

Synthesis and Characterization of New Pyrano[2,3-c]pyrazole Derivatives as 3-Hydroxyflavone Analogues

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. NMR Spectroscopic Investigations

2.3. Single-Crystal X-ray Diffraction Analysis
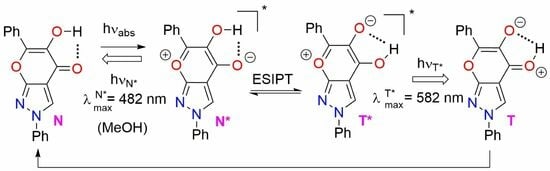
2.4. Optical Investigations
3. Materials and Methods
3.1. General
3.2. Synthetic Procedures
3.2.1. (2E)-3-(3,4-Dimethoxyphenyl)-1-(3-hydroxy-1-phenyl-1H-pyrazol-4-yl)prop-2-en-1-one (2d)
3.2.2. General Procedure for the Synthesis of 3a–h
- 5-Hydroxy-2,6-diphenylpyrano[2,3-c]pyrazol-4(2H)-one (3a). Off white solid; yield 58% (177 mg); m.p. 183–184 °C. 1H NMR (700 MHz, DMSO-d6): δH ppm 7.46 (t, J = 7.4 Hz, 1H, NPh 4-H), 7.49 (t, J = 7.4 Hz, 1H, 6-CPh 4-H), 7.55–7.58 (m, 2H, 6-CPh 3,5-H), 7.58–7.61 (m, 2H, NPh 3,5-H), 8.01–8.03 (m, 2H, NPh 2,6-H), 8.13–8.14 (m, 2H, 6-CPh 2,6-H), 9.38 (s, 1H, 3-H), 9.44 (s, 1H, OH). 13C NMR (176 MHz, DMSO-d6): δC ppm 108.3 (C-3a), 119.9 (NPh C-2,6), 126.6 (C-3), 127.9 (6-CPh C-2,6), 128.5 (NPh C-4), 129.0 (6-CPh C-3,5), 130.0 (6-CPh C-4), 130.2 (NPh C-3,5), 131.8 (6-CPh C-1), 139.16 (C-5), 139.19 (NPh C-1), 144.4 (C-6), 161.2 (C-7a), 171.8 (C-4). 15N NMR (71 MHz, DMSO-d6): δN ppm −167.7 (N-2), −117.0 (N-1). IR (νmax, cm−1): 3110, 3062, 2920, 2850, 1679 (C=O), 1568, 1489, 1199, 1110, 913, 832, 761, 696. HRMS (ESI+) for C18H12N2NaO3 ([M + Na]+) calcd 327.0740, found 327.0740.
- 6-(4-Chlorophenyl)-5-hydroxy-2-phenylpyrano[2,3-c]pyrazol-4(2H)-one (3b). Light yellow solid; yield 63% (213 mg); m.p. 262–263 °C. 1H NMR (700 MHz, DMSO-d6): δH ppm 7.45 (t, J = 7.3 Hz, 1H, NPh 4-H), 7.58 (t, J = 7.8 Hz, 2H, NPh 3,5-H), 7.62 (d, J = 8.6 Hz, 2H, 6-CPh 3,5-H), 8.01 (d, J = 7.9 Hz, 2H, NPh 2,6-H), 8.15 (d, J = 8.6 Hz, 2H, 6-CPh 2,6-H), 9.38 (s, 1H, 3-H), 9.70 (s, 1H, OH). 13C NMR (176 MHz, DMSO-d6): δC ppm 107.9 (C-3a), 119.5 (NPh C-2,6), 126.3 (C-3), 128.1 (NPh C-4), 128.6 (6-CPh C-3,5), 129.1 (6-CPh C-2,6), 129.8 (NPh C-3,5), 130.2 (6-CPh C-1), 134.1 (6-CPh C-4), 138.7 (NPh C-1), 139.1 (C-5), 142.7 (C-6), 160.6 (C-7a), 171.3 (C-4). 15N NMR (71 MHz, DMSO-d6): δN ppm −167.5 (N-2), −117.2 (N-1). IR (νmax, cm−1): 3348, 3289, 3100, 1645 (C=O), 1576, 1495, 1442, 1098, 825, 752, 679. HRMS (ESI+) for C18H11ClN2NaO3 ([M + Na]+) calcd 361.0350, found 361.0350.
- 5-Hydroxy-6-(4-methoxyphenyl)-2-phenylpyrano[2,3-c]pyrazol-4(2H)-one (3c). Yellow solid; yield 51% (171 mg); m.p. 263–264 °C. 1H NMR (700 MHz, DMSO-d6): δH ppm 3.85 (s, 3H, CH3), 7.12–7.13 (m, 2H, 6-CPh 3,5-H), 7.45 (t, J = 7.4 Hz, 1H, NPh 4-H), 7.57–7.60 (m, 2H, NPh 3,5-H), 8.00–8.02 (m, 2H, NPh 2,6-H), 8.09–8.12 (m, 2H, 6-CPh 2,6-H), 9.27 (s, 1H, 3-H), 9.36 (s, 1H, OH). 13C NMR (176 MHz, DMSO-d6): δC ppm 55.3 (CH3), 107.9 (C-3a), 114.2 (6-CPh C-3,5), 119.4 (NPh C-2,6), 123.6 (6-CPh C-1), 126.0 (C-3), 128.0 (NPh C-4), 129.2 (6-CPh C-2,6), 129.8 (NPh C-3,5), 137.8 (C-5), 138.8 (NPh C-1), 144.4 (C-6), 160.2 (6-CPh C-4), 160.7 (C-7a), 171.2 (C-4). 15N NMR (71 MHz, DMSO-d6): δN ppm −168.3 (N-2). IR (νmax, cm−1): 3286, 3134, 1642 (C=O), 1580, 1509, 1441, 1256, 1108, 821, 748, 680. HRMS (ESI+) for C19H14N2NaO4 ([M + Na]+) calcd 357.0846, found 357.0841.
- 6-(3,4-Dimethoxyphenyl)-5-hydroxy-2-phenylpyrano[2,3-c]pyrazol-4(2H)-one (3d). Orange solid; yield 67% (245 mg); m.p. 252–253 °C. 1H NMR (700 MHz, DMSO-d6): δH ppm 3.84 (s, 3H, 6-CPh 3-OCH3), 3.85 (s, 3H, 6-CPh 4-OCH3), 7.15 (d, J = 8.7 Hz, 1H, 6-CPh 5-H), 7.45 (t, J = 7.4 Hz, 1H, NPh 4-H), 7.58 (t, J = 8.0 Hz, 2H, NPh 3,5-H), 7.71 (d, J = 2.1 Hz, 1H, 6-CPh 2-H), 7.78 (dd, J = 8.6, 2.1 Hz, 1H, 6-CPh 6-H), 8.02 (d, J = 7.7 Hz, 2H, NPh 2,6-H), 9.28 (s, 1H, OH), 9.35 (s, 1H, 3-H). 13C NMR (176 MHz, DMSO-d6): δC ppm 55.6 (6-CPh 3,4-OCH3), 107.8 (C-3a), 110.7 (6-CPh C-2), 111.5 (6-CPh C-5), 119.4 (NPh C-2,6), 121.3 (6-CPh C-6), 123.7 (6-CPh C-1), 126.0 (C-3), 128.0 (NPh C-4), 129.8 (NPh C-3,5), 137.9 (C-5), 138.8 (NPh C-1), 144.3 (C-6), 148.3 (6-CPh C-3), 150.0 (6-CPh C-4), 160.6 (C-7a), 171.1 (C-4). 15N NMR (71 MHz, DMSO-d6): δN ppm −168.2 (N-2), −117.3 (N-1). IR (νmax, cm−1): 3281, 2963, 1632 (C=O), 1583, 1515, 1439, 1106, 754, 657. HRMS (ESI+) for C20H16N2NaO5 ([M + Na]+) calcd 387.0951, found 387.0953.
