
The Copper(II)-Thiodiacetate (tda) Chelate as Efficient Receptor of N9-(2-Hydroxyethyl)Adenine (9heade): Synthesis, Molecular and Crystal Structures, Physical Properties and DFT Calculations of [Cu(tda)(9heade)(H2O)]·2H2O
 ,
,  ,
,  , and
, and
Abstract
:1. Introduction
2. Results and Discussions
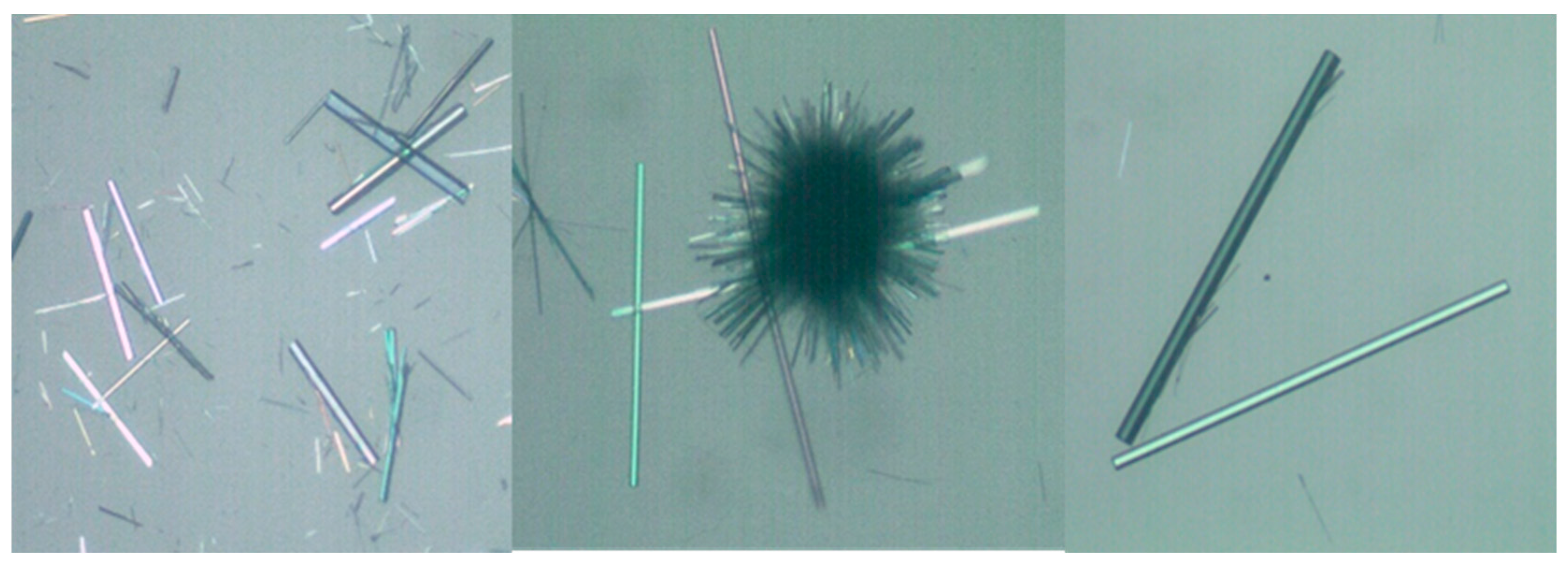
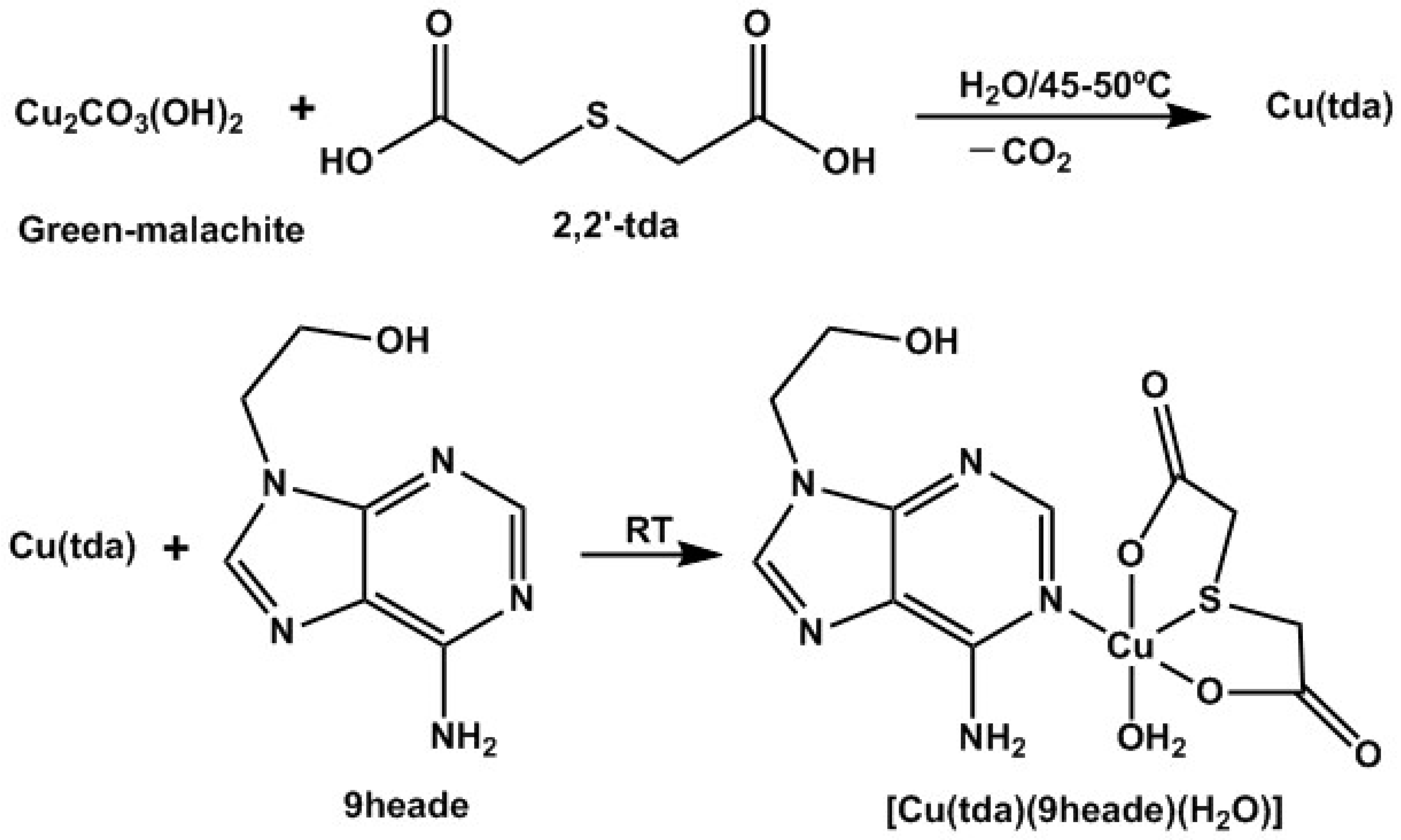
2.1. About the Strategy of Synthesis
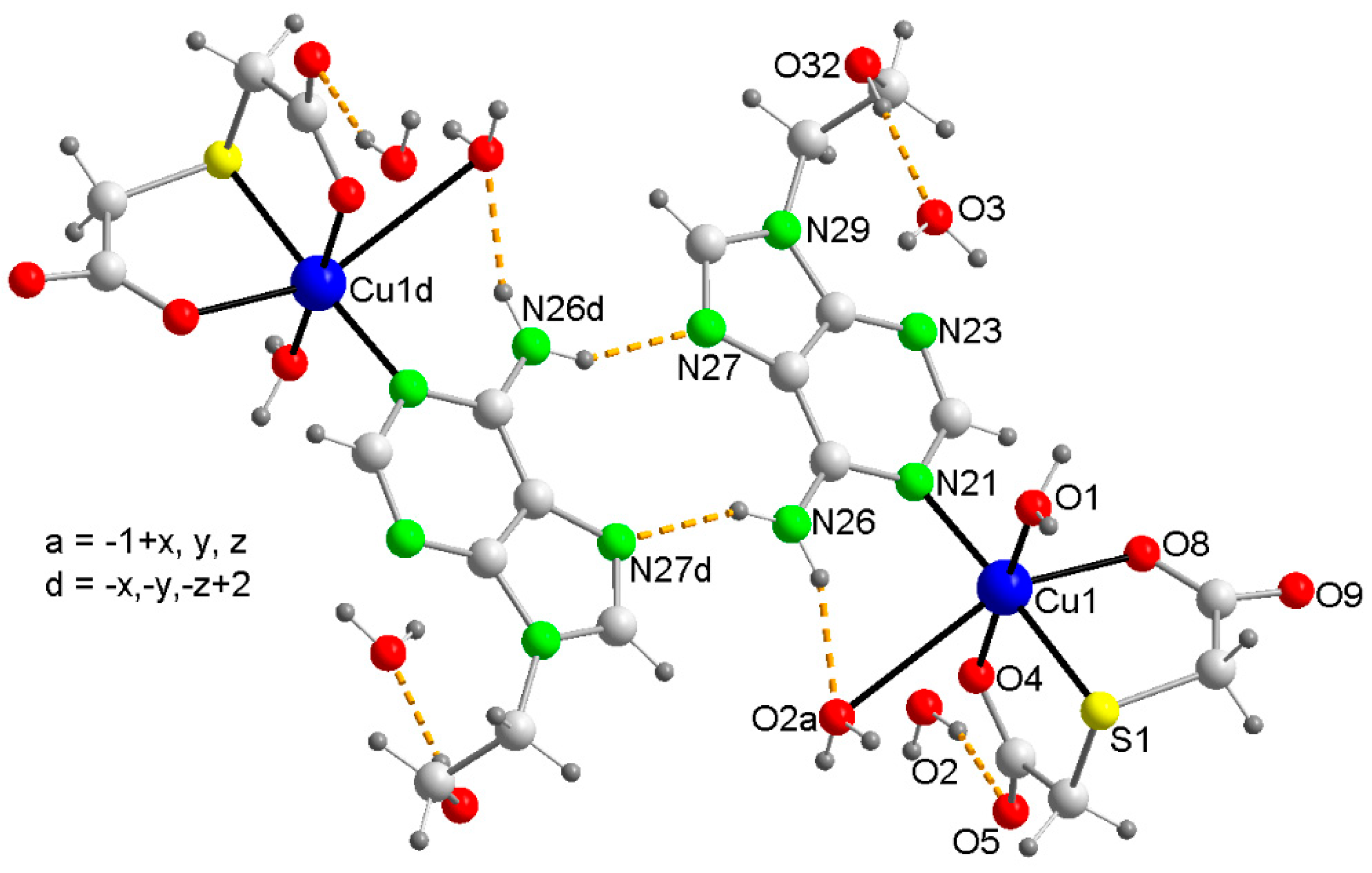
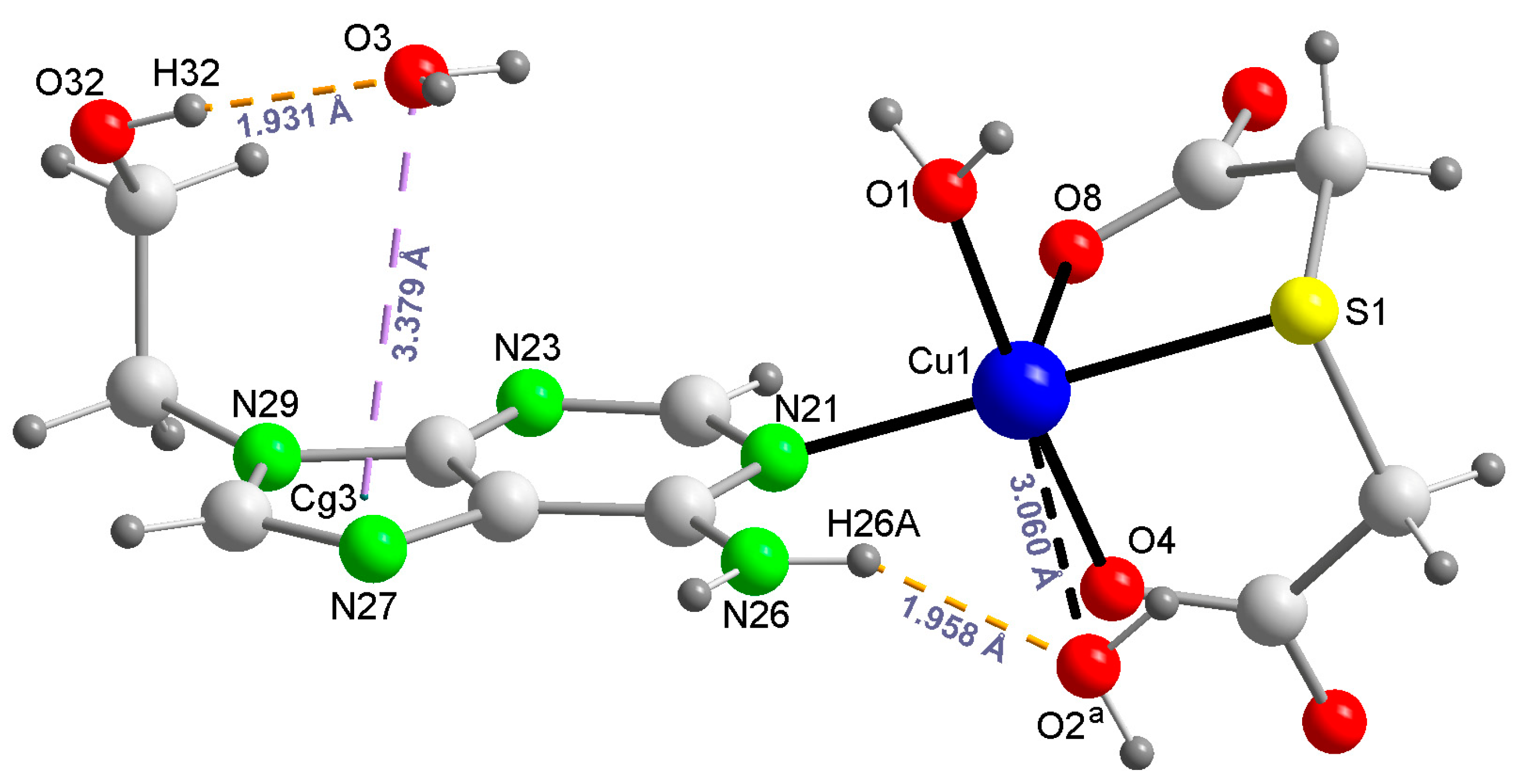
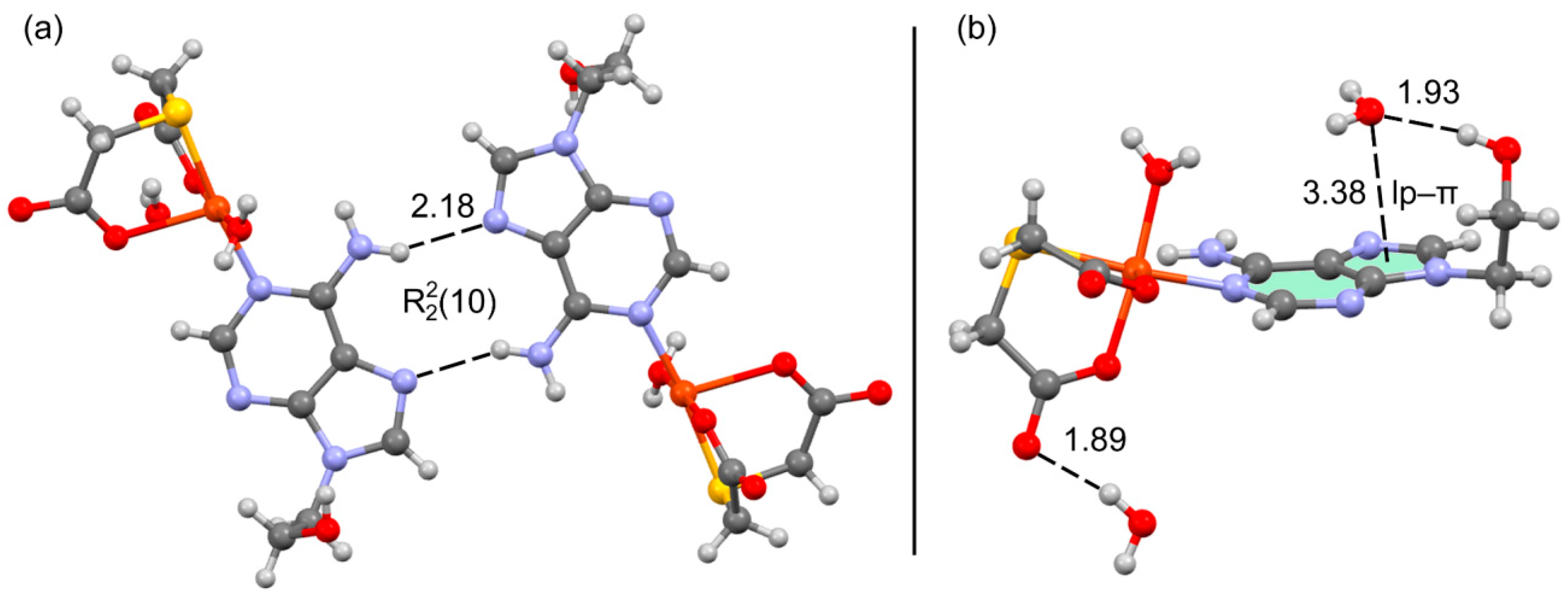
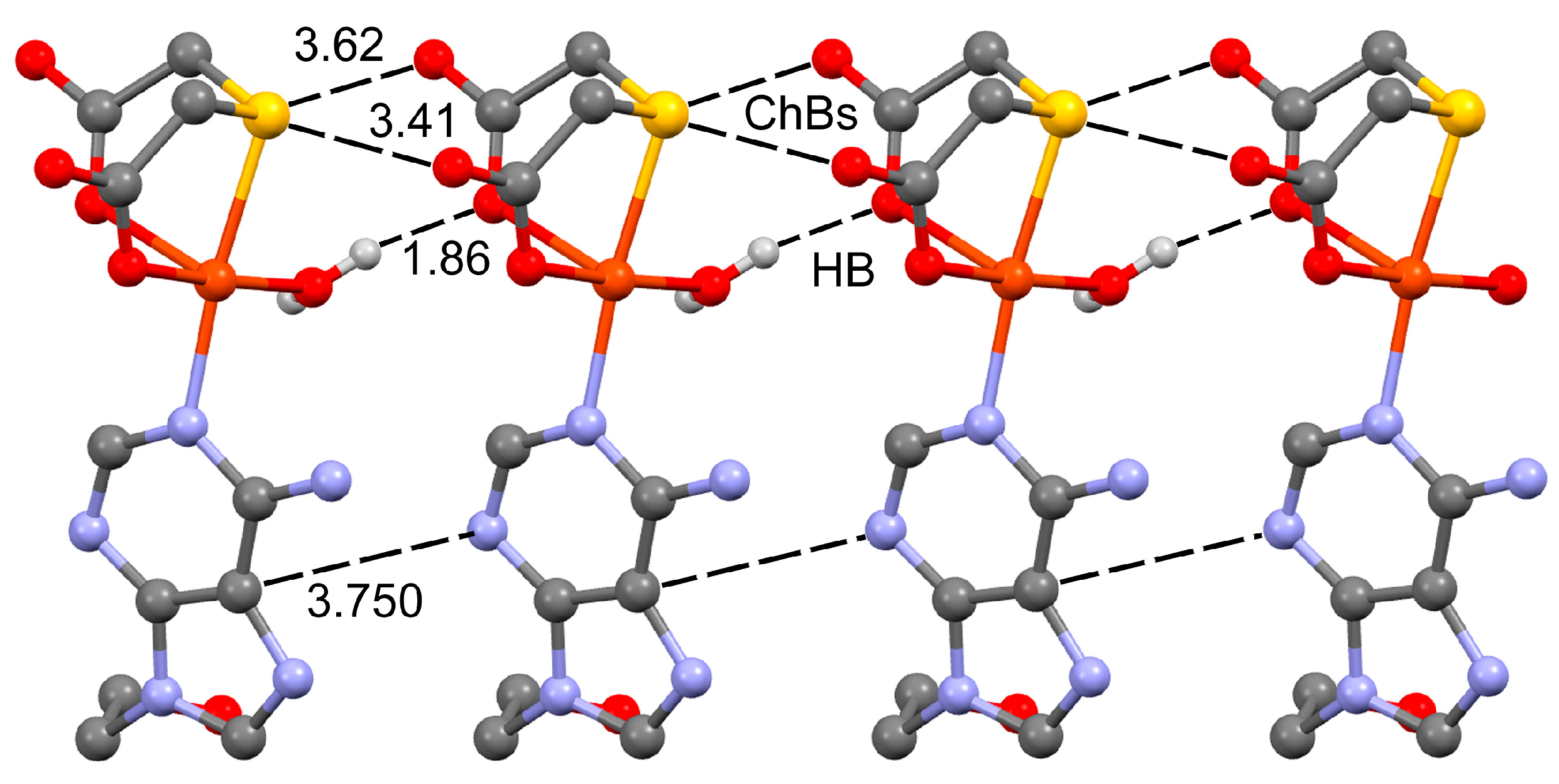
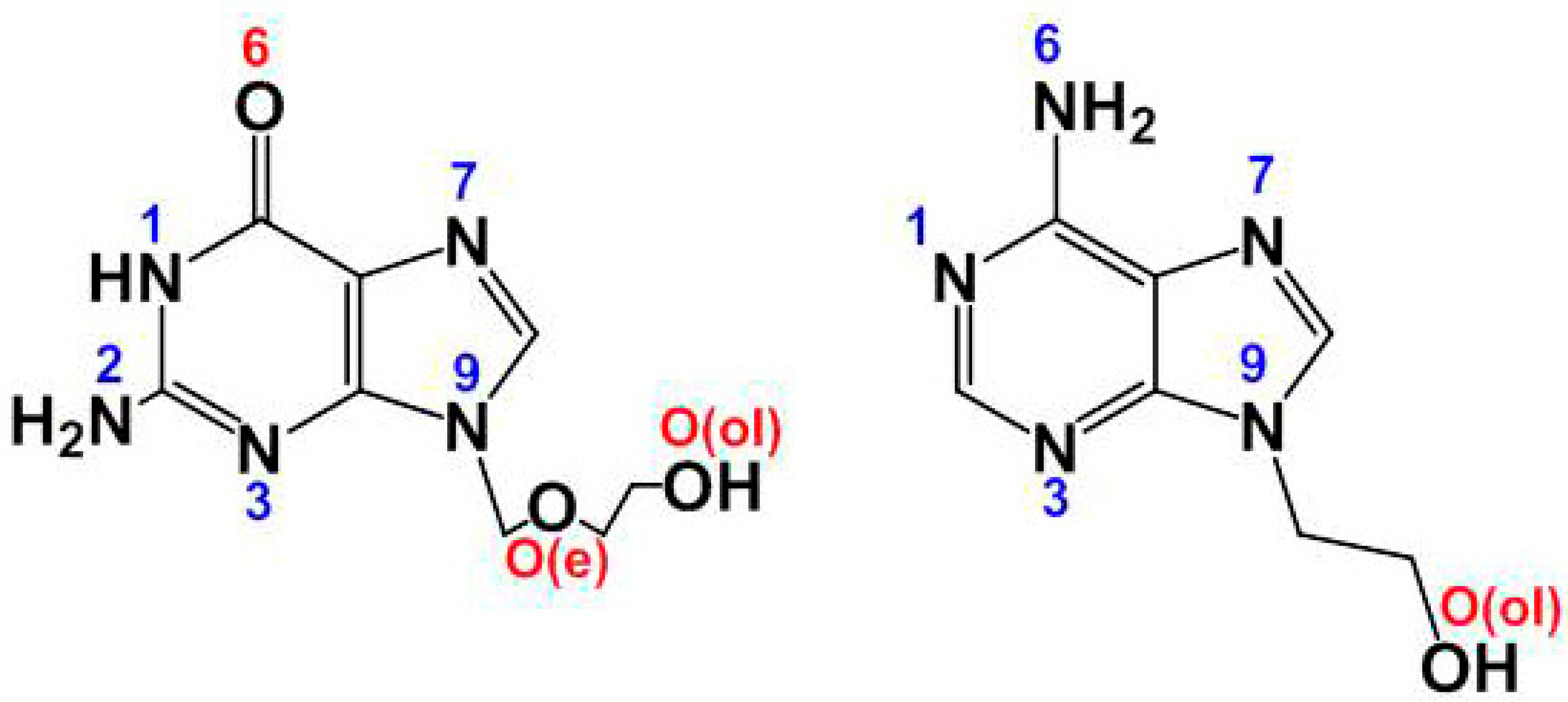
2.2. Molecular and Crystal Structure of Compound 1 and Their Relevant Significance for Molecular and Supra-Molecular Recognition
2.3. Molecular and Crystal Structures of Compound [Cu(tda)(9heade)(H2O)]·2H2O (1)
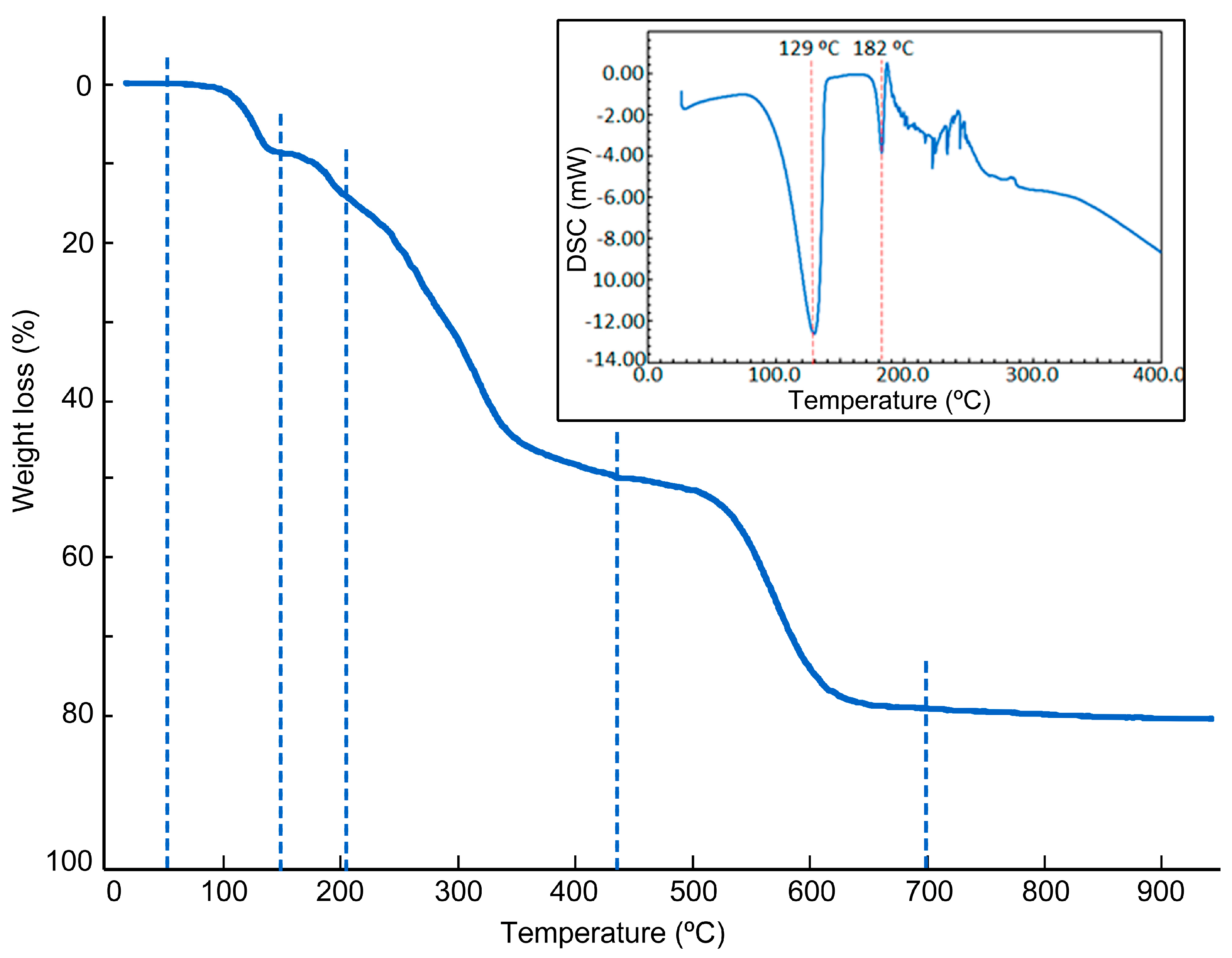
2.4. Physical Properties
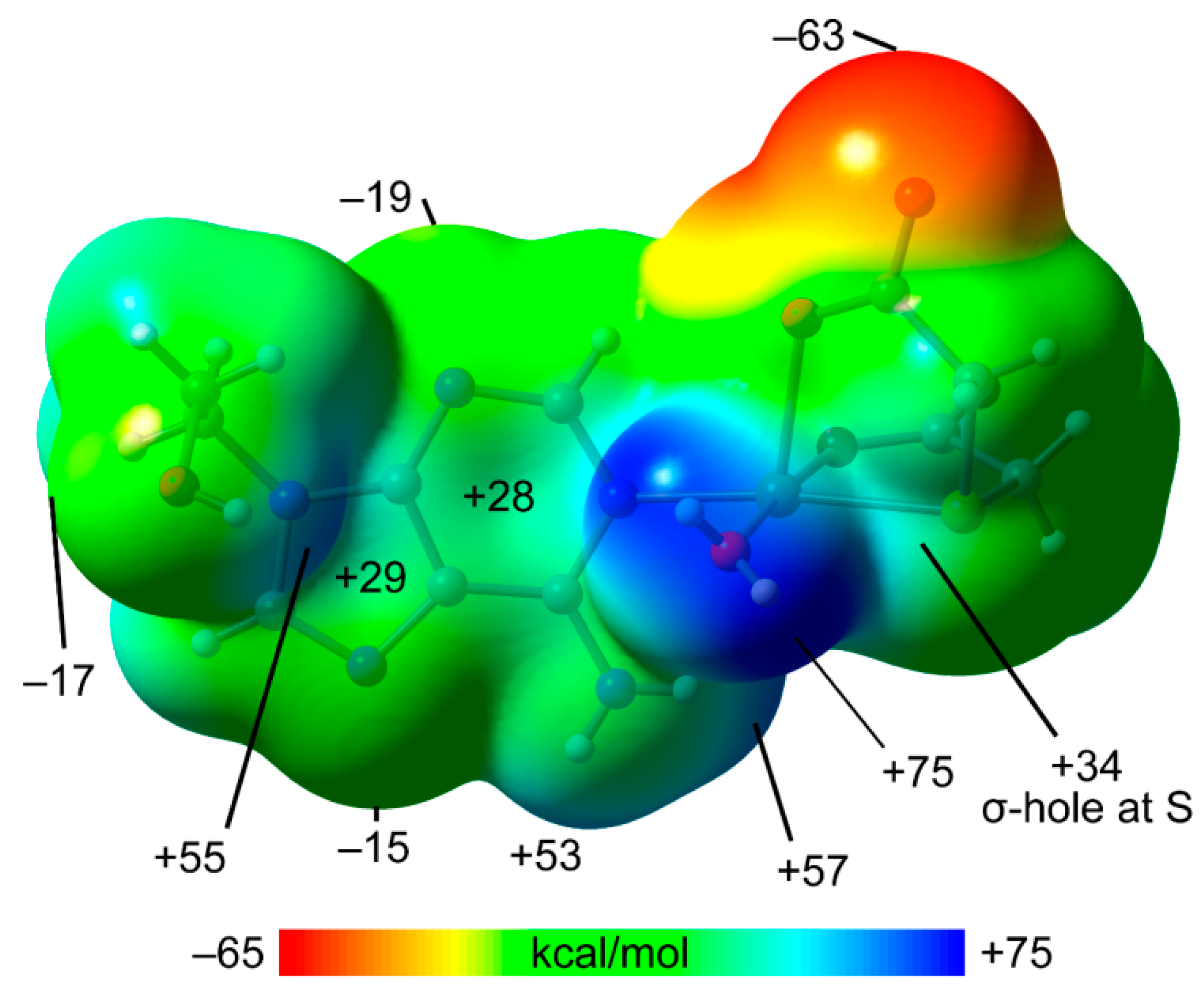
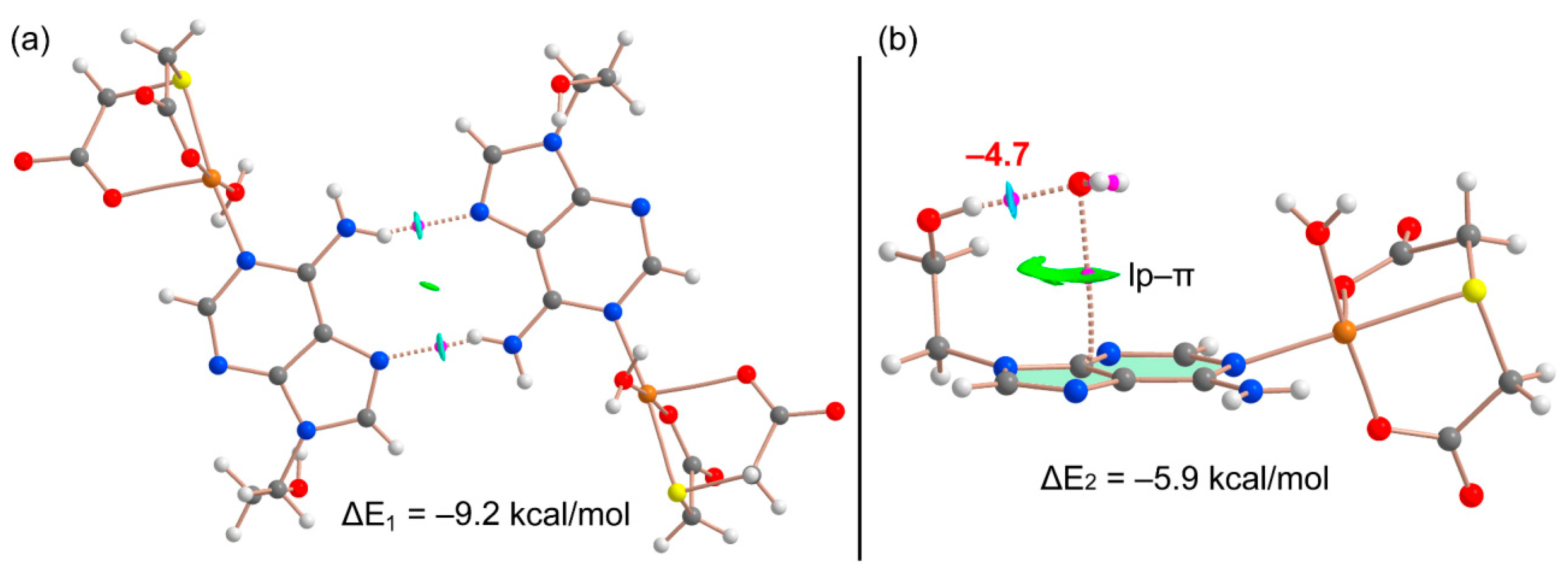
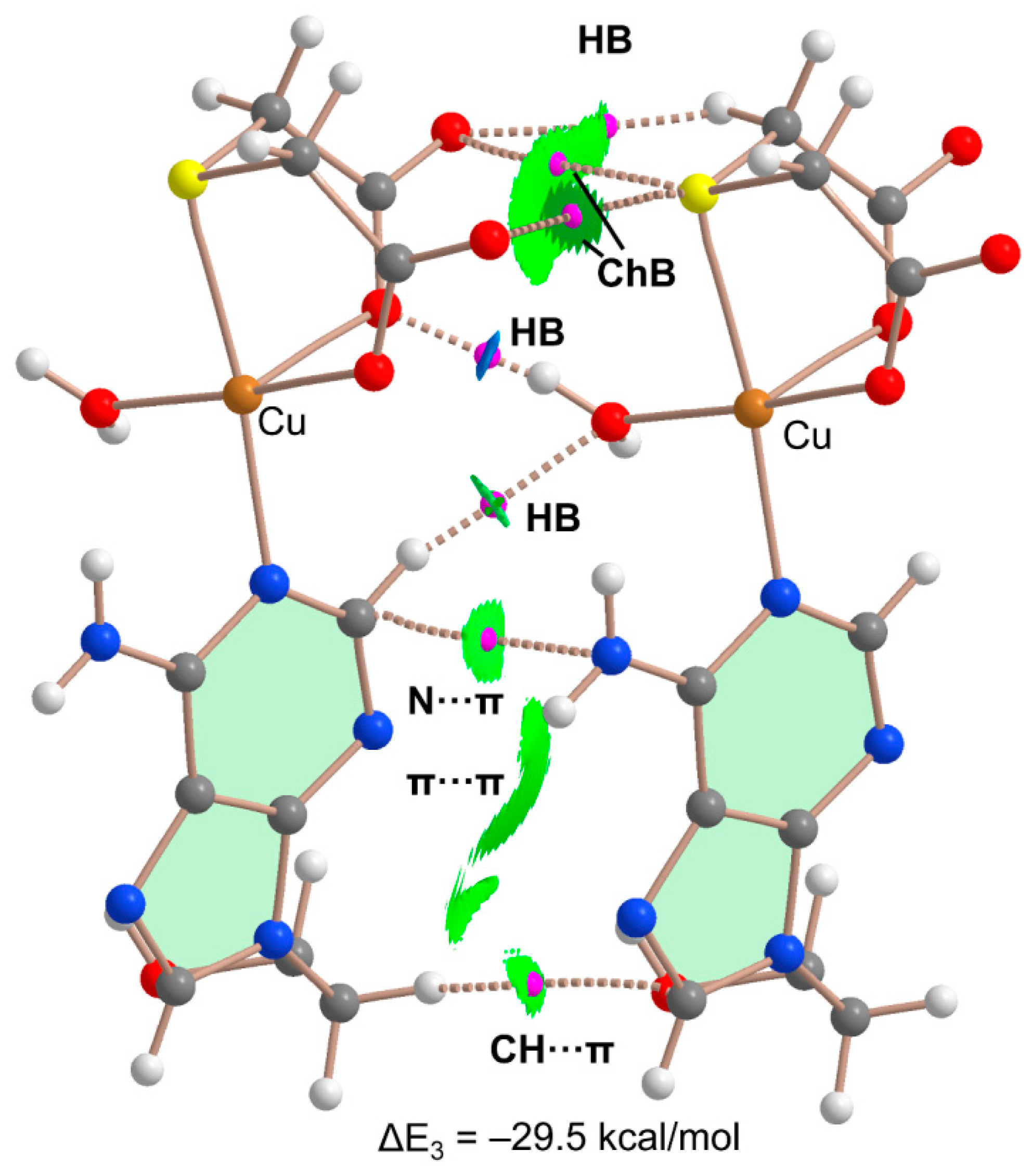
2.5. DFT Calculations
3. Concluding Remarks
4. Materials and Methods
4.1. Reagents and Synthesis of Compound 1
4.2. Physical Measurements
4.3. Crystallography
4.4. Computational Details
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Lehn, J.M. Perspectives in Supramolecular Chemistry-From Molecular Recognition towards Molecular Information Processing and Self-Organization. Angew. Chem. Int. Ed. 1990, 29, 1304–1319. [Google Scholar] [CrossRef]
- Persch, E.; Dumele, O.; Diederich, F. Molecular Recognition in Chemical and Biological Systems. Angew. Chem. Int. Ed. 2015, 54, 3290–3327. [Google Scholar] [CrossRef] [PubMed]
- Rebek, J. Molecular Recognition with Model Systems. Angew. Chem. Int. Ed. 1990, 29, 245–255. [Google Scholar] [CrossRef]
- Belmont-Sánchez, J.C.; Choquesillo-Lazarte, D.; Navarrete-Casas, R.; Frontera, A.; Castiñeiras, A.; Niclós-Gutiérrez, J.; Matilla-Hernández, A. A tetranuclear Ni(II)-cubane cluster molecule build by four µ3-O-methanolate (MeO) ligands, externally cohesive by four unprecedented bridging µ2-N7,O6-acyclovirate (acv-H) anions. Crystals 2023, 13, 7. [Google Scholar] [CrossRef]
