
N-Heterocyclic Carbene-Iridium Complexes as Photosensitizers for In Vitro Photodynamic Therapy to Trigger Non-Apoptotic Cell Death in Cancer Cells
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results and Discussion
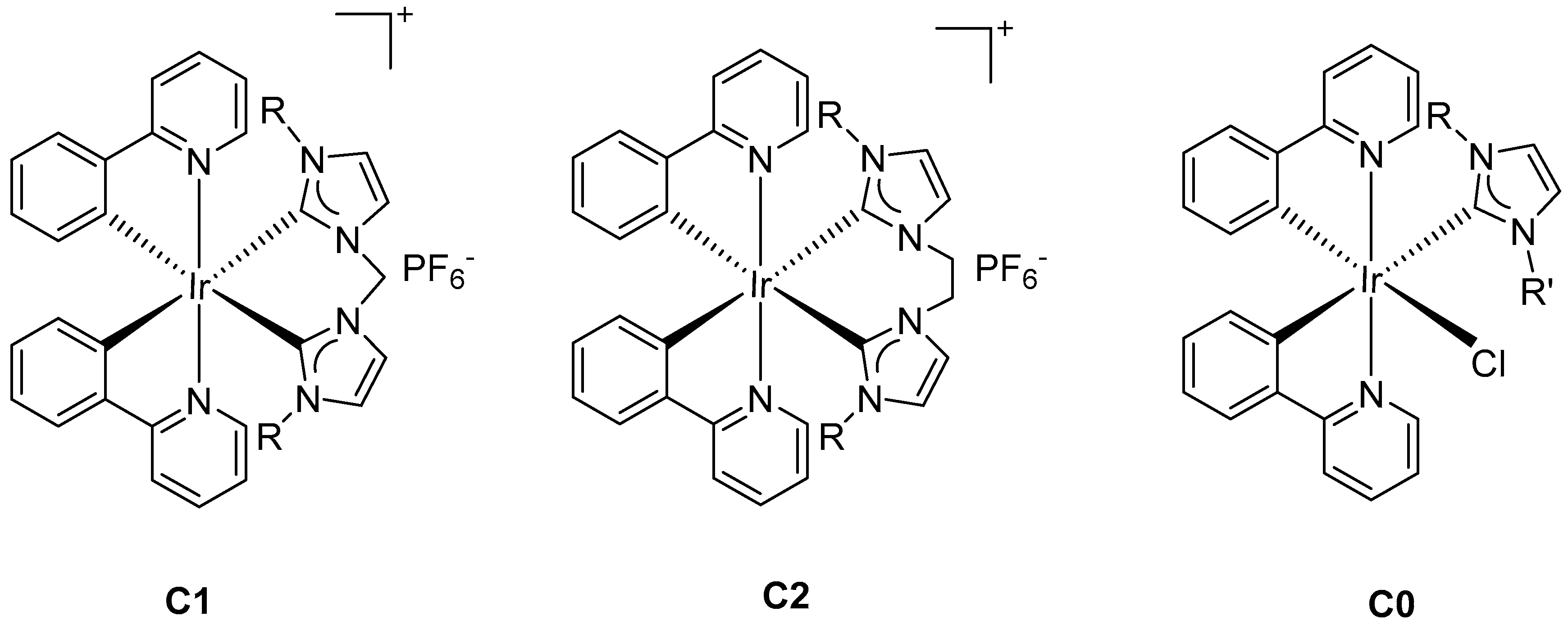
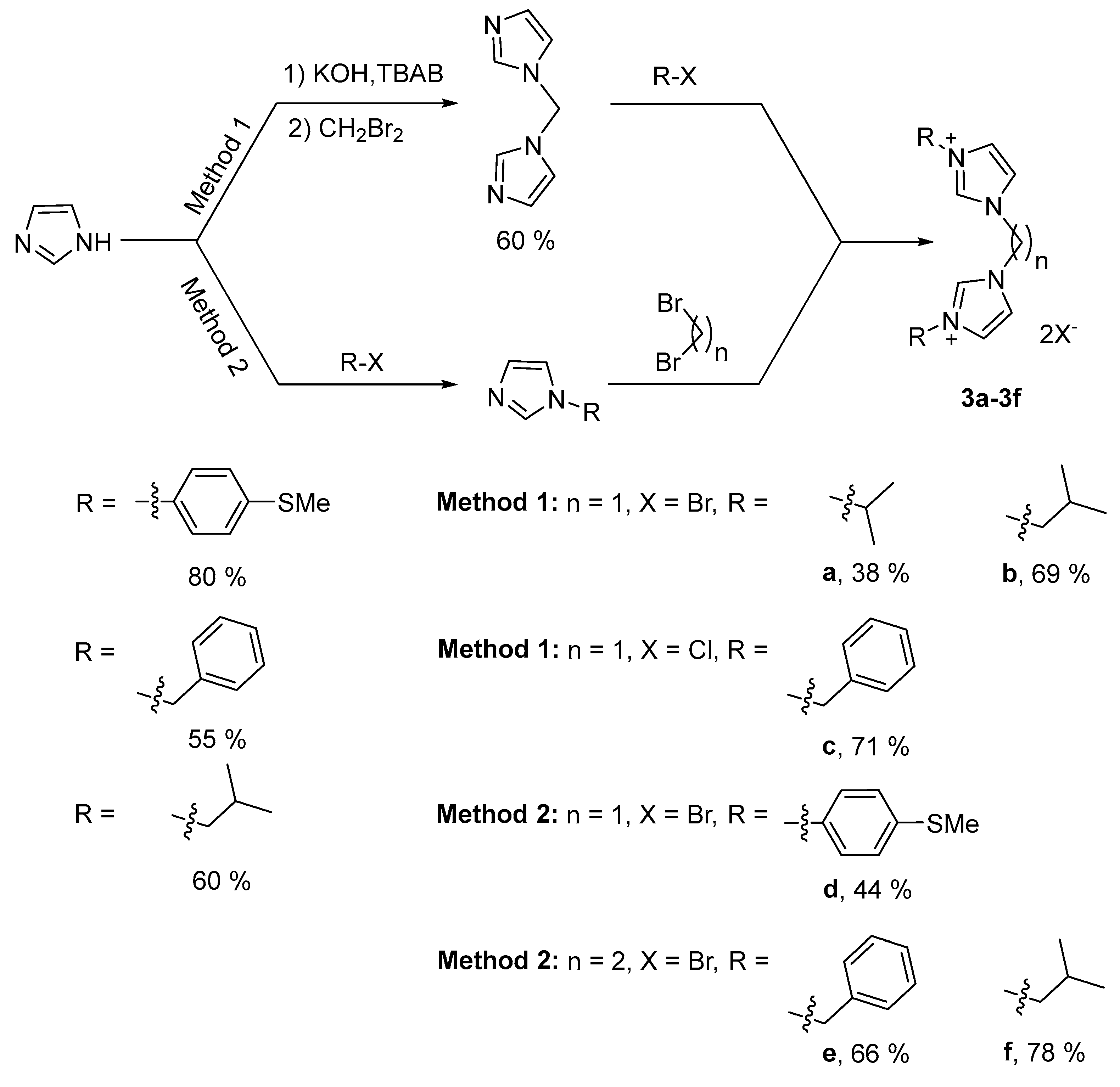
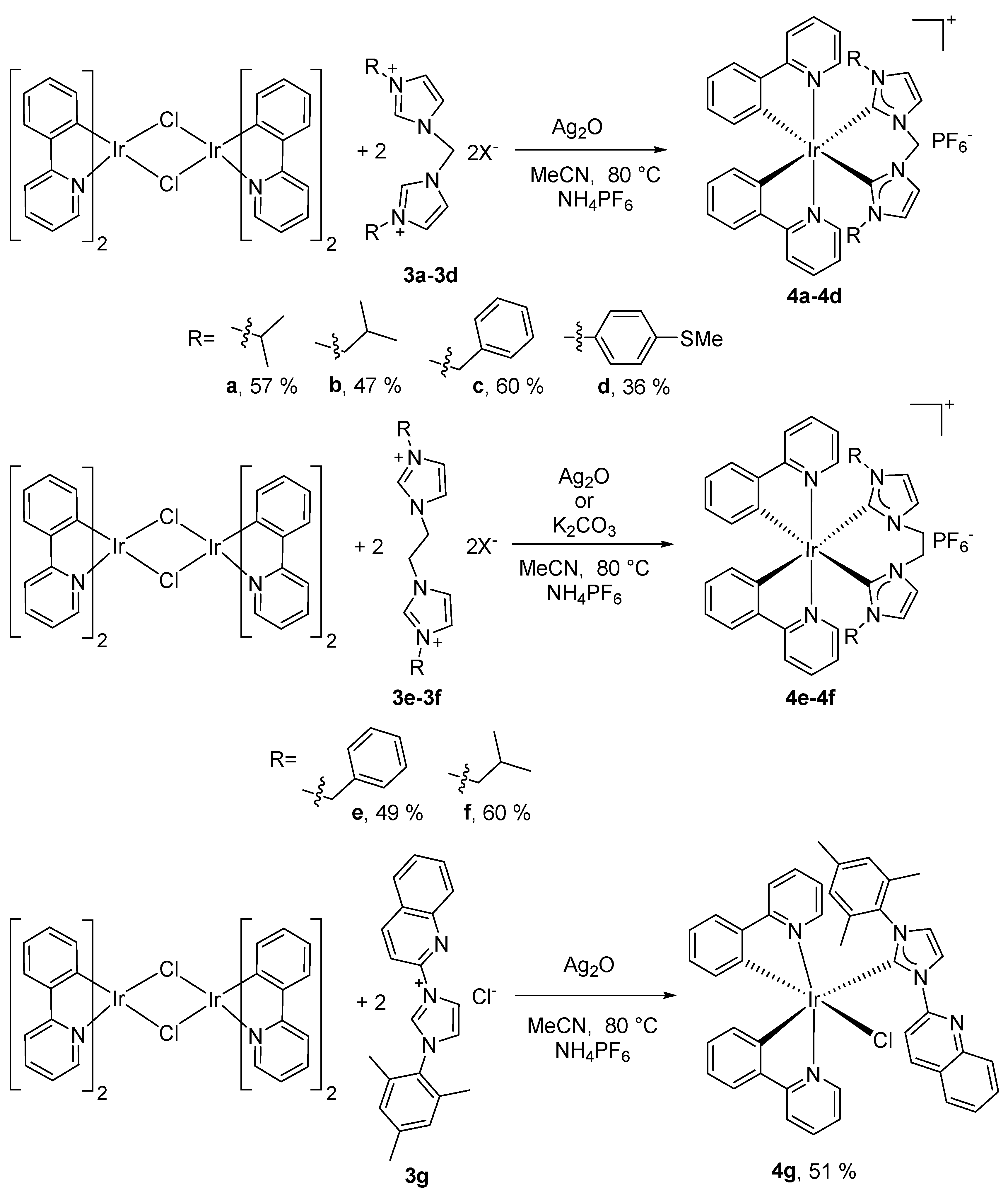
2.1. Synthesis and Characterization of Cyclometalated Iridium (NHC) Complexes
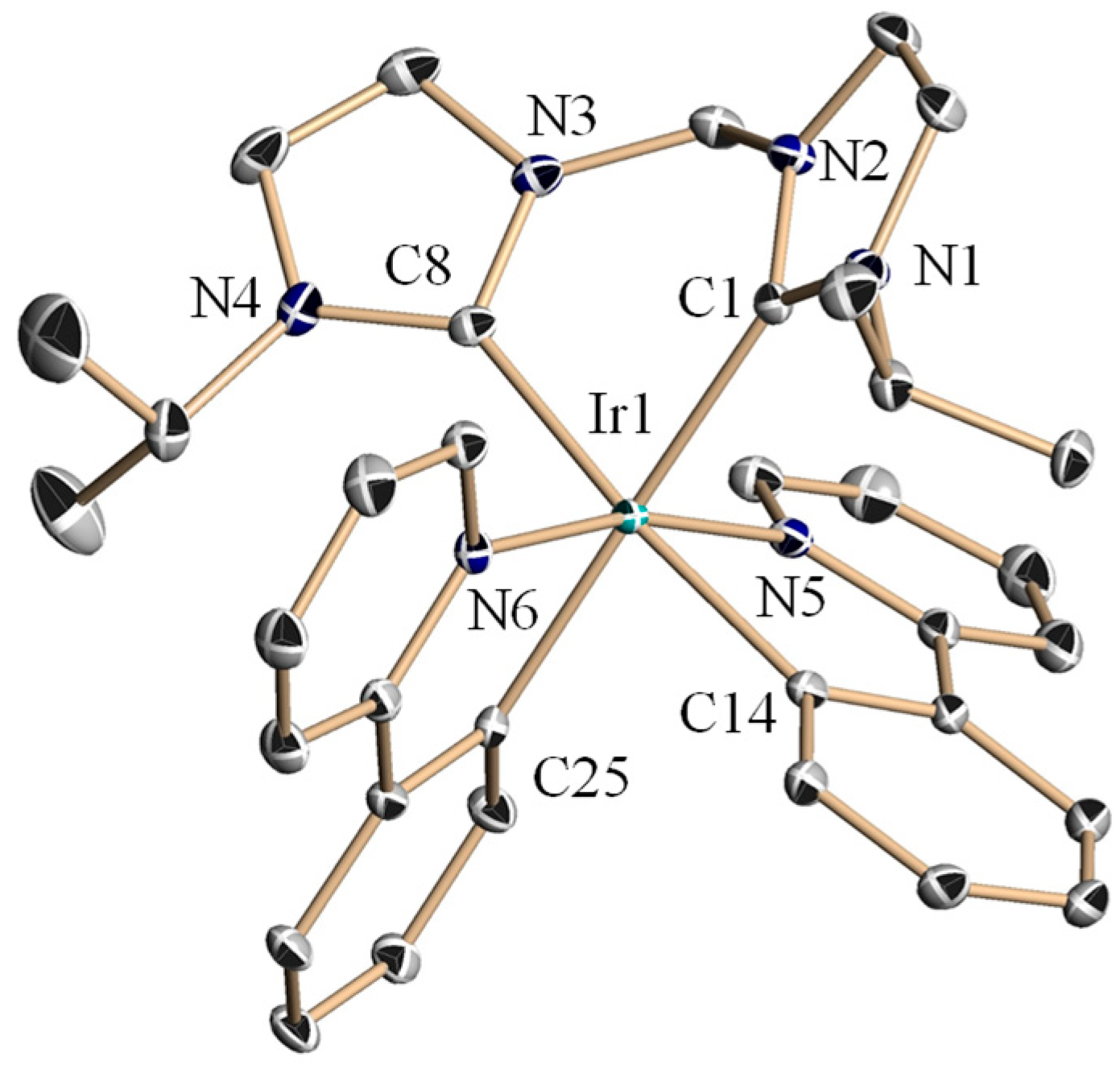
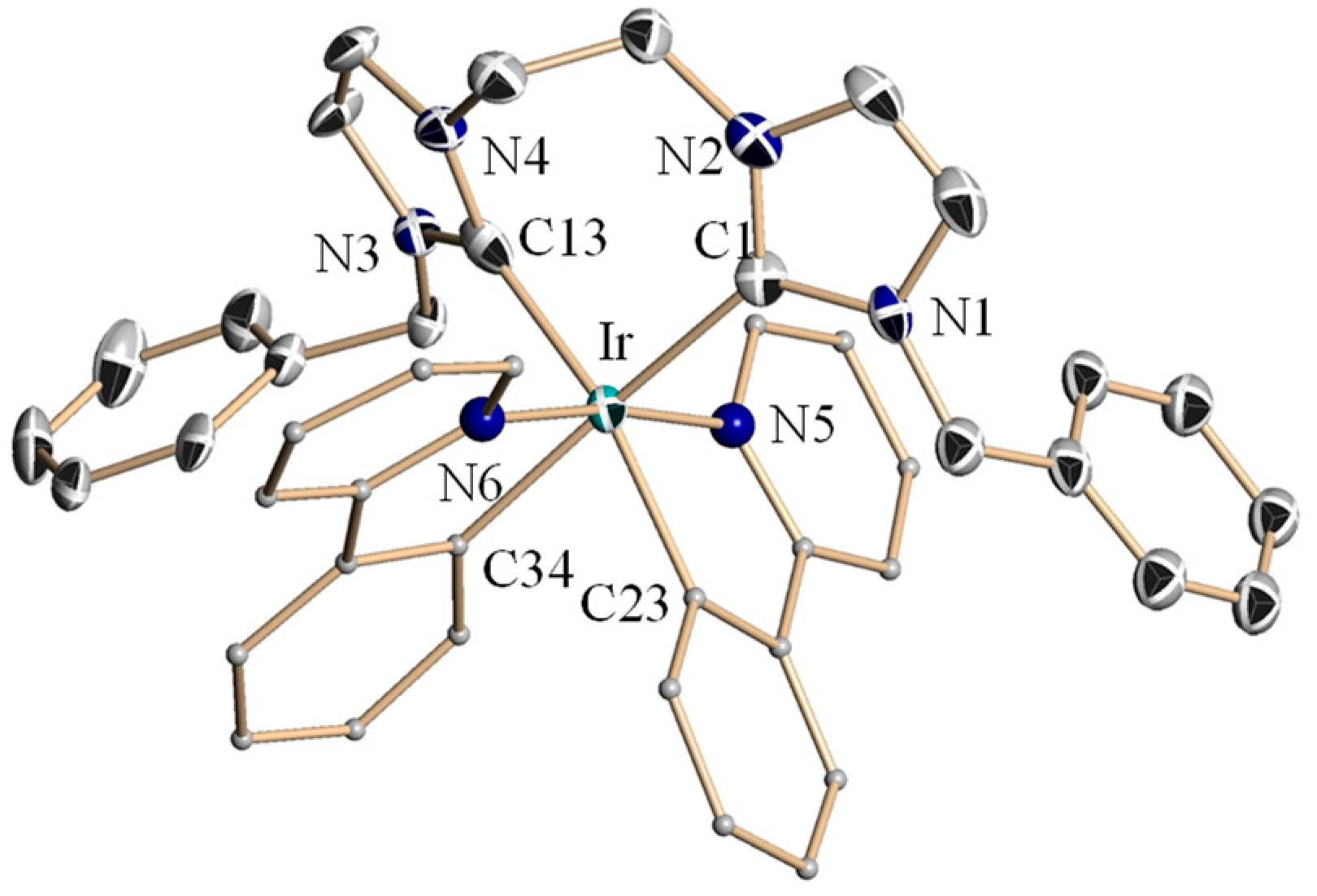
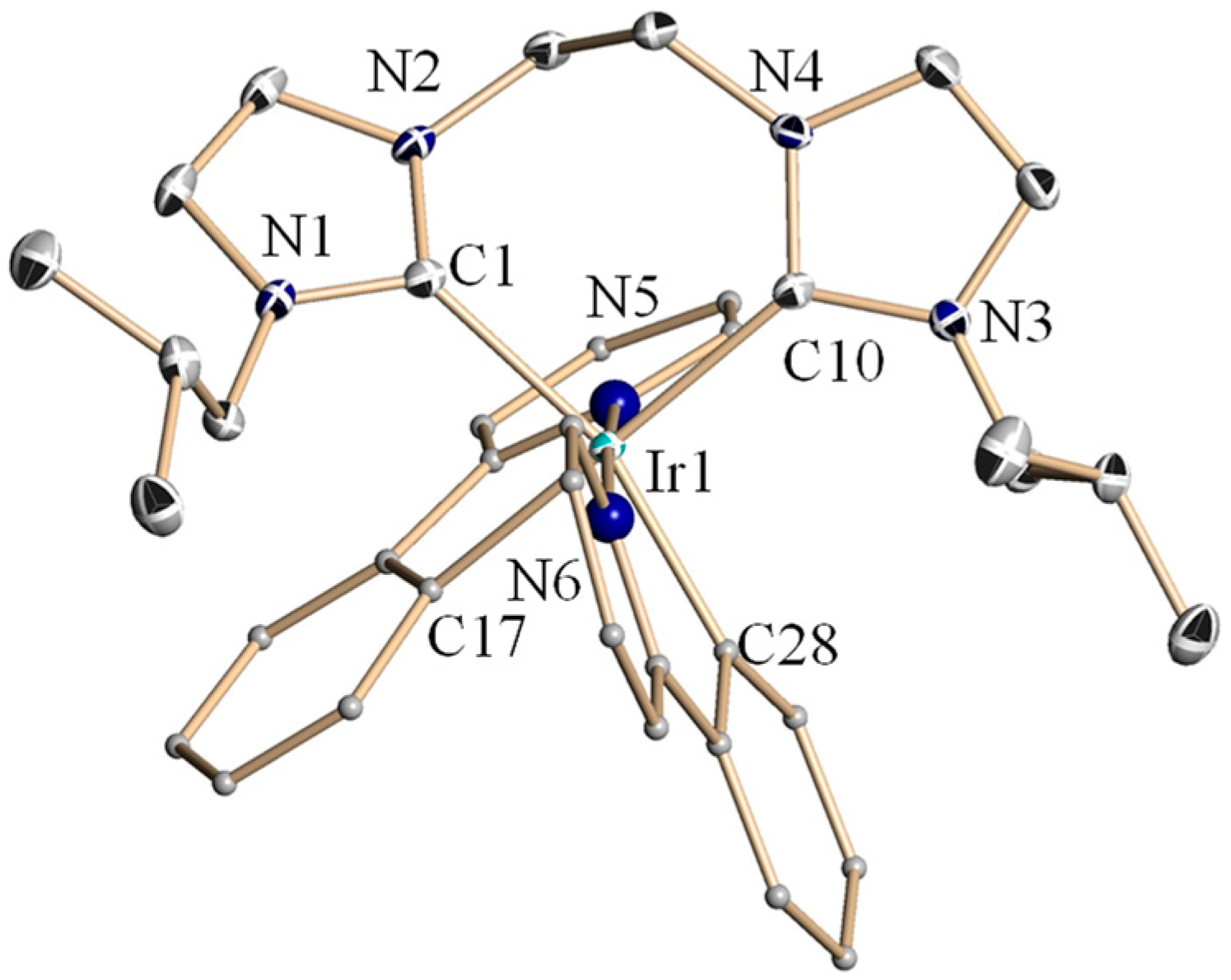
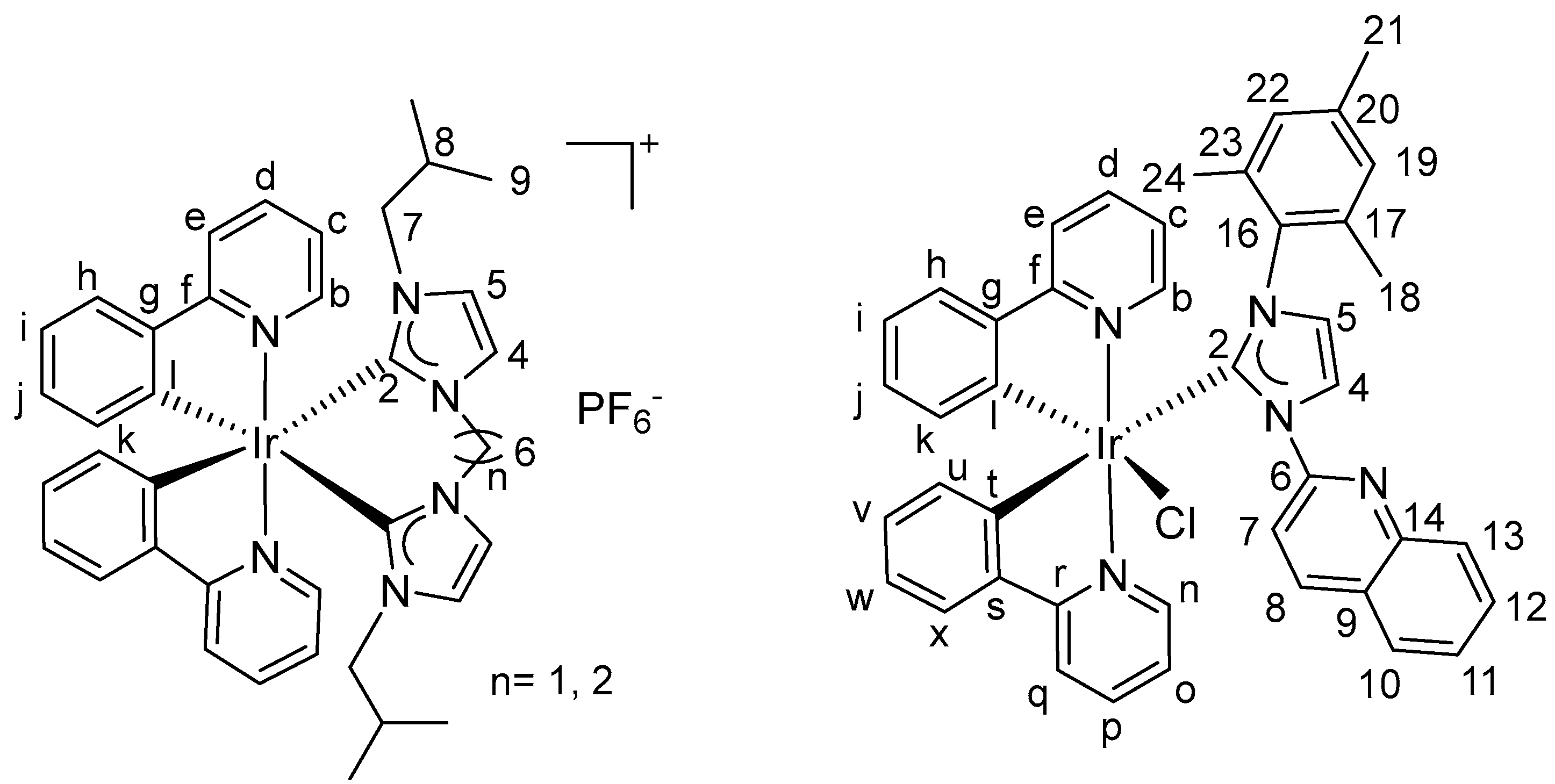
2.2. Crystal Structures of Complexes 4a–4c and 4e–4f
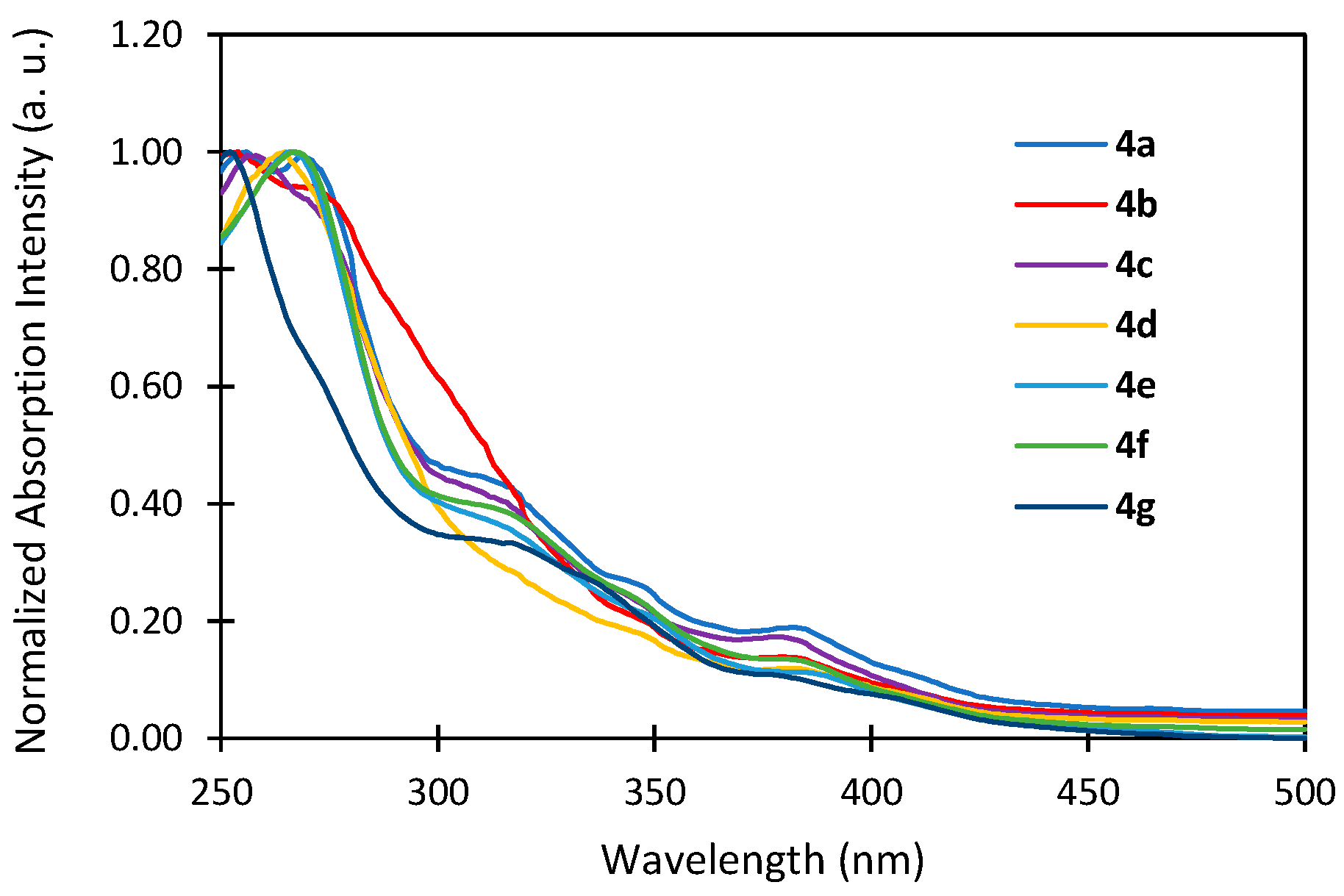
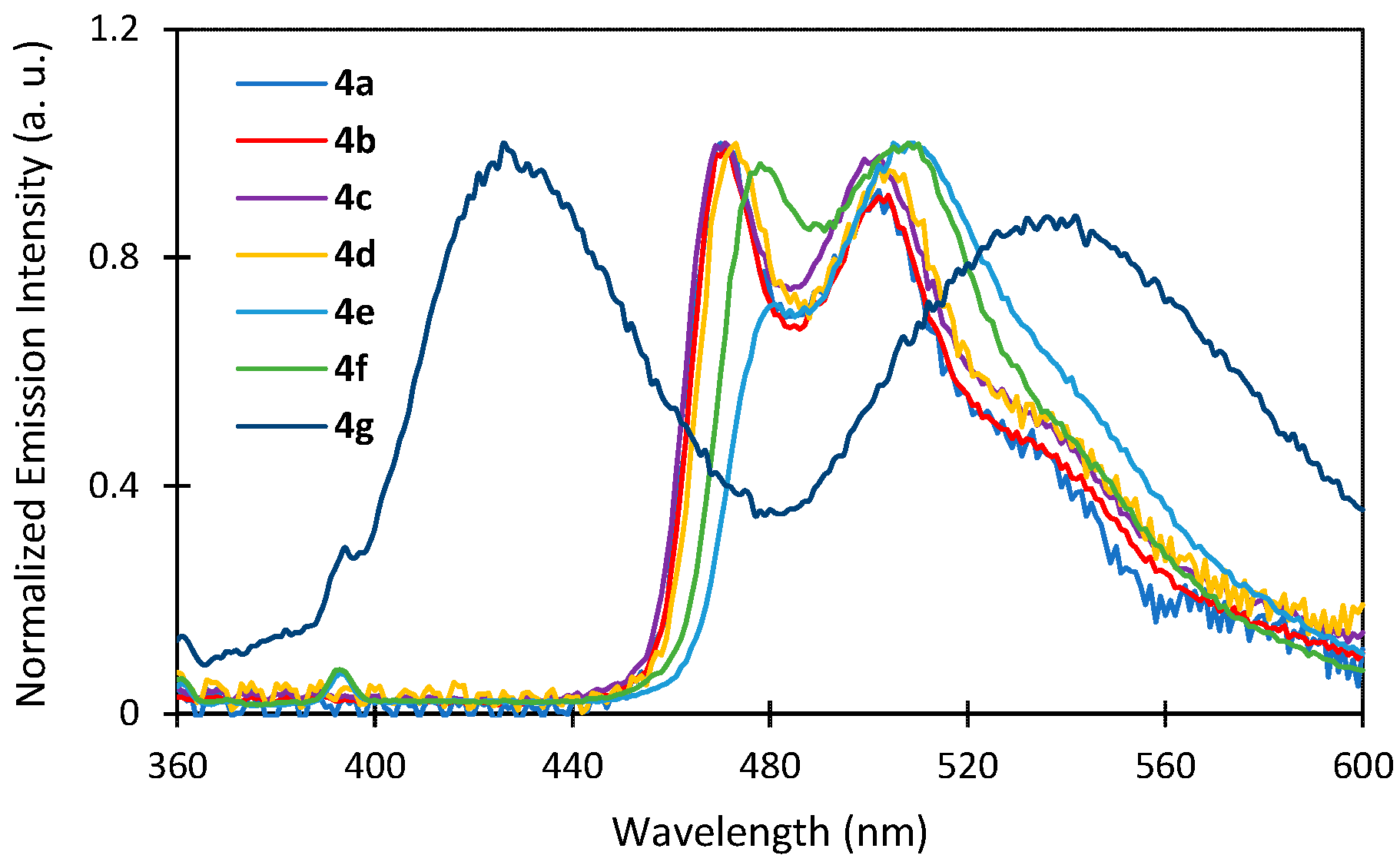
2.3. Photophysical Properties
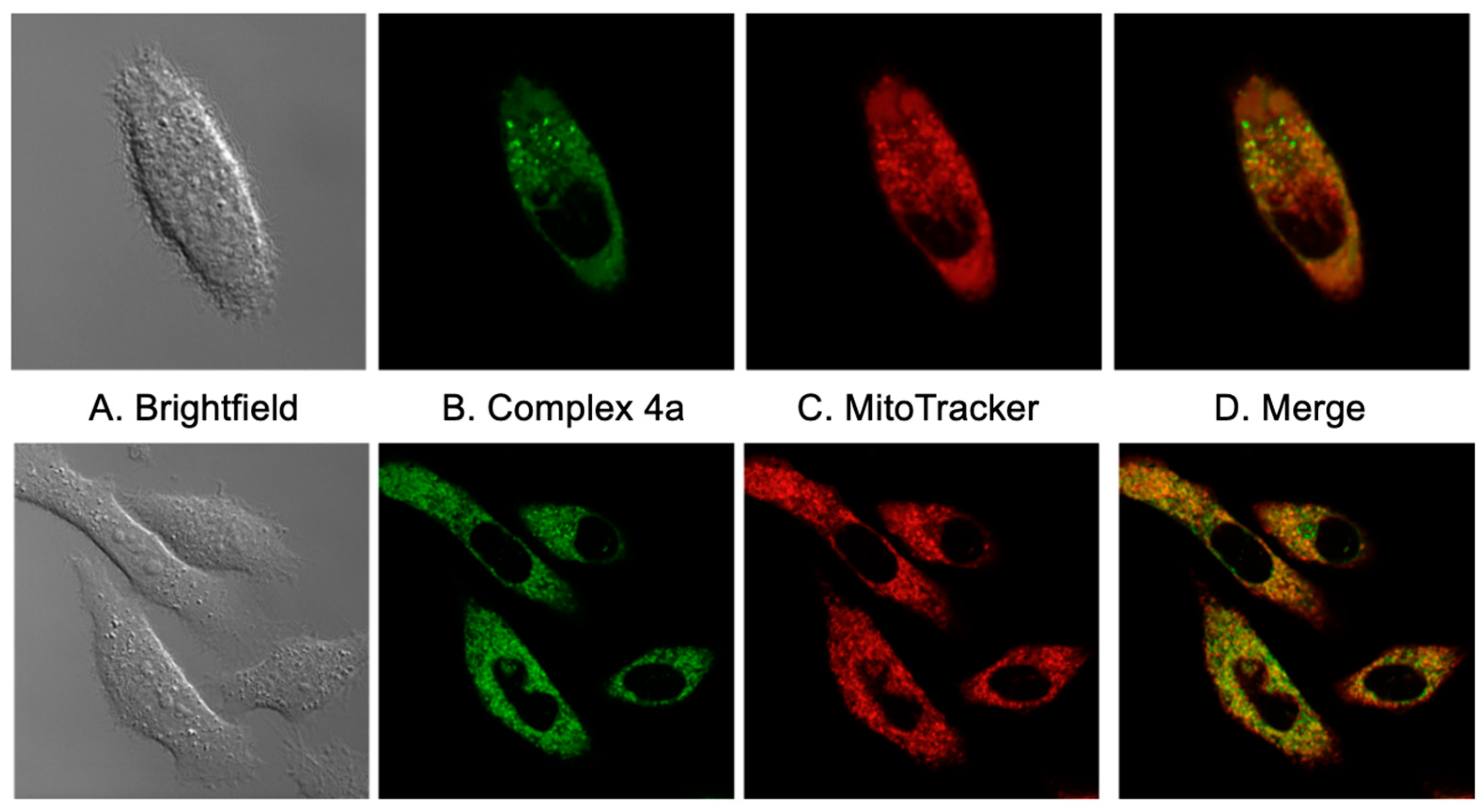
2.4. Subcellular Localization
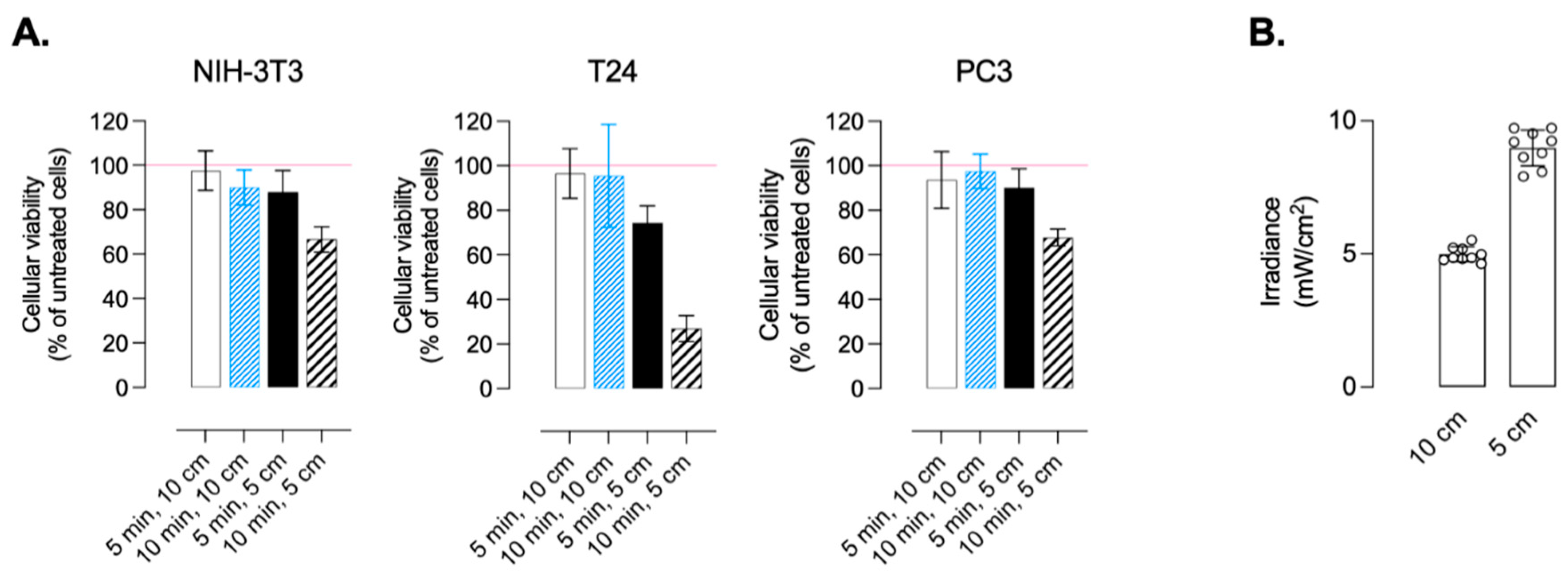
2.5. In Vitro Cytotoxicity
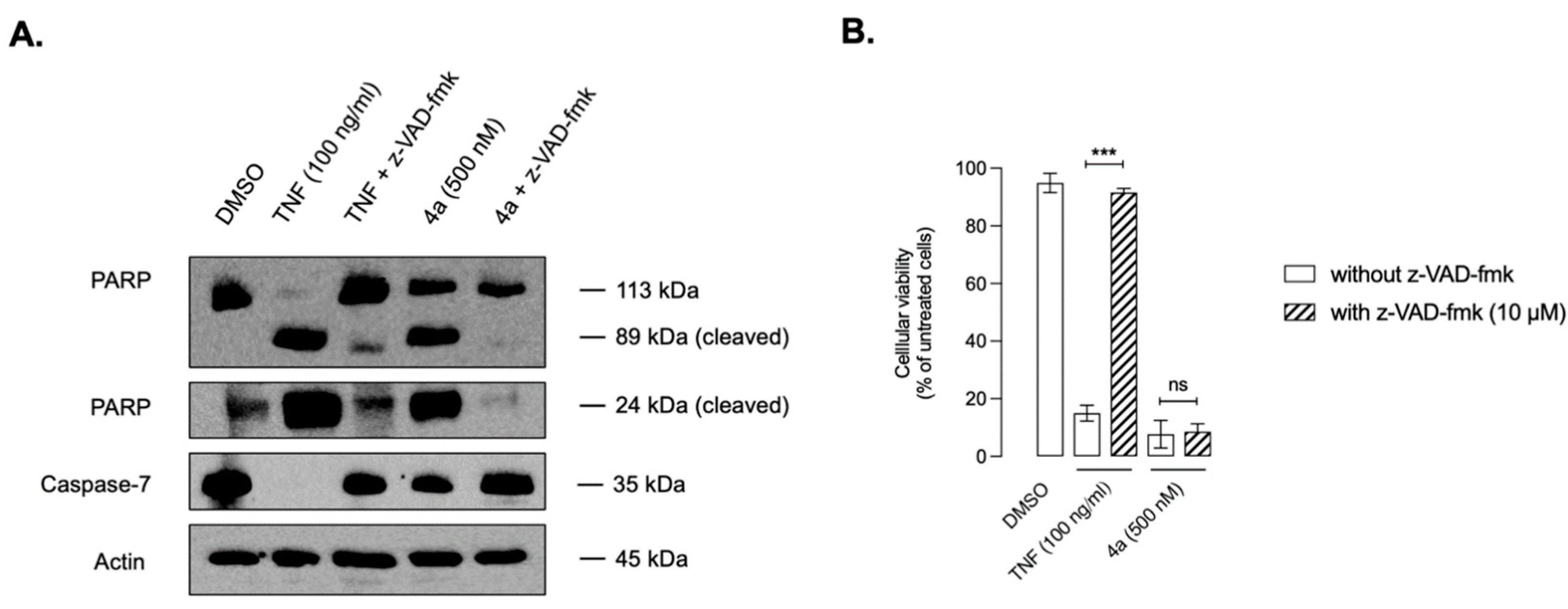
2.6. Mechanistic Studies
3. Material and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. Preparation of Imidazolium Salts
3.1.3. Synthesis of Iridium(III) Complexes
3.1.4. Crystallographic Data for 4a, 4b, 4c, 4e and 4f
3.1.5. Measurement of Lipophilicity
3.1.6. Photophysical Measurements
3.2. Biology
3.2.1. Cell Culture and Reagents
3.2.2. Treatments and Conditions of Irradiation
3.2.3. Cell Viability Assays
3.2.4. ATP and GSH Quantification
3.2.5. Preparation of Cellular Extracts and Western Blot Analysis
3.2.6. Mitochondrial Subcellular Localization
3.2.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- World Health Organisation. WHO Report on Cancer Settings Priorities, Investing Wysely and Providing Care for All. World Health Organ. 2020, 1–160. [Google Scholar]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef] [Green Version]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Kang, Y.; Chen, L.; Wang, H.; Liu, J.; Zeng, S.; Yu, L. The Drug-Resistance Mechanisms of Five Platinum-Based Anticancer Agents. Front. Pharmacol. 