- 5-Hydroxy-6-(naphthalen-2-yl)-2-phenylpyrano[2,3-c]pyrazol-4(2H)-one (3e). Yellow solid; yield 32% (113 mg); m.p. 256–257 °C. 1H NMR (700 MHz, DMSO-d6): δH ppm 7.46 (t, J = 7.4 Hz, 1H, NPh 4-H), 7.59–7.63 (m, 4H, NPh 3,5-H and Naph 6,7-H), 7.99 (d, J = 7.8 Hz, 1H, Naph 5-H), 8.04 (d, J = 7.9 Hz, 2H, NPh 2,6-H), 8.06–8.09 (m, 1H, Naph 4-H), 8.09–8.10 (m, 1H, Naph 8-H), 8.27 (dd, J = 8.7, 1.8 Hz, 1H, Naph 3-H), 8.71 (s, 1H, Naph 1-H), 9.41 (s, 1H, 3-H), 9.59 (s, 1H, OH). 13C NMR (176 MHz, DMSO-d6): δC ppm 107.8 (C-3a), 119.3 (NPh C-2,6), 124.3 (Naph C-3), 126.1 (C-3), 126.7 (Naph C-7), 127.38 (Naph C-1 and Naph C-6), 127.44 (Naph C-5), 127.82 (Naph C-4), 128.01 (NPh C-4), 128.76 (Naph C-8), 128.79 (Naph C-2), 129.7 (NPh C-3,5), 132.4 (Naph C-8a), 132.9 (Naph C-4a), 138.7 (NPh C-1), 139.0 (C-5), 143.8 (C-6), 160.7 (C-7a), 171.2 (C-4). 15N NMR (71 MHz, DMSO-d6): δN ppm −167.7 (N-2), −117.4 (N-1). IR (νmax, cm−1): 3240, 1629 (C=O), 1576, 1564, 1441, 1386, 1216, 1096, 753, 685. HRMS (ESI+) for C22H14N2NaO3 ([M + Na]+) calcd 377.0897, found 377.0908.
- 5-Hydroxy-2-phenyl-6-(thiophen-2-yl)pyrano[2,3-c]pyrazol-4(2H)-one (3f). Yellow solid; yield 62% (193 mg); m.p. 187–188 °C. 1H NMR (700 MHz, DMSO-d6): δH ppm 7.29 (dd, J = 5.0, 3.8 Hz, 1H, Th 5-H), 7.44–7.46 (m, 1H, NPh 4-H), 7.57–7.60 (m, 2H, NPh 3,5-H), 7.87 (dd, J = 3.8, 1.2 Hz, 1H, Th 3-H), 7.88 (dd, J = 5.0, 1.2 Hz, 1H, Th 4-H), 8.00–8.02 (m, 2H, NPh 2,6-H), 9.35 (s, 1H, 3-H), 10.12 (s, 1H, OH). 13C NMR (176 MHz, DMSO-d6): δC ppm 108.1 (C-3a), 119.3 (NPh C-2,6), 126.1 (C-3), 127.66 (Th C-3), 127.70 (Th C-5), 127.9 (NPh C-4), 129.6 (NPh C-3,5), 130.4 (Th C-4), 132.3 (Th C-2), 136.5 (C-5), 138.6 (NPh C-1), 141.9 (C-6), 160.2 (C-7a), 170.4 (C-4). 15N NMR (71 MHz, DMSO-d6): δN ppm −168.6 (N-2), −117.2 (N-1). IR (νmax, cm−1): 3259, 3113, 1629 (C=O), 1575, 1503, 1217, 1103, 826, 753, 685. HRMS (ESI+) for C16H10N2NaO3S ([M + Na]+) calcd 333.0304, found 333.0309.
- 6-(Furan-3-yl)-5-hydroxy-2-phenylpyrano[2,3-c]pyrazol-4(2H)-one (3g). Beige solid; yield 30% (89 mg); m.p. 228–229 °C. 1H NMR (700 MHz, DMSO-d6): δH ppm 6.78 (dd, J = 3.4, 1.7 Hz, 1H, Furanyl 5-H), 7.23 (d, J = 3.4 Hz, 1H, Furanyl 4-H), 7.45 (t, J = 7.4 Hz, 1H, NPh 4-H), 7.59 (t, J = 7.9 Hz, 2H, NPh 3,5-H), 8.00 (d, J = 7.8 Hz, 2H, NPh 2,6-H), 8.02 (d, J = 1.0 Hz, 1H, Furanyl 2-H), 9.36 (s, 1H, 3-H), 9.86 (s, 1H, OH). 13C NMR (176 MHz, DMSO-d6): δC ppm 108.2 (C-3a), 112.7 (Furanyl C-5), 114.5 (Furanyl C-4), 119.3 (NPh C-2,6), 126.1 (C-3), 127.9 (NPh C-4), 129.7 (NPh C-3,5), 136.9 (C-5), 138.0 (Furanyl C-3), 138.6 (NPh C-1), 144.0 (C-6), 144.9 (Furanyl C-2), 160.2 (C-7a), 170.3 (C-4). 15N NMR (71 MHz, DMSO-d6): δN ppm −168.4 (N-2), −117.1 (N-1). IR (νmax, cm−1): 3246, 3138, 2957, 2856, 1633 (C=O), 1576, 1483, 1221, 1124, 934, 845, 755, 681. HRMS (ESI+) for C16H10N2NaO4 ([M + Na]+) calcd 317.0533, found 317.0534.
- 5-Hydroxy-2-phenyl-6-(pyridin-4-yl)pyrano[2,3-c]pyrazol-4(2H)-one (3h). Yellow solid; yield 53% (163 mg); m.p. 298–299 °C. 1H NMR (700 MHz, DMSO-d6) δ 7.45–7.48 (m, 1H, Ph 4-H), 7.56–7.61 (m, 2H, Ph 3,5-H), 8.00–8.03 (m, 2H, Ph 2,6-H), 8.04–8.07 (m, 2H, Pyr 3,5-H), 8.74–8.77 (m, 2H, Pyr 2,4-H), 9.41 (s, 1H, 3-H), 10.13 (s, 1H, OH). 13C NMR (176 MHz, DMSO-d6) δ 107.8 (C-3a), 119.4 (Ph C-2,6), 120.6 (Pyr C-3,5), 126.4 (C-3), 128.1 (Ph C-4), 129.7 (Ph C-3,5), 138.45 (Pyr C-4), 138.53 (Ph C-1), 140.5 (C-6), 140.9 (C-5), 150.0 (Pyr C-2,6), 160.5 (C-7a), 171.2 (C-4). 15N NMR (71 MHz, DMSO-d6): δN ppm −166.9 (N-2), −117.1 (N-1), −62.2 (Pyr N). IR (νmax, cm−1): 3112, 3087, 1648 (C=O), 1571, 1500, 1442, 1228, 1026, 834, 754, 629. HRMS (ESI+) for C17H11N3O3 ([M + H]+) calcd 306.0873, found 306.0871.