- Domínguez-Martín, A.; Brandi-Blanco, M.P.; Matilla-Hernández, A.; El Bakkali, H.; Nurchi, V.M.; González-Párez, J.M.; Castiñeiras, A.; Niclós-Gutiérrez, J. Unravelling the versatile metal binding modes of adenine: Looking at the molecular recognition patterns of deaza- and aza-adenines in mixed ligand metal complexes. Coord. Chem. Rev. 2013, 257, 2814–2838. [Google Scholar] [CrossRef]
- Amo-Ochoa, P.; Zamora, F. Coordination polymers with nucleobases: From structural aspects to potential applications. Coord. Chem. Rev. 2014, 276, 34–58. [Google Scholar] [CrossRef]
- Brandi-Blanco, M.P.; Choquesillo-Lazarte, D.; Domínguez-Martín, A.; Matilla-Hernández, A.; González-Pérez, J.M.; Castiñeiras, A.; Niclós-Gutiérrez, J. Molecular recognition modes between adenine or adeniniun(1+) ion and binary MII(pdc) chelates (M = Co-Zn; pdc = pyridine-2,6-dicarboxylate(2-) ion). J. Inorg. Biochem. 2013, 127, 211–219. [Google Scholar] [CrossRef]
- Khaled Hassanein, K.; Castillo, O.; Gómez-García, C.J.; Zamora, F.; Amo-Ochoa, P. Asymmetric and symmetric dicopper(II) paddle-wheel units with modified nucleobases. Cryst. Growth Des. 2015, 15, 5485–5494. [Google Scholar] [CrossRef]
- Pérez-Toro, I.; Domínguez-Martín, A.; Choquesillo-Lazarte, D.; González-Pérez, J.M.; Castiñeiras, A.; Niclós-Gutiérrez, J. Highest reported denticity of a synthetic nucleoside in the unprecedented tetradentate mode of acyclovir. Cryst. Growth Des. 2018, 18, 4282–4286. [Google Scholar] [CrossRef]
- Hammud, H.H.; Travis Holman, K.; Al-Noaimi, M.; Sadiq Sheikh, N.; Ghannoum, A.M.; Bouhadir, K.H.; Masoud, M.S.; Karnati, R.K. Structures of selected transition metal complexes with 9-(2-hydroxyethyl)adenine: Potentiometric complexation and DFT studies. J. Mol. Struct. 2020, 1205, 127548. [Google Scholar] [CrossRef]
