diglycoside and its mammalian lignan metabolites enterodiol and enterolactone. Mol. Cell. Biochem. 1999, 202, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Yu, X.; McClements, D.J.; Huang, Q.; Tang, H.; Yu, K.; Xia, X.; Chen, P.; Wang, X.T.; Deng, Q.C. Effect of flaxseed polyphenols on physical stability and oxidative stability of flaxseed oil-in-water nanoemulsions. Food Chem. 2019, 301, 125207. [Google Scholar] [CrossRef] [PubMed]
- Murkies, A.; Wilcox, G.; Davis, S. Phytoestrogens-review. J. Clin. Endocrinol. Metab. 1998, 83, 297–303. [Google Scholar]
- Thompson, L.U. Experimental studies on lignans and cancer. Bailliere’s Clin. Endocrinol. Metab. 1998, 12, 691–705. [Google Scholar] [CrossRef]
- Asad, B.; Khan, T.; Gul, F.Z.; Ullah, M.A.; Drouet, S.; Mikac, S.; Garros, L.; Ferrier, M.; Bose, S.; Munsch, T.; et al. Scarlet flax Linum grandiflorum (L.) in vitro cultures as a new source of antioxidant and anti-inflammatory lignans. Molecules 2021, 26, 4511. [Google Scholar]
- Adlercreutz, H.; Mazur, W.; Bartels, P.; Elomaa, V.-V.; Watanabe, S.; Wahala, K.; LandstoM, M.; Lundin, E.; Bergh, A.; Damber, J.E. Phytoestrogens and prostate disease. J. Nutr. 2000, 130, 658S–659S. [Google Scholar] [CrossRef] [Green Version]
- Kajla, P.; Sharma, A.; Sood, D.R. Flaxseed—A potential functional food source. J. Food Sci. Technol. 2015, 52, 1857–1871. [Google Scholar] [CrossRef] [Green Version]
- Bloedon, L.T.; Balikai, S.; Chittams, J.; Cunnane, S.C.; Berlin, J.A.; Rader, D.J.; Szapary, P.O. Flaxseed and cardiovascular risk factors: Results from a double blind, randomized, controlled clinical trial. J. Am. Coll. Nutr. 2008, 27, 65–74. [Google Scholar]
- Imran, M.; Ahmad, N.; Anjum, F.M.; Khan, M.K.; Mushtaq, Z.; Nadeem, M.; Hussain, S. Potential protective properties of flax lignan secoisolariciresinol diglucoside. Nutr. J. 2015, 14, 71. [Google Scholar] [CrossRef] [Green Version]
- Hafiz, H.; Commane, D.; Walton, G.; Jackson, K. Effect of Whole Flaxseed on Bone Turnover Markers and Gut Microbiota in Menopausal-Related Bone Loss. In Bioactive Compounds from Multifarious Natural Foods for Human Health; Apple Academic Press: Palm Bay, FL, USA, 2022; pp. 229–253. [Google Scholar]
- Xiao, H.H.; Chan, C.; Wong, K.; Kam-wah, D.; Chan, S.; Cooper, R. Lignans from Sambucus williasmii Hance against osteoporosis: A pharmacodynamic and pharmacokinetic study. Planta Medica 2015, 81, PX56. [Google Scholar] [CrossRef]
- Xiao, H.H.; Wong, M.S.; Yao, X.S. A Lignan-Rich Bioactive Fraction of Sambucus williamsii Hance Exerts Oestrogen-Like Bone Protective Effects in Aged Ovariectomized Rats and Osteoblastic Cells. In Nutritional Influences on Bone Health: 9th International Symposium; Springer: Berlin/Heidelberg, Germany, 2016; pp. 137–143. [Google Scholar]
- Peñalvo, J.L.; Nurmi, T.; Haajanen, K.; Al-Maharik, N.; Botting, N.; Adlercreutz, H. Determination of lignans in human plasma by liquid chromatography with coulometric electrode array detection. Anal. Biochem. 2004, 332, 384–393. [Google Scholar]
- Nurmi, T.; Voutilainen, S.; Nyyssönen, K.; Adlercreutz, H.; Salonen, J.T. Liquid chromatography method for plant and mammalian lignans in human urine. J. Chromatogr. B 2003, 798, 101–110. [Google Scholar] [CrossRef]
- Bannwart, C.; Adlercreutz, H.; Wähälä, K.; Brunow, G.; Hase, T. Detection and identification of the plant lignans lariciresinol, isolariciresinol and secoisolariciresinol in human urine. Clin. Chim. Acta 1989, 180, 293–301. [Google Scholar] [CrossRef]