3.2.3. Procedure for the Synthesis of 5-Methoxy-2,6-diphenylpyrano[2,3-c]pyrazol-4(2H)-one (4)
3.2.4. Procedure for the Synthesis of 6-(1-Methylpyridin-1-ium-4-yl)-4-oxo-2-phenyl-2,4-dihydropyrano[2,3-c]pyrazol-5-olate (5)
3.2.5. Procedure for the Synthesis of 4-(5-Hydroxy-4-oxo-2-phenyl-2,4-dihydropyrano[2,3-c]pyrazol-6-yl)-1-methylpyridin-1-ium Iodide (6)
3.2.6. Procedure for the Synthesis of 4-Oxo-2,6-diphenyl-2,4-dihydropyrano[2,3-c]pyrazol-5-yl Trifluoromethanesulfonate (7)
3.2.7. General Procedure for the Synthesis of 5-(Hetero)aryl-2,6-diphenylpyrano[2,3-c]pyrazol-4(2H)-ones 8a–e
- 2,5,6-Triphenylpyrano[2,3-c]pyrazol-4(2H)-one (8a). Yellow solid; yield 95% (346 mg); m.p. 265–266 °C. 1H NMR (700 MHz, CDCl3): δH ppm 7.20–7.21 (m, 2H, 5-CPh 2,6-H), 7.24–7.25 (m, 2H, 6-CPh 3,5-H), 7.28–7.33 (m, 4H, 5-CPh 3,4,5-H and 6-CPh 4-H), 7.39 (d, J = 7.6 Hz, 2H, 6-CPh 2,6-H), 7.42 (t, J = 7.4 Hz, 1H, NPh 4-H), 7.53 (t, J = 7.9 Hz, 2H, NPh 3,5-H), 7.80 (d, J = 8.0 Hz, 2H, NPh 2,6-H), 8.55 (s, 1H, 3-H). 13C NMR (176 MHz, CDCl3): δC ppm 109.7 (C-3a), 119.9 (NPh C-2,6), 123.0 (C-5), 124.7 (C-3), 127.7 (5-CPh C-4), 128.0 (6-CPh C-3,5), 128.24 (NPh C-4), 128.29 (5-CPh C-3,5), 129.78 (6-CPh C-2,6), 129.83 (NPh C-3,5), 130.0 (6-CPh C-4), 131.4 (5-CPh C-2,6), 132.8 (5-CPh C-1), 133.0 (6-CPh C-1), 139.2 (NPh C-1), 160.8 (C-6), 162.4 (C-7a), 175.5 (C-4). 15N NMR (71 MHz, CDCl3): δN ppm −169.5 (N-2), −115.3 (N-1). IR (νmax, cm−1): 3093, 2922, 1642 (C=O), 1578, 1561, 1493, 1349, 1224, 1056, 755, 730, 694, 683. HRMS (ESI+) for C24H16N2NaO2 ([M + Na]+) calcd 387.1104, found 387.1107.
- 5-(4-Methylphenyl)-2,6-diphenylpyrano[2,3-c]pyrazol-4(2H)-one (8b). White solid; yield 62% (235 mg); m.p. 256–257 °C. 1H NMR (700 MHz, CDCl3): δH ppm 2.34 (s, 3H, CH3), 7.08 (m, 2H, 5-CPh 2,6-H), 7.11 (m, 2H, 5-CPh 3,5-H), 7.24–7.27 (m, 2H, 6-CPh 3,5-H), 7.32 (m, 2H, 6-CPh 4-H), 7.40–7.43 (m, 3H, 6-CPh 2,6-H, NPh 4-H), 7.53 (m, 2H, NPh 3,5-H), 7.78–7.81 (m, 2H, NPh 2,6-H), 8.55 (s, 1H, 3-H). 13C NMR (176 MHz, CDCl3): δC ppm 21.3 (CH3), 109.7 (C-3a), 119.8 (NPh C-2,6), 122.9 (C-5), 124.6 (C-3), 128.0 (6-CPh C-3,5), 128.2 (NPh C-4), 129.1 (5-CPh C-3,5), 129.6 (5-CPh C-1), 129.7 (6-CPh C-2,6), 129.8 (NPh C-3,5), 129.9 (6-Ph C-4), 131.2 (5-CPh C-2,6), 133.1 (6-CPh C-1), 137.4 (5-CPh C-4), 139.2 (NPh C-1), 160.5 (C-6), 162.3 (C-7a), 175.7 (C-4). 15N NMR (71 MHz, CDCl3): δN ppm −169.7 (N-2), −115.3 (N-1). IR (νmax, cm−1): 3098, 3023, 1649 (C=O), 1578, 1565, 1348, 1181, 1021, 755, 742, 732, 683. HRMS (ESI+) for C25H18N2O2 ([M + Na]+) calcd 401.1260, found 401.1262.
- 5-(4-Methoxyphenyl)-2,6-diphenylpyrano[2,3-c]pyrazol-4(2H)-one (8c). White solid; yield 77% (304 mg); m.p. 236–237 °C. 1H NMR (700 MHz, CDCl3): δH ppm 3.80 (s, 3H, CH3), 6.85 (m, 2H, 5-CPh 3,5-H), 7.12 (m, 2H, 5-CPh 2,6-H), 7.24–7.28 (m, 2H, 6-CPh 3,5-H), 7.32 (m, 2H, 6-CPh 4-H), 7.40–7.44 (m, 3H, 6-CPh 2,6-H, NPh 4-H), 7.54 (m, 2H, NPh 3,5-H), 7.80 (m, 2H, NPh 2,6-H), 8.54 (s, 1H, 3-H). 13C NMR (176 MHz, CDCl3): δC ppm 55.2 (CH3), 109.7 (C-3a), 113.9 (5-CPh C-3,5), 119.8 (NPh C-2,6), 122.5 (C-5), 124.6 (C-3), 124.8 (5-CPh C-1), 128.0 (6-CPh C-3,5), 128.2 (NPh C-4), 129.7 (6-CPh C-2,6), 129.8 (NPh C-3,5), 129.9 (6-Ph C-4), 132.5 (5-CPh C-2,6), 133.2 (6-CPh C-1), 139.2 (NPh C-1), 159.1 (5-CPh C-4), 160.5 (C-6), 162.3 (C-7a), 175.8 (C-4). 15N NMR (71 MHz, CDCl3): δN ppm −169.7 (N-2), −115.6 (N-1). IR (νmax, cm−1): 3102, 3024, 1650 (C=O), 1598, 1567, 1335, 1241, 1167, 1023, 748, 686, 549. HRMS (ESI+) for C25H18N2O3 ([M + Na]+) calcd 417.1210, found 417.1208.