- Ruiz-González, N.; García-Rubiño, M.E.; Domínguez-Martín, A.; Choquesillo-Lazarte, D.; Franconetti, A.; Frontera, A.; Castiñeiras, A.; González-Pérez, J.M.; Niclós-Gutiérrez, J. Molecular and supra-molecular recognition patterns in ternary copper(II) or zinc(II) complexes with selected rigid-planar chelators and a synthetic adenine-nucleoside. J. Inorg. Biochem. 2020, 203, 110920. [Google Scholar] [CrossRef]
- García-Rubiño, M.E.; Matilla-Hernández, A.; Frontera, A.; Lezama, L.; Niclós-Gutiérrez, J.; Choquesillo-Lazarte, D. Dicopper(II)-EDTA Chelate as a bicephalic receptor model for a synthetic adenine nucleoside. Pharmaceuticals 2021, 14, 426. [Google Scholar] [CrossRef] [PubMed]
- Belmont-Sánchez, J.C.; García-Rubiño, M.E.; Frontera, A.; Matilla-Hernández, A.; Castiñeiras, A.; Niclós-Gutiérrez, J. Novel Cd(II) coordination polymers afforded with EDTA or trans-1,2-CDTA chelators and imidazole, adenine, or 9-(2-hydroxyethyl)adenine coligands. Crystals 2020, 10, 391. [Google Scholar] [CrossRef]
- Sushrutha, S.R.; Hota, R.; Natarajan, S. Adenine-Based Coordination Polymers: Synthesis, Structure, and Properties. Eur. J. Inorg. Chem. 2016, 2016, 2962–2964. [Google Scholar] [CrossRef]
- Pimentel, G.C.; McClellan, A.L. The Hydrogen Bond; W.H. Freeman & Co.: San Francisco, CA, USA, 1960. [Google Scholar]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond. In Structural Chemistry and Biology; International Union of Crystallography, Oxford Science Publications: Oxford, UK, 1999. [Google Scholar]
- Gilli, G.; Gilli, P. The Nature of the Hydrogen Bond: Outline of a Comprehensive Hydrogen Bond Theory; International Union of Crystallography, Oxford Science Publications: Oxford, UK, 2009. [Google Scholar]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. The bright future of unconventional σ/π-hole interactions. ChemPhysChem 2015, 16, 2496–2517. [Google Scholar] [CrossRef] [PubMed]
- Aakeröy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the chalcogen bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef]