2020, 11, 343. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.J.S.A.; Gois, P.M.P.; Gasser, G. Unveiling the Potential of Transition Metal Complexes for Medicine: Translational in Situ Activation of Metal-Based Drugs from Bench to in Vivo Applications. ChemBioChem 2021, 22, 1740–1742. [Google Scholar] [CrossRef] [PubMed]
- Yusoh, N.A.; Ahmad, H.; Gill, M.R. Combining PARP Inhibition with Platinum, Ruthenium or Gold Complexes for Cancer Therapy. ChemMedChem 2020, 15, 2121–2135. [Google Scholar] [CrossRef] [PubMed]
- Ceramella, J.; Mariconda, A.; Iacopetta, D.; Saturnino, C.; Barbarossa, A.; Caruso, A.; Rosano, C.; Sinicropi, M.S.; Longo, P. From coins to cancer therapy: Gold, silver and copper complexes targeting human topoisomerases. Bioorg. Med. Chem. Lett. 2020, 30, 126905. [Google Scholar] [CrossRef]
- Liu, J.; Lai, H.; Xiong, Z.; Chen, B.; Chen, T. Functionalization and cancer-targeting design of ruthenium complexes for precise cancer therapy. Chem. Commun. 2019, 55, 9904–9914. [Google Scholar] [CrossRef]
- Simpson, P.V.; Desai, N.M.; Casari, I.; Massi, M.; Falasca, M. Metal-Based Anticancer Compounds: Beyond Cisplatin. Future Med. Chem. 2019, 11, 119–135. [Google Scholar] [CrossRef]
- O’Connor, A.E.; Gallagher, W.M.; Byrne, A.T. Porphyrin and Nonporphyrin Photosensitizers in Oncology: Preclinical and Clinical Advances in Photodynamic Therapy. Photochem. Photobiol. 2009, 85, 1053–1074. [Google Scholar] [CrossRef] [PubMed]
- Smithen, D.A.; Yin, H.; Beh, M.H.R.; Hetu, M.; Cameron, T.S.; McFarland, S.A.; Thompson, A. Synthesis and Photobiological Activity of Ru(II) Dyads Derived from Pyrrole-2-carboxylate Thionoesters. Inorg. Chem. 2017, 56, 4121–4132. [Google Scholar] [CrossRef] [PubMed]