- Setchell, K.; Lawson, A.; Mitchell, F.; Adlercreutz, H.; Kirk, D.; Axelson, M. Lignans in man and in animal species. Nature 1980, 287, 740–742. [Google Scholar] [CrossRef]
- Setchell, K.; Borriello, S.; Gordon, H.; Lawson, A.; Harkness, R.; Morgan, D. Lignan formation in man—Microbial involvement and possible roles in relation to cancer. Lancet 1981, 318, 4–7. [Google Scholar]
- Axelson, M.; Sjövall, J.; Gustafsson, B.; Setchell, K. Origin of lignans in mammals and identification of a precursor from plants. Nature 1982, 298, 659–660. [Google Scholar] [CrossRef]
- Mueller, S.O.; Simon, S.; Chae, K.; Metzler, M.; Korach, K.S. Phytoestrogens and their human metabolites show distinct agonistic and antagonistic properties on estrogen receptor α (ERα) and ERβ in human cells. Toxicol. Sci. 2004, 80, 14–25. [Google Scholar] [PubMed] [Green Version]
- Brooks, J.D.; Thompson, L.U. Mammalian lignans and genistein decrease the activities of aromatase and 17β-hydroxysteroid dehydrogenase in MCF-7 cells. J. Steroid Biochem. Mol. Biol. 2005, 94, 461–467. [Google Scholar]
- Jacobs, M.N.; Nolan, G.T.; Hood, S.R. Lignans, bacteriocides and organochlorine compounds activate the human pregnane X receptor (PXR). Toxicol. Appl. Pharmacol. 2005, 209, 123–133. [Google Scholar]
- Xiao, H.H.; Sham, T.T.; Chan, C.; Li, M.H.; Chen, X.; Wu, Q.C.; Mok, D.K.W.; Yao, X.S.; Wong, M.S. A metabolomics study on the bone protective effects of a lignan-rich fraction from Sambucus williamsii Ramulus in aged rats. Front. Pharmacol. 2018, 9, 932. [Google Scholar]
- Xiao, H.H.; Dai, Y.; Wan, H.Y.; Wong, M.S.; Yao, X.S. Bone-protective effects of bioactive fractions and ingredients in Sambucus williamsii HANCE. Br. J. Nutr. 2011, 106, 1802–1809. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.H.; Zhu, Y.X.; Lu, L.; Zhou, L.P.; Poon, C.C.; Chan, C.O.; Wang, L.J.; Cao, S.; Yu, W.X.; Wong, K.Y.; et al. The Lignan-Rich Fraction from Sambucus williamsii Hance Exerts Bone Protective Effects via Altering Circulating Serotonin and Gut Microbiota in Rats. Nutrients 2022, 14, 4718. [Google Scholar]
- Quartieri, A.; García-Villalba, R.; Amaretti, A.; Raimondi, S.; Leonardi, A.; Rossi, M. Detection of novel metabolites of flaxseed lignans in vitro and in vivo. Mol. Nutr. Food Res. 2016, 60, 1590–1601. [Google Scholar] [CrossRef]
- Setchell, K.D.; Brown, N.M.; Zimmer-Nechemias, L.; Wolfe, B.; Jha, P.; Heubi, J.E. Metabolism of secoisolariciresinol-diglycoside the dietary precursor to the intestinally derived lignan enterolactone in humans. Food Funct. 2014, 5, 491–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemeyer, H.B.; Honig, D.M.; Kulling, S.E.; Metzler, M. Studies on the metabolism of the plant lignans secoisolariciresinol and matairesinol. J. Agric. Food Chem. 2003, 51, 6317–6325. [Google Scholar] [CrossRef]
- Wishart, D.S.; Tian, S.; Allen, D.; Oler, E.; Peters, H.; Lui, V.W. BioTransformer 3.0—A web server for accurately predicting metabolic transformation products. Nucleic Acids Res. 2022, 50, W115–W123. [Google Scholar] [CrossRef]
- Senizza, A.; Rocchetti, G.; Mosele, J.I.; Patrone, V.; Callegari, M.L.; Morelli, L. Lignans and gut microbiota: An interplay revealing potential health implications. Molecules 2020, 25, 5709. [Google Scholar] [PubMed]