- Priimagi, A.; Cavallo, G.; Metrangolo, P.; Resnati, G. The halogen bond in the design of functional supramolecular materials: Recent advances. Acc. Chem. Res. 2013, 46, 2686–2695. [Google Scholar] [CrossRef] [Green Version]
- Frontera, A.; Bauzá, A. Metal coordination enhances chalcogen bonds: CSD survey and theoretical calculations. Int. J. Mol. Sci. 2022, 23, 4188. [Google Scholar] [CrossRef]
- Kopel, P.; Travnicek, Z.; Marek, J.; Korabik, M.; Mrozinski, J. Syntheses and properties of binuclear copper (II) mixed-ligand complexes involving thiodiglycolic acid.: The crystal structures of [(phen)2Cu(μ-tdga)Cu(phen)](NO3)2·5H2O and [(H2O)(pmdien)Cu(μ-tdga)Cu(pmdien)(H2O)](ClO4)2. Polyhedron 2003, 22, 411–418. [Google Scholar] [CrossRef]
- Patel, D.K.; Choquesillo-Lazarte, D.; Domínguez-Martín, A.; Brandi-Blanco, M.P.; González-Pérez, J.M.; Castiñeiras, A.; Niclós-Gutiérrez, J. Chelating Ligand Conformation Driving the Hypoxanthine Metal Binding Patterns. Inorg. Chem. 2011, 50, 10549–10551. [Google Scholar] [CrossRef]
- Abbaszadeh, A.; Safari, N.; Amani, V.; Notash, B.; Raei, F.; Eftekhar, F. Mononuclear and Dinuclear Copper(II) Complexes Containing N, O and S Donor Ligands: Synthesis, Characterization, Crystal Structure Determination and Antimicrobial Activity of [Cu(phen)(tda)]·2H2O and [(phen)2Cu(µ-tda)Cu(phen)](ClO4)2.1·5H2O. Iran J. Chem. Chem. Eng. 2014, 33, 1–13. [Google Scholar] [CrossRef]
- Baggio, R.; Garland, M.T.; Manzur, J.; Peña, O.; Perec, M.; Spodine, E.; Vega, A. A dinuclear copper(II) complex involving monoatomic O-carboxylate bridging and Cu–S(thioether) bonds: [Cu(tda)(phen)]2·H2tda (tda-thiodiacetate, phen-phenanthroline). Inorg. Chim. Acta 1999, 286, 74–79. [Google Scholar] [CrossRef]
- Shahwaz Ahmad, M.; Khalid, M.; Shahnawaz Khan, M.; Shahid, M.; Ahmad, M.; Monika; Ansaric, A.; Ashafaq, M. Exploring catecholase activity in dinuclear Mn(II) and Cu(II) complexes: An experimental and theoretical approach. New J. Chem. 2020, 44, 7998–8009. [Google Scholar] [CrossRef]