- 5-(4-Chlorophenyl)-2,6-diphenylpyrano[2,3-c]pyrazol-4(2H)-one (8d). White solid; yield 44% (176 mg); m.p. 255–256 °C. 1H NMR (700 MHz, CDCl3): δH ppm 7.14 (m, 2H, 5-CPh 2,6-H), 7.27–7.31 (m, 4H, 5-CPh 3,5-H, 6-CPh 3,5-H), 7.35 (m, 2H, 6-CPh 4-H), 7.39 (m, 2H, 6-CPh 2,6-H), 7.43 (m, 1H, NPh 4-H), 7.54 (m, 2H, NPh 3,5-H), 7.80 (m, 2H, NPh 2,6-H), 8.55 (s, 1H, 3-H). 13C NMR (176 MHz, CDCl3): δC ppm 109.5 (C-3a), 119.9 (NPh C-2,6), 121.8 (C-5), 124.7 (C-3), 128.2 (6-CPh C-3,5), 128.4 (NPh C-4), 128.6 (5-CPh C-3,5), 129.7 (6-CPh C-2,6), 129.8 (NPh C-3,5), 130.3 (6-Ph C-4), 131.3 (5-CPh C-1), 132.7 (6-CPh C-1), 132.8 (5-CPh C-2,6), 133.7 (5-CPh C-4), 139.1 (NPh C-1), 161.0 (C-6), 162.32 (C-7a), 175.2 (C-4). 15N NMR (71 MHz, CDCl3): δN ppm −169.1 (N-2), −115.0 (N-1). IR (νmax, cm−1): 3206, 3105, 1650 (C=O), 1568, 1422, 1348, 1211, 1135, 757, 731, 686. HRMS (ESI+) for C24H15ClN2O2 ([M + Na]+) calcd 421.0714, found 421.0711.
- 2,6-Diphenyl-5-(thiophen-3-yl)pyrano[2,3-c]pyrazol-4(2H)-one (8e). Beige solid; yield 80% (297 mg); m.p. 265–266 °C. 1H NMR (700 MHz, CDCl3): δH ppm 6.88 (m, 1H, Th 4-H), 7.20 (m, 1H, Th 2-H), 7.24 (m, 1H, Th 5-H), 7.31 (m, 2H, 6-CPh 3,5-H), 7.37 (m, 2H, 6-CPh 4-H), 7.42 (m, 1H, NPh 4-H), 7.44 (m, 2H, 6-CPh 2,6-H), 7.54 (m, 2H, NPh 3,5-H), 7.80 (m, 2H, NPh 2,6-H), 8.54 (s, 1H, 3-H). 13C NMR (176 MHz, CDCl3): δC ppm 109.6 (C-3a), 118.1 (C-5), 119.9 (NPh C-2,6), 124.6 (C-3), 124.7 (Th C-5), 126.4 (Th C-2), 128.1 (6-CPh C-3,5), 128.3 (NPh C-4), 129.8 (Th C-4), 129.5 (6-CPh C-2,6), 129.8 (NPh C-3,5), 130.2 (6-Ph C-4), 131.9 (Th C-3), 132.2 (6-CPh C-1), 139.1 (NPh C-1), 160.8 (C-6), 162.2 (C-7a), 175.3 (C-4). 15N NMR (71 MHz, CDCl3): δN ppm −169.4 (N-2), −115.2 (N-1). IR (νmax, cm−1): 3100, 1644 (C=O), 1577, 1564, 1441, 1328, 1218, 753, 739, 685. HRMS (ESI+) for C22H14N2O2S ([M + H]+) calcd 393.0668, found 393.0669.
3.2.8. Procedure for the Synthesis of tert-Butyl (2E)-3-(4-oxo-2,6-diphenyl-2,4-dihydropyrano[2,3-c]pyrazol-5-yl)prop-2-enoate (8f)
3.2.9. Procedure for the Synthesis of 2,6-Diphenyl-5-(phenylethynyl)pyrano[2,3-c]pyrazol-4(2H)-one (8g)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Li, M.-M.; Huang, H.; Pu, Y.; Tian, W.; Deng, Y.; Lu, J. A close look into the biological and synthetic aspects of fused pyrazole derivatives. Eur. J. Med. Chem. 2022, 243, 114739. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.Y.; Mohamed, M.A.; Abdel-Aziem, A.; Hussain, A.O. Synthesis and Anticancer Activity of Some Fused Heterocyclic Compounds Containing Pyrazole Ring. Polycycl. Aromat. Compd. 2020, 40, 1280–1290. [Google Scholar] [CrossRef]
- Bondock, S.; Fadaly, W.; Metwally, M.A. Synthesis and antimicrobial activity of some new thiazole, thiophene and pyrazole derivatives containing benzothiazole moiety. Eur. J. Med. Chem. 2010, 45, 3692–3701. [Google Scholar] [CrossRef]
- Han, C.; Guo, Y.-C.; Wang, D.-D.; Dai, X.-Y.; Wu, F.-J.; Liu, H.-F.; Dai, G.-F.; Tao, J.-C. Novel pyrazole fused heterocyclic ligands: Synthesis, characterization, DNA binding/cleavage activity and anti-BVDV activity. Chin. Chem. Lett. 2015, 26, 534–538. [Google Scholar] [CrossRef]
- Pinto, D.J.P.; Orwat, M.J.; Koch, S.; Rossi, K.A.; Alexander, R.S.; Smallwood, A.; Wong, P.C.; Rendina, A.R.; Luettgen, J.M.; Knabb, R.M.; et al. Discovery of 1-(4-Methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (Apixaban, BMS-562247), a Highly Potent, Selective, Efficacious, and Orally Bioavailable Inhibitor of Blood Coagulation Factor Xa. J. Med. Chem. 2007, 50, 5339–5356. [Google Scholar] [PubMed]
- Xu, Y.; Zhang, Z.; Jiang, X.; Chen, X.; Wang, Z.; Alsulami, H.; Qin, H.-L.; Tang, W. Discovery of δ-sultone-fused pyrazoles for treating Alzheimer’s disease: Design, synthesis, biological evaluation and SAR studies. Eur. J. Med. Chem. 2019, 181, 111598. [Google Scholar] [CrossRef]
- Syed, Y.Y. Futibatinib: First Approval. Drugs 2022, 82, 1737–1743. [Google Scholar] [CrossRef]
- Kumar, A.; Lohan, P.; Aneja, D.K.; Gupta, G.K.; Kaushik, D.; Prakash, O. Design, synthesis, computational and biological evaluation of some new hydrazino derivatives of DHA and pyranopyrazoles. Eur. J. Med. Chem. 2012, 50, 81–89. [Google Scholar] [CrossRef]
- Parikh, P.H.; Timaniya, J.B.; Patel, M.J.; Patel, K.P. Microwave-assisted synthesis of pyrano[2,3-c]-pyrazole derivatives and their anti-microbial, anti-malarial, anti-tubercular, and anti-cancer activities. J. Mol. Struct. 2022, 1249, 131605. [Google Scholar] [CrossRef]
- Parshad, M.; Verma, V.; Kumar, D. Iodine-mediated efficient synthesis of pyrano[2,3-c]pyrazoles and their antimicrobial activity. Monatsh. Chem. 2014, 145, 1857–1865. [Google Scholar] [CrossRef]
- Wang, J.; Liu, D.; Zheng, Z.; Shan, S.; Han, X.; Srinivasula, S.M.; Croce, C.M.; Alnemri, E.S.; Huang, Z. Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc. Natl. Acad. Sci. USA 2000, 97, 7124–7129. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhang, L.; Gao, M.; Que, X.; Zhou, C.; Zhu, D.; Cai, Y. Nanoformulation of a Novel Pyrano[2,3-c]Pyrazole Heterocyclic Compound AMDPC Exhibits Anti-Cancer Activity via Blocking the Cell Cycle through a P53-Independent Pathway. Molecules 2019, 24, 624. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Truong, M.-N.H.; Le, T.V.; Vo, N.T.; Nguyen, H.D.; Tran, P.H. A New Pathway for the Preparation of Pyrano[2,3-c]pyrazoles and molecular Docking as Inhibitors of p38 MAP Kinase. ACS Omega 2022, 7, 17432–17443. [Google Scholar] [CrossRef] [PubMed]