- Monro, S.; Colón, K.L.; Yin, H.; Roque, J., III; Konda, P.; Gujar, S.; Thummel, R.P.; Lilge, L.; Cameron, C.G.; McFarland, S.A. Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev. 2019, 119, 797–828. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.-L.; Lin, S.; Wang, W.; Yang, C.; Leung, C.-H. Luminescent chemosensors by using cyclometalated iridium(iii) complexes and their applications. Chem. Sci. 2017, 8, 878–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, Y.; Chang, T.-K.; Ganesan, P.; Rajakannu, P. Emissive bis-tridentate Ir(III) metal complexes: Tactics, photophysics and applications. Coord. Chem. Rev. 2017, 346, 91–100. [Google Scholar] [CrossRef]
- McKenzie, L.K.; Bryant, H.E.; Weinstein, J.A. Transition metal complexes as photosensitisers in one- and two-photon photodynamic therapy. Coord. Chem. Rev. 2019, 379, 2–29. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.-Y.; Ho, C.-L.; Wong, W.-Y. Recent advances of iridium(III) metallophosphors for health-related applications. Coord. Chem. Rev. 2020, 413, 213267. [Google Scholar] [CrossRef]
- Zamora, A.; Vigueras, G.; Rodríguez, V.; Santana, M.D.; Ruiz, J. Cyclometalated iridium(III) luminescent complexes in therapy and phototherapy. Coord. Chem. Rev. 2018, 360, 34–76. [Google Scholar] [CrossRef]
- de Almeida, A.; Bonsignore, R. Fluorescent metal-based complexes as cancer probes. Bioorg. Med. Chem. Lett. 2020, 30, 127219. [Google Scholar] [CrossRef]
- Imberti, C.; Zhang, P.; Huang, H.; Sadler, P.J. New Designs for Phototherapeutic Transition Metal Complexes. Angew. Chem. Int. Ed. 2020, 59, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Sudhindra, P.; Roy, N.; Paira, P. Advances in novel iridium (III) based complexes for anticancer applications: A review. Inorg. Chim. Acta 2020, 513, 119925. [Google Scholar] [CrossRef]
- Guan, R.; Xie, L.; Ji, L.; Chao, H. Phosphorescent Iridium(III) Complexes for Anticancer Applications. Eur. J. Inorg. Chem. 2020, 2020, 3978–3986. [Google Scholar] [CrossRef]
- Tan, C.-P.; Zhong, Y.-M.; Ji, L.-N.; Mao, Z.-W. Phosphorescent metal complexes as theranostic anticancer agents: Combining imaging and therapy in a single molecule. Chem. Sci. 2021, 12, 2357–2367. [Google Scholar] [CrossRef] [PubMed]