- Orimo, H. The mechanism of mineralization and the role of alkaline phosphatase in health and disease. J. Nippon. Med. Sch. 2010, 77, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Ho, M.X.; Poon, C.C.W.; Wong, K.C.; Qiu, Z.C.; Wong, M.S. Icariin, but not genistein, exerts osteogenic and anti-apoptotic effects in osteoblastic cells by selective activation of non-genomic ERα signaling. Front. Pharmacol. 2018, 9, 474. [Google Scholar]
- Tham, D.M.; Gardner, C.D.; Haskell, W.L. Potential health benefits of dietary phytoestrogens: A review of the clinical, epidemiological, and mechanistic evidence. J. Clin. Endocrinol. Metab. 1998, 83, 2223–2235. [Google Scholar]
- Wang, L.Q.; Meselhy, M.R.; Li, Y.; Qin, G.W.; Hattori, M. Human intestinal bacteria capable of transforming secoisolariciresinol diglucoside to mammalian lignans, enterodiol and enterolactone. Chem. Pharm. Bull. 2000, 48, 1606–1610. [Google Scholar] [CrossRef] [Green Version]
- Clavel, T.; Henderson, G.; Engst, W.; Dore, J.; Blaut, M. Phylogeny of human intestinal bacteria that activate the dietary lignan secoisolariciresinol diglucoside. Fems Microbiol. Ecol. 2006, 55, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Clavel, T.; Lippman, R.; Gavini, F.; Dore, J.; Blaut, M. Clostridium saccharogumia sp nov and Lactonifactor longoviformis gen. nov., sp nov., two novel human faecal bacteria involved in the conversion of the dietary phytoestrogen secoisolariciresinol diglucoside. Syst. Appl. Microbiol. 2007, 30, 16–26. [Google Scholar] [CrossRef]
- Jin, J.S.; Kakiuchi, J.; Hattori, M. Enantioselective oxidation of enterodiol to enterolactone by human intestinal bacteria. Biol. Pharm. Bull. 2007, 30, 2204–2206. [Google Scholar] [CrossRef] [Green Version]
- Struijs, K.; Vincken, J.P.; Gruppen, H. Bacterial conversion of secoisolariciresinol and anhydrosecoisolariciresinol. J. Appl. Microbiol. 2009, 107, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.S.; Zhao, Y.F.; Nakamura, N.; Akao, T.; Kakiuchi, N.; Min, B.S. Enantioselective dehydroxylation of enterodiol and enterolactone precursors by human intestinal bacteria. Biol. Pharm. Bull. 2007, 30, 2113–2119. [Google Scholar] [CrossRef] [Green Version]
- Corona, G.; Kreimes, A.; Barone, M.; Turroni, S.; Brigidi, P.; Keleszade, E. Impact of lignans in oilseed mix on gut microbiome composition and enterolignan production in younger healthy and premenopausal women: An in vitro pilot study. Microb. Cell Factories 2020, 19, 82. [Google Scholar]
- Peiroten, A.; Gaya, P.; Alvarez, I.; Bravo, D.; Landete, J.M. Influence of different lignan compounds on enterolignan production by Bifidobacterium and Lactobacillus strains. Int. J. Food Microbiol. 2019, 289, 17–23. [Google Scholar] [CrossRef]
- Bravo, D.; Peirotén, Á.; Álvarez, I.; Landete, J.M. Phytoestrogen metabolism by lactic acid bacteria: Enterolignan production by Lactobacillus salivarius and Lactobacillus gasseri strains. J. Funct. Foods 2017, 37, 373–378. [Google Scholar]
- Feng, J.; Shi, Z.L.; Ye, Z.M. Effects of metabolites of the lignans enterolactone and enterodiol on osteoblastic differentiation of MG-63 cells. Biol. Pharm. Bull. 2008, 31, 1067–1070. [Google Scholar] [CrossRef] [Green Version]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef]
- Sun, L.J.; Li, C.; Wen, X.H.; Guo, L.; Guo, Z.F.; Liao, L.Q. Icariin stimulates hFOB 1.19 osteoblast proliferation and differentiation via OPG/RANKL mediated by the estrogen receptor. Curr. Pharm. Biotechnol. 2021, 22, 168–175. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolites | Formula | +ESI | −ESI | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Rt (min) | m/z | Adduct | MS2 Fragments (m/z) | Rt (min) | m/z | Adduct | MS2 Fragments (m/z) | ||