- Bieliauskas, A.; Krikštolaitytė, S.; Holzer, W.; Šačkus, A. Ring-closing metathesis as a key step to construct 2,6-dihydropyrano[2,3-c]pyrazole ring system. Arkivoc 2018, 2018, 296–307. [Google Scholar] [CrossRef]
- Milišiūnaitė, V.; Kadlecová, A.; Žukauskaitė, A.; Doležal, K.; Strnad, M.; Voller, J.; Arbačiauskienė, E.; Holzer, W.; Šačkus, A. Synthesis and Anthelmintic Activity of Benzopyrano[2,3-c]Pyrazol-4(2H)-One Derivatives. Mol. Divers. 2020, 24, 1025–1042. [Google Scholar] [CrossRef]
- Al-Khayri, J.M.; Sahana, G.R.; Nagella, P.; Joseph, B.V.; Alessa, F.M.; Al-Mssallem, M.Q. Flavonoids as Potential Anti-Inflammatory. Molecules 2022, 27, 2901. [Google Scholar] [CrossRef]
- Panche, A.; Diwan, A.; Chandra, S. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, E47. [Google Scholar] [CrossRef]
- Ullah, A.; Munir, S.; Badshah, S.L.; Khan, N.; Ghani, L.; Poulson, B.G.; Mews, A.-H.; Jaremko, M. Important Flavonoids and Their Role as a Therapeutic Agent. Molecules 2020, 25, 5243. [Google Scholar] [CrossRef]
- Jan, R.; Khan, M.; Asaf, S.; Lubna; Asif, S.; Kim, K.-M. Bioactivity and Therapeutic Potential of Kaempferol and Quercetin: New Insights for Plant and Human Health. Plants 2022, 11, 2623. [Google Scholar] [CrossRef]
- Nejabati, H.R.; Roshangar, L. Kaempferol: A potential agent in the prevention of colorectal cancer. Physiol. Rep. 2022, 10, e15488. [Google Scholar] [CrossRef]
- Borsari, C.; Jiménez-Antón, M.D.; Eick, J.; Bifeld, E.; Torrado, J.J.; Olías-Molero, A.I.; Corral, M.J.; Santarem, N.; Baptista, C.; Severi, L.; et al. Discovery of a benzothiophene-flavonol halting miltefosine and antimonial drug resistance in Leishmania parasites through the application of medicinal chemistry, screening and genomics. Eur. J. Med. Chem. 2019, 183, 111676. [Google Scholar] [CrossRef]
- Kishore, N.R.; Ashok, D.; Sarasija, M.; Murthy, N.Y.S. One-pot synthesis of spirochromanone-based 3-hydroxy-4H-chromen-4-ones by a modified Algar–Flynn–Oyamada reaction and evaluation of their antimicrobial activity. Chem. Heterocycl. Compd. 2017, 53, 1187–1191. [Google Scholar] [CrossRef]
- Ashok, D.; Kifah, M.A.; Lakshmi, B.V.; Sarasija, M.; Adam, S. Microwave-assisted one-pot synthesis of some new flavonols by modified Algar–Flynn–Oyamada reaction and their antimicrobial activity. Chem. Heterocycl. Compd. 2016, 52, 172–176. [Google Scholar] [CrossRef]
- Lee, J.; Park, T.; Jeong, S.; Kim, K.-H.; Hong, C. 3-Hydroxychromones as cyclin-dependent kinase inhibitors: Synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2007, 17, 1284–1287. [Google Scholar] [CrossRef] [PubMed]
- Joshi, H.C.; Antonov, L. Excited-State Intramolecular Proton Transfer: A Short Introductory Review. Molecules 2021, 26, 1475. [Google Scholar] [CrossRef]
- Ameer-Beg, S.; Ormson, S.M.; Brown, R.G.; Matousek, P.; Towrie, M.; Nibbering, E.T.J.; Foggi, P.; Neuwahl, F.V.R. Ultrafast Measurements of Excited State Intramolecular Proton Transfer (ESIPT) in Room Temperature Solutions of 3-Hydroxyflavone and Derivatives. J. Phys. Chem. A 2001, 105, 3709–3718. [Google Scholar] [CrossRef]
- Sarkar, M.; Ray, J.G.; Sengupta, P.K. Effect of reverse micelles on the intramolecular excited state proton transfer (ESPT) and dual luminescence behaviour of 3-hydroxyflavone. Spectrochim. Acta A Mol. Biomol. 1996, 52, 275–278. [Google Scholar] [CrossRef]
- Zhao, X.; Li, X.; Liang, S.; Dong, X.; Zhang, Z. 3-Hydroxyflavone derivatives: Promising scaffolds for fluorescent imaging in cells. RSC Adv. 2021, 11, 28851. [Google Scholar] [CrossRef]
- Butun, B.; Topcu, G.; Ozturk, T. Recent Advances on 3-Hydroxyflavone Derivatives: Structures and Properties. Mini Rev. Med. Chem. 2018, 18, 98–103. [Google Scholar] [CrossRef]
- Russo, M.; Orel, V.; Takko, P.; Šranková, M.; Muchová, L.; Vítek, L.; Klán, P. Structure–Photoreactivity Relationship of 3-Hydroxyflavone-Based CO-Releasing Molecules. J. Org. Chem. 2022, 87, 4750–4763. [Google Scholar] [CrossRef]
- Jiang, G.; Jin, Y.; Li, M.; Wang, H.; Xiong, M.; Zeng, W.; Yuan, H.; Liu, C.; Ren, Z.; Liu, C. Faster and More Specific: Excited-State Intramolecular Proton Transfer-Based Dyes for High-Fidelity Dynamic Imaging of Lipid Droplets within Cells and Tissues. Anal. Chem. 2020, 92, 10342–10349. [Google Scholar] [CrossRef] [PubMed]
- Kamariza, M.; Keyser, S.G.L.; Utz, A.; Knapp, B.D.; Ealand, C.; Ahn, G.; Cambier, C.J.; Chen, T.; Kana, B.; Huang, K.C.; et al. Toward Point-of-Care Detection of Mycobacterium tuberculosis: A Brighter Solvatochromic Probe Detects Mycobacteria within Minutes. JACS Au 2021, 1, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Bernini, R.; Crisante, F.; Ginnasi, M.C. A Convenient and Safe O-Methylation of Flavonoids with Dimethyl Carbonate (DMC). Molecules 2011, 16, 1418–1425. [Google Scholar] [CrossRef] [PubMed]