- Caporale, C.; Massi, M. Cyclometalated iridium(III) complexes for life science. Coord. Chem. Rev. 2018, 363, 71–91. [Google Scholar] [CrossRef] [Green Version]
- Lv, W.; Zhang, Z.; Zhang, K.Y.; Yang, H.; Liu, S.; Xu, A.; Guo, S.; Zhao, Q.; Huang, W. A Mitochondria-Targeted Photosensitizer Showing Improved Photodynamic Therapy Effects Under Hypoxia. Angew. Chem. Int. Ed. 2016, 55, 9947–9951. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, M.; Zeng, L.; Qiu, K.; Chen, Y.; Ji, L.; Chao, H. Cyclometalated Ir III Complexes as Mitochondria-Targeted Photodynamic Anticancer Agents. Eur. J. Inorg. Chem. 2017, 2017, 1764–1771. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, B.; Lu, X.-R.; Li, M.-F.; Ji, L.-N.; Mao, Z.-W. Cyclometalated iridium(iii) N-heterocyclic carbene complexes as potential mitochondrial anticancer and photodynamic agents. Dalton Trans. 2017, 46, 11363–11371. [Google Scholar] [CrossRef]
- Zhang, C.; Lai, S.-H.; Yang, H.-H.; Xing, D.-G.; Zeng, C.-C.; Tang, B.; Wan, D.; Liu, Y.-J. Photoinduced ROS regulation of apoptosis and mechanism studies of iridium(iii) complex against SGC-7901 cells. RSC Adv. 2017, 7, 17752–17762. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, M.; Zeng, L.; Huang, H.; Jin, C.; Liu, J.; Chen, Y.; Ji, L.; Chao, H. Fluorinated cyclometalated iridium(iii) complexes as mitochondria-targeted theranostic anticancer agents. Dalton Trans. 2017, 46, 6734–6744. [Google Scholar] [CrossRef]
- Liu, J.; Jin, C.; Yuan, B.; Chen, Y.; Liu, X.; Ji, L.; Chao, H. Enhanced cancer therapy by the marriage of metabolic alteration and mitochondrial-targeted photodynamic therapy using cyclometalated Ir(iii) complexes. Chem. Commun. 2017, 53, 9878–9881. [Google Scholar] [CrossRef]