- Bonomo, R.P.; Rizzarelli, E.; Bresciani-Pahor, N.; Nardin, G. Properties and X-ray crystal structures of copper(II) mixed complexes with thiodiacetate and 2,2′-bipyridyl or 2,2′:6′.2″-terpyridyl. J. Chem. Soc. Dalton Trans. 1982, 4, 681–685. [Google Scholar] [CrossRef]
- Alarcón-Payer, C.; Pivetta, T.; Choquesillo-Lazarte, D.; González-Pérez, J.M.; Crisponi, G.; Castiñeiras, A.; Niclós-Gutiérrez, J. Thiodiacetato-copper(II) chelates with or without N-heterocyclic donor ligands: Molecular and/or crystal structures of [Cu(tda)]n, [Cu(tda)(Him)2(H2O)] and [Cu(tda)(5Mphen)]·2H2O (Him = imidazole, 5Mphen = 5-methyl-1,10-phenanthroline). Inorg. Chim. Acta 2005, 358, 1918–1926. [Google Scholar] [CrossRef]
- Gu, N.X.; Oyala, P.H.; Peters, J.C. H2 Evolution from a Thiolate Ni(III) Hydride. J. Am. Chem. Soc. 2020, 142, 7827–7835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khullar, S.; Mandal, S.K. Effect of spacer atoms in the dicarboxylate linkers on the formation of coordination architecture. Molecular rectangles vs 1D coordination polymers: Synthesis, crystal structures, vapor/gas adsorption studies, and magnetic properties. Cryst. Growth Des. 2014, 14, 6433–6444. [Google Scholar] [CrossRef]
- Coates, J. Interpretation of infrared spectra, A practical approach. In Encyclopedia of Analytical Chemistry; Meyers, R.A., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2000; pp. 10815–10837. [Google Scholar]
- Coates, J.P. The interpretation of infrared spectra: Published reference sources. Appied Spectrosc. Rev. 2006, 31, 179–192. [Google Scholar] [CrossRef]
- Hathaway, B.J.; Billing, D.E. The Electronic Properties and Stereochemistry of Mono-Nuclear Complexes of the Copper(II) Ion. Coord. Chem. Rev. 1970, 5, 143–207. [Google Scholar] [CrossRef]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring nature and predicting strength of hydrogen bonds: A correlation analysis between atoms-in-molecules descriptors, binding energies, and energy components of symmetry-adapted perturbation theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef] [PubMed]