- Koirala, N.; Thuan, N.H.; Ghimire, G.P.; Thang, D.V.; Sohng, J.K. Methylation of flavonoids: Chemical structures, bioactivities, progress and perspectives for biotechnological production. Enzym. Microb. 2016, 86, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fernie, A.R.; Tohge, T. Diversification of Chemical Structures of Methoxylated Flavonoids and Genes Encoding Flavonoid-O-Methyltransferases. Plants 2022, 11, 564. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, H.; Ikegawa, T.; Honda, Y.; Kohyama, N.; Morimoto, S.; Shoyama, Y.; Juichi, M.; Naito, M.; Tsuruo, T.; Sawada, T. Effects of various methoxyflavones on vincristine uptake and multidrug resistance to vincristine in P-gp-overexpressing K562/ADM cells. Pharm. Res. 2007, 24, 1936–1943. [Google Scholar] [CrossRef]
- Juvale, K.; Stefan, K.; Wiese, M. Synthesis and biological evaluation of flavones and benzoflavones as inhibitors of BCRP/ABCG. Eur. J. Med. Chem. 2013, 67, 115–126. [Google Scholar] [CrossRef]
- Khan, D.; Parveen, I.; Shaily, S.S. Design, Synthesis and Characterization of Aurone Based α,β-unsaturated Carbonyl-Amino Ligands and their Application in Microwave Assisted Suzuki, Heck and Buchwald Reactions. Asian J. Org. Chem. 2022, 11, e202100638. [Google Scholar] [CrossRef]
- Khan, D.; Parveen, I. Chroman-4-one-Based Amino Bidentate Ligand: An Efficient Ligand for Suzuki-Miyaura and Mizoroki-Heck Coupling Reactions in Aqueous Medium. Eur. J. Org. Chem. 2021, 35, 4946–4957. [Google Scholar] [CrossRef]
- Joo, Y.H.; Kim, J.K.; Kang, S.-H.; Noh, M.-S.; Ha, J.Y.; Choi, J.C.; Lim, K.M.; Lee, C.H.; Chung, S. 2,3-Diarylbenzopyran derivatives as a novel class of selective cyclooxygenase-2 inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 413–417. [Google Scholar] [CrossRef]
- Prasanna, S.; Manivannan, E.; Chaturvedi, S.C. Quantitative structure–activity relationship analysis of a series of 2,3-diaryl benzopyran analogues as novel selective cyclooxygenase-2 inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 4005–4011. [Google Scholar] [CrossRef]
- O’Brien, D.F.; Gates, J.W., Jr. Some Reactions of 3-Hydroxy-1-phenylpyrazole. J. Org. Chem. 1966, 31, 1538–1542. [Google Scholar] [CrossRef]
- Milišiūnaitė, V.; Arbačiauskienė, E.; Řezníčková, E.; Jorda, R.; Malínková, V.; Žukauskaitė, A.; Holzer, W.; Šačkus, A.; Kryštof, V. Synthesis and anti-mitotic activity of 2,4- or 2,6-disubstituted- and 2,4,6-trisubstituted-2H-pyrazolo[4,3-c]pyridines. Eur. J. Med. Chem. 2018, 150, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Urbonavičius, A.; Fortunato, G.; Ambrazaitytė, E.; Plytninkienė, E.; Bieliauskas, A.; Milišiūnaitė, V.; Luisi, R.; Arbačiauskienė, E.; Krikštolaitytė, S.; Šačkus, A. Synthesis and Characterization of Novel Heterocyclic Chalcones from 1-Phenyl-1H-pyrazol-3-ol. Molecules 2022, 27, 3752. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhou, Q.; Xiong, W.; Pu, W.; Zhang, W.; Zhang, G.; Wang, C. Synthesis of 5-subsituted flavonols via the Algar-Flynn-Oyamada (AFO) reaction: The mechanistic implication. Tetrahedron 2017, 73, 4822–4829. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Hatua, K. Computational insight of the mechanism of Algar–Flynn–Oyamada (AFO) reaction. RSC Adv. 2014, 4, 18702–18709. [Google Scholar] [CrossRef]
- Ferreira, D.; Brandt, E.V.; Volsteedt, F.D.R.; Roux, D.G. Parameters regulating the α- and β-cyclization of chalcones. J. Chem. Soc. Perkin Trans 1975, 1, 1437–1446. [Google Scholar] [CrossRef]
- Pati, S.K.; Marks, T.J.; Ratner, M.A. Conformationally Tuned Large Two-Photon Absorption Cross Sections in Simple Molecular Chromophores. J. Am. Chem. Soc. 2001, 123, 7287–7291. [Google Scholar] [CrossRef]
- Jutand, A.; Mosleh, A. Rate and Mechanism of Oxidative Addition of Aryl Triflates to Zerovalent Palladium Complexes. Evidence for the Formation of Cationic (.sigma.-Aryl)palladium Complexes. Organometallics 1995, 14, 1810–1817. [Google Scholar] [CrossRef]
- Kumar, A.; Rao, M.L.N. Pot-economic synthesis of diarylpyrazoles and pyrimidines involving Pd-catalyzed cross-coupling of 3-trifloxychromone and triarylbismuth. J. Chem. Sci. 2018, 130, 165. [Google Scholar] [CrossRef]
- Dahlén, K.; Wallén, E.A.A.; Grøtli, M.; Luthman, K. Synthesis of 2,3,6,8-Tetrasubstituted Chromone Scaffolds. J. Org. Chem. 2006, 71, 6863–6871. [Google Scholar] [CrossRef] [PubMed]
- Akrawi, D.A.; Patonay, T.; Kónya, K.; Langer, P. Chemoselective Suzuki–Miyaura Cross-Coupling Reactions of 6-Bromo-3-(trifluoromethylsulfonyloxy)flavone. Synlett 2013, 24, 860–864. [Google Scholar] [CrossRef]
- Nuzillard, J.-M. Use of carbon-13 NMR to identify known natural products by querying a nuclear magnetic resonance database—An assessment. Magn Reson Chem 2023, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dolbier, W.R. Guide to Fluorine NMR for Organic Chemists; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016. [Google Scholar]
- Arbačiauskienė, E.; Martynaitis, V.; Krikštolaitytė, S.; Holzer, W.; Šačkus, A. Synthesis of 3-substituted 1-phenyl-1H-pyrazole-4-carbaldehydes and the corresponding ethanones by Pd-catalysed cross-coupling reactions. ARKIVOC 2011, 11, 1–21. [Google Scholar] [CrossRef]
- Solum, M.S.; Altmann, K.L.; Strohmeier, M.; Berges, D.A.; Zhang, Y.; Facelli, J.C.; Pugmire, R.J.; Grant, D.M. 15N Chemical Shift Principal Values in Nitrogen Heterocycles. J. Am. Chem. Soc. 1997, 119, 9804–9809. [Google Scholar] [CrossRef]