- Chen, Y.; Rees, T.W.; Ji, L.; Chao, H. Mitochondrial dynamics tracking with iridium(III) complexes. Curr. Opin. Chem. Biol. 2018, 43, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.-L.; Wu, C.; Wu, K.-J.; Leung, C.-H. Iridium(III) Complexes Targeting Apoptotic Cell Death in Cancer Cells. Molecules 2019, 24, 2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.-J.; Zheng, Y.; Wu, X.-W.; Tan, C.-P.; Chen, M.-H.; Wu, N.; Ji, L.-N.; Mao, Z.-W. Anticancer Cyclometalated Iridium(III) Complexes with Planar Ligands: Mitochondrial DNA Damage and Metabolism Disturbance. J. Med. Chem. 2019, 62, 3311–3322. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Zhang, M.-F.; Pan, Z.-Y.; Wang, K.-N.; Zhao, Z.-J.; Li, Y.; Mao, Z.-W. A mitochondria-targeted iridium(iii)-based photoacid generator induces dual-mode photodynamic damage within cancer cells. Chem. Commun. 2019, 55, 10472–10475. [Google Scholar] [CrossRef]
- Li, Y.; Wang, K.-N.; He, L.; Ji, L.-N.; Mao, Z.-W. Synthesis, photophysical and anticancer properties of mitochondria-targeted phosphorescent cyclometalated iridium(III) N-heterocyclic carbene complexes. J. Inorg. Biochem. 2020, 205, 110976. [Google Scholar] [CrossRef]
- Qin, W.-W.; Pan, Z.-Y.; Cai, D.-H.; Li, Y.; He, L. Cyclometalated iridium(iii) complexes for mitochondria-targeted combined chemo-photodynamic therapy. Dalton Trans. 2020, 49, 3562–3569. [Google Scholar] [CrossRef]
- Li, Y.; Liu, B.; Xu, C.-X.; He, L.; Wan, Y.-C.; Ji, L.-N.; Mao, Z.-W. Mitochondria-targeted phosphorescent cyclometalated iridium(III) complexes: Synthesis, characterization, and anticancer properties. JBIC J. Biol. Inorg. Chem. 2020, 25, 597–607. [Google Scholar] [CrossRef]
- Ye, R.-R.; Peng, W.; Chen, B.-C.; Jiang, N.; Chen, X.-Q.; Mao, Z.-W.; Li, R.-T. Mitochondria-targeted artesunate conjugated cyclometalated iridium(iii) complexes as potent anti-HepG2 hepatocellular carcinoma agents. Metallomics 2020, 12, 1131–1141. [Google Scholar] [CrossRef]