- Ostojić, B.D.; Janjića, G.V.; Zarić, S.D. Parallel alignment of water and aryl rings—Crystallographic and theoretical evidence for the interaction. Chem. Commun. 2008, 6546–6548. [Google Scholar] [CrossRef]
- Andrić, J.M.; Janjić, G.V.; Ninkovića, D.B.; Zarić, S.D. The influence of water molecule coordination to a metal ion on water hydrogen bonds. Phys. Chem. Chem. Phys. 2012, 14, 10896–10898. [Google Scholar] [CrossRef]
- Bruker. APEX3 Software; v2018.7-2; Bruker AXS Inc.: Madison, WI, USA, 2019. [Google Scholar]
- Sheldrick, G.M. SADABS. Program for Empirical Absorption Correction of Area Detector Data; University of Goettingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, A.J.C. International Tables for Crystallography; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1995; Volume C. [Google Scholar]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. 2009, D65, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F. Accurate coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Bader, R.F.W. A Bond Path: A Universal Indicator of Bonded Interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll (Version 13.05.06); TK Gristmill Software: Overland Park, KS, USA, 2013. [Google Scholar]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting noncovalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MBP | Complexes * (CSD Reference Codes Indicated) | Observations and Binding Sites | Ref. |
|---|---|---|---|
| MBP-1 | IPUXIB, MENJOE, RALPLAY, RUMGAH, SIMMOQ, UNAPEG, ZAYBIK | M-N7(acv) | [4] |
| MBP-2 | BETSOL, BETSUR, BETTAY, BETTEC, BETTUS, BETVAA | M-N7(acv) + N(amine)-H⋯O6(acv) | [4] |
| MBP-2 | CAFVUD, HOPBOD, HOPBUJ, HPPCAQ, JAJPOA, JAJPUG, LUFGIC | M-N7(acv) + O(aqua)-H⋯O6(acv) | [4] |
| MBP-2 | ARAMOV, LUFGEY | M-N7(acv) + O(alcohol)-H⋯O6(acv) | [4] |
| MBP-3 | BETTIG, BETTOM, HOSQUB | Chelating-N7,O6 | [4] |
| MBP-4 | HOPCAD | Bridging (acv) μ-N7,O(alcohol) | [4] |