| SECO | C20H26O6 | 4.49 | 345.169 | [M − H2O + H]+ | 295.1319, 263.1059, 235.1111, 189.0904, 137.0593 | 4.49 | 361.1657 | [M − H]− | 209.0815, 179.0705, 165.0549, 136.0520, 122.0364 |
| M1 | C19H24O6 | 4.25 | 313.143 | [M − 2H2O + H]+ | 295.1332, 263.1066, 235.1113, 189.0905, 149.0592, 137.0435 | 4.24 | 347.1500 | [M − H]− | 209.0799, 179.0694, 165.0542, 136.0525, 122.0360 |
| M2 | C18H22O6 | 4.04 | 299.127 | [M − 2H2O + H]+ | 263.1064, 235.1115, 149.0590 | 4.04 | 315.1238 | [M − H2O − H]− | 122.0360 |
| M3 | C18H22O5 | 4.33 | 283.133 | [M − 2H2O + H]+ | 189.0905, 137.0589 | 4.33 | 317.1394 | [M − H]− | 122.0369, 107.0484 |
| M4 (Enterodiol) | C18H22O4 | 4.68 | 303.159 | [M + H]+ | 159.0800, 133.0644, 107.0489 | 4.69 | 301.1445 | [M − H]− | 107.0494 |
| M5 (Enterolactone) | C18H18O4 | 5.40 | 299.127 | [M + H]+ | 263.1059, 235.1108, 133.0640, 107.0484 | 5.40 | 297.1132 | [M − H]− | 165.0549, 107.0494 |
| M6 | C18H18O2 | 4.70 | 267.138 | [M + H]+ | N.D | N.D | N.D | N.D | N.D |
| M7 | C20H24O6 | N.D | N.D | N.D | N.D | 5.24 | 343.1551 | [M − H]− | N.D |
| M8 | C19H21O5 | 4.62 | 297.149 | [M − 2H2O + H]+ | N.D | N.D | N.D | N.D | N.D |
| M9 | C18H20O4 | 4.95 | 265.123 | [M − 2H2O + H]+ | 263.1052, 133.0639, 159.0704, 107.0489 | 4.95 | 299.1289 | [M − H]− | 107.0495 |
| Metabolites | OPG | RANKL | OPG/RANKL |
|---|---|---|---|
| M1 |  |  |  |
| M2 |  |  |  |
| M3 |  |  |  |
| M4 (Enterodiol) |  |  |  |
| M5 (Enterolactone) |  |  |  |
| M6 |  |  |  |
| M7 |  |  |  |
| M8 |  |  |  |
| M9 |  |  |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, W.-X.; Tang, H.-H.; Ye, J.-J.; Xiao, H.-H.; Lam, C.-Y.; Shum, T.-F.; Sun, Z.-K.; Li, Y.-Z.; Zang, X.-Y.; Du, W.-C.; et al. Identification of the Microbial Transformation Products of Secoisolariciresinol Using an Untargeted Metabolomics Approach and Evaluation of the Osteogenic Activities of the Metabolites. Molecules 2023, 28, 5742. https://doi.org/10.3390/molecules28155742
Yu W-X, Tang H-H, Ye J-J, Xiao H-H, Lam C-Y, Shum T-F, Sun Z-K, Li Y-Z, Zang X-Y, Du W-C, et al. Identification of the Microbial Transformation Products of Secoisolariciresinol Using an Untargeted Metabolomics Approach and Evaluation of the Osteogenic Activities of the Metabolites. Molecules. 2023; 28(15):5742. https://doi.org/10.3390/molecules28155742
Chicago/Turabian StyleYu, Wen-Xuan, Hok-Him Tang, Jun-Jie Ye, Hui-Hui Xiao, Chung-Yan Lam, Tim-Fat Shum, Zhi-Kang Sun, Yuan-Zhen Li, Xin-Yu Zang, Wen-Chao Du, and et al. 2023. "Identification of the Microbial Transformation Products of Secoisolariciresinol Using an Untargeted Metabolomics Approach and Evaluation of the Osteogenic Activities of the Metabolites" Molecules 28, no. 15: 5742. https://doi.org/10.3390/molecules28155742
APA StyleYu, W. -X., Tang, H. -H., Ye, J. -J., Xiao, H. -H., Lam, C. -Y., Shum, T. -F., Sun, Z. -K., Li, Y. -Z., Zang, X. -Y., Du, W. -C., Zhang, J. -P., Kong, T. -H., Zhou, L. -P., Chiou, J. -C., Kung, C. -F., Mok, K. -W., Hu, J., & Wong, M. -S. (2023). Identification of the Microbial Transformation Products of Secoisolariciresinol Using an Untargeted Metabolomics Approach and Evaluation of the Osteogenic Activities of the Metabolites. Molecules, 28(15), 5742. https://doi.org/10.3390/molecules28155742