- Li, X.; Wu, J.; Wang, L.; He, C.; Chen, L.; Jiao, Y.; Duan, C. Mitochondrial-DNA-Targeted Ir III -Containing Metallohelices with Tunable Photodynamic Therapy Efficacy in Cancer Cells. Angew. Chem. Int. Ed. 2020, 59, 6420–6427. [Google Scholar] [CrossRef]
- Gu, Y.; Wen, H.; Bai, L.; Zhou, Y.; Zhang, H.; Tian, L.; Zhang, Y.; Hao, J.; Liu, Y. Exploring anticancer efficiency of mitochondria-targeted cyclometalated iridium(III) complexes. J. Inorg. Biochem. 2020, 212, 111215. [Google Scholar] [CrossRef]
- Wu, Y.; Wu, J.; Wong, W.-Y. A new near-infrared phosphorescent iridium(iii) complex conjugated to a xanthene dye for mitochondria-targeted photodynamic therapy. Biomater. Sci. 2021, 9, 4843–4853. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Xiong, K.; Wang, L.; Guan, R.; Chen, Y.; Ji, L.; Chao, H. Iridium(iii) complexes as mitochondrial topoisomerase inhibitors against cisplatin-resistant cancer cells. Chem. Commun. 2021, 57, 8308–8311. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Tan, C.-P.; Ye, R.-R.; Zhao, Y.-Z.; Liu, Y.-H.; Zhao, Q.; Ji, L.-N.; Mao, Z.-W. Theranostic Iridium(III) Complexes as One- and Two-Photon Phosphorescent Trackers to Monitor Autophagic Lysosomes. Angew. Chem. Int. Ed. 2014, 53, 12137–12141. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Li, Y.; Tan, C.-P.; Ye, R.-R.; Chen, M.-H.; Cao, J.-J.; Ji, L.-N.; Mao, Z.-W. Cyclometalated iridium(iii) complexes as lysosome-targeted photodynamic anticancer and real-time tracking agents. Chem. Sci. 2015, 6, 5409–5418. [Google Scholar] [CrossRef] [Green Version]
- Qiu, K.; Huang, H.; Liu, B.; Liu, Y.; Huang, Z.; Chen, Y.; Ji, L.; Chao, H. Long-Term Lysosomes Tracking with a Water-Soluble Two-Photon Phosphorescent Iridium(III) Complex. ACS Appl. Mater. Interfaces 2016, 8, 12702–12710. [Google Scholar] [CrossRef]