| MBP-5 | DIDJUY | N7,O6,O(e),O(ol)-Tetradentate O(e),O(ol)-chelating, μ3-bridging | [9] |
| MBP-2 | RUFBAY, [Zn(9heade)2Cl2] | M-N7(9heade) + (9heade)N6-H⋯Cl | [10] |
| MBP-2 | RUFBEC, trans-[CoII(acac)(9heade)2]·5H2O | M-N7(9heade) | [10] |
| MBP-2 | MOTBOP, [Cu(pdc)(9heade)(H2O)]·H2O | M-N7(9heade) | [11] |
| MBP-2 | MOTBIJ, trans-[Cu(pdc)(9heade)(H2O)2]·3H2O | M-N7(9heade) | [11] |
| MBP-2 | MOTBUV, [Zn(pdc)(9heade)(H2O)] | M-N7(9heade) | [11] |
| MBP-2 | OSEKIJ, [Cu2(µ2-EDTA)(9heade)2(H2O)2]·5H2O | M-N7(9heade) | [12] |
| MBP-2 | PUXQEH, {[Cd2(µ2-EDTA)(9heade)(H2O)]·4H2O}n | M-N7(9heade) | [13] |
| MBP-2 | ANEHOS, {[Cd(µ-(cis-1,2-chdca)(μ2-N1,N7-9heade)(H2O)]·4H2O}n | Bridging μ2-N1,N7 | [14] |
| MBP-2 + MBP-3 | MOTCAC, [Cu2(glygly)2(µ2-9heade)(H2O)]·8H2O | M-N7(9heade) + μ2-N7,O(ol) | [11] |
| Empirical formula | C11H19CuN5O8S |
| Formula weight | 444.91 |
| Temperature | 298 (2) K |
| Wavelength | 1.54178 Å |
| Crystal system, space group | Triclinic, |
| Unit cell dimensions | a = 5.3908 (3) Å |
| b = 12.9325 (7) Å | |
| c = 13.4804 (7) Å | |
| α = 62.939 (3)° | |
| = 86.260 (3)° | |
| γ = 81.195 (4)° | |
| Volume | 827.05 (8) Å3 |
| Z, Calculated density | 2, 1.787 Mg/m3 |
| Reflections collected/unique | 10,657/2881 |
| Data/parameters | 2881/238 |
| Goodness-of-fit on F2 | 1.132 |
| Final R indices [I > 2σ(I)] | R1 = 0.0415, wR2 = 0.1173 |
| R indices (all data) | R1 = 0.0447, wR2 = 0.1198 |
| Largest diff. peak and hole | 0.323 and −0.452 e.Å−3 |
| CCDC number | 2267019 |
| Cu(1)-O(4) | 1.933 (2) |
| Cu(1)-O(1) | 1.962 (2) |
| Cu(1)-N(21) | 2.025 (2) |
| Cu(1)-O(8) | 2.262 (2) |
| Cu(1)-S(1) | 2.3625 (8) |
| Cu(1)-O2 a | 3.060 (4) |
| O(4)-Cu(1)-O(1) | 175.04 (10) |
| N(21)-Cu(1)-S(1) | 173.58 (7) |
| Step or R | Temp. (°C) | Time (min) | Weight (%) | Evolved Gases or Residue (R) | |
|---|---|---|---|---|---|
| Exp. | Cal. | ||||
| 1 | ~50–155 | 2–13 | 8.911 | 8.098 | 2 H2O *, CO2 (t *) |
| 2 | 155–220 | 13–20 | 6.387 | 4.049 | 1 H2O, CO2 |