- Williamson, R.T.; Buevich, A.V.; Martin, G.E.; Parella, T. LR-HSQMBC: A Sensitive NMR Technique To Probe Very Long-Range Heteronuclear Coupling Pathways. J. Org. Chem. 2014, 79, 3887–3894. [Google Scholar] [CrossRef]
- Barczyński, P.; Szafran, M.; Ratajczak-Sitarz, M.; Nowaczyk, Ł.; Dega-Szafran, Z.; Katrusiak, A. Structure of 2,3-dicarboxy-1-methylpyridinium chloride studied by X-ray diffraction, DFT calculation, NMR, FTIR and Raman spectra. J. Mol. Struct. 2012, 1018, 21–27. [Google Scholar] [CrossRef]
- Iwatsuki, S.; Kanamitsu, Y.; Ohara, H.; Kawahata, M.; Danjo, H.; Ishihara, K. Crystal Structure of a Methanesulfonate Salt of 4-(N-Methyl)pyridinium Boronic Acid. X-ray Struct. Anal. Online 2012, 28, 63–64. [Google Scholar] [CrossRef]
- Macdonald, A.L.; James Trotter, J. Crystal and molecular structure of o-benzoquinone. J. Chem. Soc. Perkin Trans. 1973, 2, 476–480. [Google Scholar] [CrossRef]
- Allinger, N.L.; Chen, K.-H.; Lii, J.-H.; Durkin, K.A. Alcohols, ethers, carbohydrates, and related compounds. I. The MM4 force field for simple compounds. J. Comput. Chem. 2003, 24, 1447–1472. [Google Scholar]
- Li, P.; Su, W.; Lei, X.; Xiao, Q.; Huang, S. Synthesis, characterization and anticancer activity of a series of curcuminoids and their half-sandwich ruthenium(II) complexes. Appl. Organomet. Chem. 2017, 31, e3685. [Google Scholar] [CrossRef]
- Milišiūnaitė, V.; Arbačiauskienė, E.; Bieliauskas, A.; Vilkauskaitė, G.; Šačkus, A.; Holzer, W. Synthesis of pyrazolo[4′,3′:3,4]pyrido[1,2-a]benzimidazoles and related new ring systems by tandem cyclisation of vic-alkynylpyrazole-4-carbaldehydes with (het)aryl-1,2-diamines and investigation of their optical properties. Tetrahedron 2015, 71, 3385–3395. [Google Scholar] [CrossRef]
- Arbačiauskienė, E.; Krikštolaitytė, S.; Mitrulevičienė, A.; Bieliauskas, A.; Martynaitis, V.; Bechmann, M.; Roller, A.; Šačkus, A.; Holzer, W. On the Tautomerism of N-Substituted Pyrazolones: 1,2-Dihydro-3H-pyrazol-3-ones versus 1H-Pyrazol-3-ols. Molecules 2018, 23, 129. [Google Scholar] [CrossRef]
- Titi, A.; Messali, M.; Alqurashy, B.A.; Touzani, R.; Shiga, T.; Oshio, H.; Fettouhi, M.; Rajabi, M.; Almalki, F.A.; Hadda, T.B. Synthesis, characterization, X-ray crystal study and bioctivities of pyrazole derivatives: Identification of antitumor, antifungal and antibacterial pharmacophore sites. J. Mol. Struct. 2020, 1205, 127625. [Google Scholar] [CrossRef]
- Sharma, S.; Brahmachari, G.; Kant, R.; Gupta, V.K. One-pot green synthesis of biologically relevant novel spiro[indolin-2-one-3,4′-pyrano[2,3-c]pyrazoles] and studies on their spectral and X-ray crystallographic behaviors. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Shynkar, V.V.; Mély, Y.; Duportail, G.; Piémont, E.; Klymchenko, A.S.; Demchenko, A.P. Picosecond Time-Resolved Fluorescence Studies Are Consistent with Reversible Excited-State Intramolecular Proton Transfer in 4′-(Dialkylamino)-3-hydroxyflavones. J. Phys. Chem. A 2003, 107, 9522–9529. [Google Scholar] [CrossRef]
- Spadafora, M.; Postupalenko, V.; Shvadchak, V.; Klymchenko, A.; Mély, Y.; Burger, A.; Benhida, R. Efficient synthesis of ratiometric fluorescent nucleosides featuring 3-hydroxychromone nucleobases. Tetrahedron 2009, 65, 7809–7816. [Google Scholar] [CrossRef]
- Klymchenko, A.S.; Ozturk, T.; Pivovarenko, V.G.; Demchenko, A.P. A 3-hydroxychromone with dramatically improved fluorescence properties. Tetrahedron Lett. 2001, 42, 7967–7970. [Google Scholar] [CrossRef]
- Klymchenko, A.S.; Kenfack, C.; Duportail, G.; Mély, Y. Effects of polar protic solvents on dual emissions of 3-hydroxychromones. J. Chem. Sci. 2007, 119, 83–89. [Google Scholar] [CrossRef]
- Voicescu, M.; Ionescu, S.; Gatea, F. Effect of pH on the fluorescence characteristics of some flavones probes. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 123, 303–308. [Google Scholar] [CrossRef]
- Klymchenko, A.S.; Pivovarenko, V.G.; Ozturk, T.; Demchenko, A.P. Modulation of the solvent-dependent dual emission in 3-hydroxychromones by substituents. New J. Chem. 2003, 27, 1336–1343. [Google Scholar] [CrossRef]
- Klymchenko, A.S.; Demchenko, A.P. Multiparametric probing of intermolecular interactions with fluorescent dye exhibiting excited state intramolecular proton transfer. Phys. Chem. Chem. Phys. 2003, 5, 461–468. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, Y.; Zhou, L.; Li, Y.; Chen, M. Time-dependent density functional theory study on excited state intramolecular proton transfer of 3-hydroxy-2-(pyridin-2-yl)-4H-chromen-4-one. J. Lumin. 2010, 130, 1431–1436. [Google Scholar] [CrossRef]
- Chen, L.; Fu, P.-Y.; Wang, H.-P.; Pan, M. Excited-State Intramolecular Proton Transfer (ESIPT) for Optical Sensing in Solid State. Adv. Optical Mater. 2021, 9, 2170097. [Google Scholar] [CrossRef]
- Ormson, S.M.; Brown, R.G.; Voller, F.; Rettig, W. Switching between charge- and proton-transfer emission in the excited state of a substituted 3-hydroxyflavone. J. Photochem. Photobiol. A Chem. 1994, 81, 65–72. [Google Scholar] [CrossRef]
- Lebeau, B.; Innocenzi, P. Hybrid materials for optics and photonics. Chem. Soc. Rev. 2011, 40, 886–906. [Google Scholar] [CrossRef]
- Mohammad-Pour, G.S.; de Coene, Y.; Wiratmo, M.; Maan, A.; Clays, K.; Masunov, A.E.; Crawford, K.E. Modular synthesis of zwitterionic, xanthene bridged, low twist angle chromophores with high hyperpolarizability. Mater. Adv. 2022, 3, 7520–7530. [Google Scholar] [CrossRef]