- Wang, F.-X.; Chen, M.-H.; Lin, Y.-N.; Zhang, H.; Tan, C.-P.; Ji, L.-N.; Mao, Z.-W. Dual Functions of Cyclometalated Iridium(III) Complexes: Anti-Metastasis and Lysosome-Damaged Photodynamic Therapy. ACS Appl. Mater. Interfaces 2017, 9, 42471–42481. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, L.; Tian, Z.; Gong, Y.; Zheng, H.; Zhang, S.; Xu, Z.; Ge, X.; Liu, Z. Novel and Versatile Imine-N-Heterocyclic Carbene Half-Sandwich Iridium(III) Complexes as Lysosome-Targeted Anticancer Agents. Inorg. Chem. 2018, 57, 11087–11098. [Google Scholar] [CrossRef]
- Ma, W.; Ge, X.; Xu, Z.; Zhang, S.; He, X.; Li, J.; Xia, X.; Chen, X.; Liu, Z. Theranostic Lysosomal Targeting Anticancer and Antimetastatic Agents: Half-Sandwich Iridium(III) Rhodamine Complexes. ACS Omega 2019, 4, 15240–15248. [Google Scholar] [CrossRef] [Green Version]
- Kuang, S.; Liao, X.; Zhang, X.; Rees, T.W.; Guan, R.; Xiong, K.; Chen, Y.; Ji, L.; Chao, H. FerriIridium: A Lysosome-Targeting Iron(III)-Activated Iridium(III) Prodrug for Chemotherapy in Gastric Cancer Cells. Angew. Chem. Int. Ed. 2020, 59, 3315–3321. [Google Scholar] [CrossRef]
- Liu, X.; Chen, S.; Ge, X.; Zhang, Y.; Xie, Y.; Hao, Y.; Wu, D.; Zhao, J.; Yuan, X.-A.; Tian, L.; et al. Dual functions of iridium(III) 2-phenylpyridine complexes: Metastasis inhibition and lysosomal damage. J. Inorg. Biochem. 2020, 205, 110983. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, L.; Tian, Z.; Ge, X.; Gong, Y.; Zheng, H.; Shi, S.; Liu, Z. Lysosome-Targeted Phosphine-Imine Half-Sandwich Iridium(III) Anticancer Complexes: Synthesis, Characterization, and Biological Activity. Organometallics 2019, 38, 1761–1769. [Google Scholar] [CrossRef]
- Nam, J.S.; Kang, M.-G.; Kang, J.; Park, S.-Y.; Lee, S.J.C.; Kim, H.-T.; Seo, J.K.; Kwon, O.-H.; Lim, M.H.; Rhee, H.-W.; et al. Endoplasmic Reticulum-Localized Iridium(III) Complexes as Efficient Photodynamic Therapy Agents via Protein Modifications. J. Am. Chem. Soc. 2016, 138, 10968–10977. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Liu, J.; Guan, R.; Jin, C.; Ji, L.; Chao, H. Endoplasmic reticulum targeted cyclometalated iridium(iii) complexes as efficient photodynamic therapy photosensitizers. Dalton Trans. 