| 3 | 220–440 | 20–43 | 34.784 | 33.296 (ξ) | CO2, H2O, SCNH(t), N2O (t *), |
| 4 | 440–700 | 43–42 | 29.077 | (∝) | CO2, H2O, CO, SCNH, N2O, NO, NO2 |
| 5 | 700–950 | 42–67 | 1.283 | (∝) | CO2, H2O, CO, N2O, NO, NO2 |
| R | 950 | 95 | 19.532 | 17.879 | CuO |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosales-Martínez, C.; Matilla-Hernádez, A.; Choquesillo-Lazarte, D.; Frontera, A.; Castiñeiras, A.; Niclós-Gutiérrez, J. The Copper(II)-Thiodiacetate (tda) Chelate as Efficient Receptor of N9-(2-Hydroxyethyl)Adenine (9heade): Synthesis, Molecular and Crystal Structures, Physical Properties and DFT Calculations of [Cu(tda)(9heade)(H2O)]·2H2O. Molecules 2023, 28, 5830. https://doi.org/10.3390/molecules28155830
Rosales-Martínez C, Matilla-Hernádez A, Choquesillo-Lazarte D, Frontera A, Castiñeiras A, Niclós-Gutiérrez J. The Copper(II)-Thiodiacetate (tda) Chelate as Efficient Receptor of N9-(2-Hydroxyethyl)Adenine (9heade): Synthesis, Molecular and Crystal Structures, Physical Properties and DFT Calculations of [Cu(tda)(9heade)(H2O)]·2H2O. Molecules. 2023; 28(15):5830. https://doi.org/10.3390/molecules28155830
Chicago/Turabian StyleRosales-Martínez, Carmen, Antonio Matilla-Hernádez, Duane Choquesillo-Lazarte, Antonio Frontera, Alfonso Castiñeiras, and Juan Niclós-Gutiérrez. 2023. "The Copper(II)-Thiodiacetate (tda) Chelate as Efficient Receptor of N9-(2-Hydroxyethyl)Adenine (9heade): Synthesis, Molecular and Crystal Structures, Physical Properties and DFT Calculations of [Cu(tda)(9heade)(H2O)]·2H2O" Molecules 28, no. 15: 5830. https://doi.org/10.3390/molecules28155830
APA StyleRosales-Martínez, C., Matilla-Hernádez, A., Choquesillo-Lazarte, D., Frontera, A., Castiñeiras, A., & Niclós-Gutiérrez, J. (2023). The Copper(II)-Thiodiacetate (tda) Chelate as Efficient Receptor of N9-(2-Hydroxyethyl)Adenine (9heade): Synthesis, Molecular and Crystal Structures, Physical Properties and DFT Calculations of [Cu(tda)(9heade)(H2O)]·2H2O. Molecules, 28(15), 5830. https://doi.org/10.3390/molecules28155830