- Andreu, R.; Carrasquer, L.; Santiago Franco, C.; Garín, J.; Orduna, J.; de Baroja, N.M.; Alicante, R.; Villacampa, B.; Allain, M. 4H-Pyran-4-ylidenes: Strong Proaromatic Donors for Organic Nonlinear Optical Chromophores. J. Org. Chem. 2009, 74, 6647–6657. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. A Complete Structure Solution, Refinement and Analysis Program. J.Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment – Olex2 dissected. Acta Cryst. 2015, A71, 59–75. [Google Scholar]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom | Atom | Length/Å | Atom | Atom | Length/Å |
|---|---|---|---|---|---|
| N1 | N2 | 1.3839(12) | C4 | C5 | 1.5140(14) |
| N1 | C7A | 1.3233(14) | C4 | O14 | 1.2265(14) |
| N2 | C3 | 1.3458(14) | C5 | C6 | 1.4047(15) |
| N2 | C8 | 1.4287(13) | C5 | O15 | 1.2723(13) |
| C3 | C3A | 1.3842(14) | C6 | O7 | 1.4135(12) |
| C3A | C4 | 1.4411(14) | C6 | C16 | 1.4308(14) |
| C3A | C7A | 1.4017(14) | O7 | C7A | 1.3387(12) |
| Atom | Atom | Atom | Angle/° | Atom | Atom | Atom | Angle/° |
|---|---|---|---|---|---|---|---|
| C7A | N1 | N2 | 102.23(8) | O14 | C4 | C5 | 120.99(10) |
| C3 | N2 | N1 | 113.26(8) | C6 | C5 | C4 | 119.08(9) |
| C8 | N2 | N1 | 119.35(8) | O15 | C5 | C4 | 116.91(9) |
| C8 | N2 | C3 | 127.39(9) | O15 | C5 | C6 | 124.01(10) |
| C3A | C3 | N2 | 106.46(9) | O7 | C6 | C5 | 123.66(9) |
| C4 | C3A | C3 | 134.37(10) | C16 | C6 | C5 | 125.60(10) |
| C7A | C3A | C3 | 104.07(9) | C16 | C6 | O7 | 110.74(9) |
| C7A | C3A | C4 | 121.56(9) | C7A | O7 | C6 | 116.70(8) |
| C5 | C4 | C3A | 113.86(9) | C3A | C7A | N1 | 113.97(9) |
| O14 | C4 | C3A | 125.15(10) | O7 | C7A | N1 | 120.98(9) |
| Entry | Comp. | λabs (nm) | ε × 103 (dm3 mol−1 cm−1) | λN*em (nm) | λT*em (nm) | IN*/IT* | Stokes Shift (cm−1) | Φf (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 3a | 337sh 311 | 70.89 78.45 | 482 | 582 | 0.102 | 8927 12491 | 59.3 |
| 2 | 3b | 341sh 317 | 112.50 119.51 | 428 | 586 | 0.009 | 5961 12261 | 42.7 |
| 3 | 3c | 355 320sh 240 | 93.74 66.27 54.91 | 446 | 582 | 0.161 | 5747 10987 | 13.4 |
| 4 | 3d | 361 311 261 | 43.82 34.60 23.65 | 479 | 580 | 0.406 | 6824 10459 | 52.7 |
| 5 | 3e | 353 321sh 310sh 293 245sh | 70.76 60.39 57.42 56.73 55.59 | 435 | 591 | 0.043 | 5340 11408 | 76.1 |
| 6 | 3f | 365 317sh 266 | 80.34 59.96 41.23 | 438 | 582 | 0.046 | 4566 10215 | 55.8 |
| 7 | 3g | 360 317 260 | 138.88 116.75 53.16 | 435 | 575 | 0.054 | 4789 10386 | 42.6 |
| 8 | 3h | 355sh 329 | 49.99 61.54 | 493 | 611 | 0.031 | 7885 11802 | 13.1 |
| Entry | Comp. | λabs (nm) | ε × 103 (dm3 mol−1 cm−1) | λN*em (nm) | λT*em (nm) | IN*/IT* | Stokes Shift (cm−1) | Φf (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 3aa | 339sh 315 | 58.67 69.15 | 466 | 588 | 0.014 | 8039 12492 | 59.2 |
| 2 | 3ba | 339 317 240 | 49.24 53.74 22.77 | 428 | 590 | 0.019 | 6134 6134 | 75.5 |
| 3 | 3ca | 353 318sh 260 | 69.71 52.84 30.30 | 428 | 591 | 0.005 | 4964 11408 | 19.2 |
| 4 | 3da | 362 311 268 | 58.61 44.19 23.99 | 429 | 594 | 0.006 | 4314 10789 | 39.6 |
| 5 | 3ea | 352 334sh 295 283 | 63.18 59.61 46.02 45.73 | 416 | 598 | 0.012 | 4371 11687 | 50.6 |
| 6 | 3fa | 357 327sh 266 | 35.90 28.75 15.87 | 423 | 593 | 0.023 | 43,701 11148 | 41.2 |
| 7 | 3ga | 352 317 262 | 82.58 75.99 32.57 | 421 | 584 | 0.020 | 4656 11286 | 55.0 |
| 8 | 3ha | 351sh 335sh 319 | 40.75 57.39 64.62 | 442 | 610 | 0.221 | 5866 12097 | 30.1 |
| 9 | 3ab | 338sh 315 | 64.22 75.82 | 428 | 589 | 0.009 | 6221 12608 | 45.5 |
| 10 | 3ac | 357sh 338 322 | 43.31 57.48 62.27 | 430 | 581 | 0.004 | 4755 10800 | 67.7 |
| Entry | Comp. | λabs (nm) | ε × 103 (dm3 mol−1 cm−1) |
|---|---|---|---|
| 1 | 5 | 528 | 0.55 |
| 499 | 0.48 | ||
| 346 | 0.39 | ||
| 299 | 0.42 | ||
| 261 | 0.30 | ||
| 2 | 6 | 528 | 0.54 |
| 499 | 0.49 | ||
| 348 | 0.52 | ||
| 297 | 0.49 | ||
| 261 | 0.37 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urbonavičius, A.; Krikštolaitytė, S.; Bieliauskas, A.; Martynaitis, V.; Solovjova, J.; Žukauskaitė, A.; Arbačiauskienė, E.; Šačkus, A. Synthesis and Characterization of New Pyrano[2,3-c]pyrazole Derivatives as 3-Hydroxyflavone Analogues. Molecules 2023, 28, 6599. https://doi.org/10.3390/molecules28186599
Urbonavičius A, Krikštolaitytė S, Bieliauskas A, Martynaitis V, Solovjova J, Žukauskaitė A, Arbačiauskienė E, Šačkus A. Synthesis and Characterization of New Pyrano[2,3-c]pyrazole Derivatives as 3-Hydroxyflavone Analogues. Molecules. 2023; 28(18):6599. https://doi.org/10.3390/molecules28186599
Chicago/Turabian StyleUrbonavičius, Arminas, Sonata Krikštolaitytė, Aurimas Bieliauskas, Vytas Martynaitis, Joana Solovjova, Asta Žukauskaitė, Eglė Arbačiauskienė, and Algirdas Šačkus. 2023. "Synthesis and Characterization of New Pyrano[2,3-c]pyrazole Derivatives as 3-Hydroxyflavone Analogues" Molecules 28, no. 18: 6599. https://doi.org/10.3390/molecules28186599
APA StyleUrbonavičius, A., Krikštolaitytė, S., Bieliauskas, A., Martynaitis, V., Solovjova, J., Žukauskaitė, A., Arbačiauskienė, E., & Šačkus, A. (2023). Synthesis and Characterization of New Pyrano[2,3-c]pyrazole Derivatives as 3-Hydroxyflavone Analogues. Molecules, 28(18), 6599. https://doi.org/10.3390/molecules28186599