2019, 48, 6408–6415. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.-L.; Tong, K.-C.; Yang, C.; Kwong, W.-L.; Guan, X.; Li, M.-D.; Lo, V.K.-Y.; Chan, S.L.-F.; Phillips, D.L.; Lok, C.-N.; et al. Luminescent ruffled iridium(iii) porphyrin complexes containing N-heterocyclic carbene ligands: Structures, spectroscopies and potent antitumor activities under dark and light irradiation conditions. Chem. Sci. 2019, 10, 293–309. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Wei, F.; Xiang, J.; Li, H.; Li, C.; Zhang, P.; Liu, C.; Gong, P.; Cai, L.; Wong, K.M.-C. Enhancing the ROS generation ability of a rhodamine-decorated iridium(iii) complex by ligand regulation for endoplasmic reticulum-targeted photodynamic therapy. Chem. Sci. 2020, 11, 12212–12220. [Google Scholar] [CrossRef]
- Zhang, P.; Huang, H.; Banerjee, S.; Clarkson, G.J.; Ge, C.; Imberti, C.; Sadler, P.J. Nucleus-Targeted Organoiridium–Albumin Conjugate for Photodynamic Cancer Therapy. Angew. Chem. Int. Ed. 2019, 58, 2350–2354. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Zhu, Y.; Zhang, M.; Luo, L.; Wu, J.; Zhou, H.; Guan, L.; Battaglia, G.; Tian, Y. Localization matters: A nuclear targeting two-photon absorption iridium complex in photodynamic therapy. Chem. Commun. 2017, 53, 3303–3306. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Wang, K.-N.; Zheng, Y.; Cao, J.-J.; Zhang, M.-F.; Tan, C.-P.; Ji, L.-N.; Mao, Z.-W. Cyclometalated iridium(iii) complexes induce mitochondria-derived paraptotic cell death and inhibit tumor growthin vivo. Dalton Trans. 2018, 47, 6942–6953. [Google Scholar] [CrossRef]
- Tripathy, S.K.; De, U.; Dehury, N.; Laha, P.; Panda, M.K.; Kim, H.S.; Patra, S. Cyclometallated iridium complexes inducing paraptotic cell death like natural products: Synthesis, structure and mechanistic aspects. Dalton Trans. 2016, 45, 15122–15136. [Google Scholar] [CrossRef]
- Yokoi, K.; Balachandran, C.; Umezawa, M.; Tsuchiya, K.; Mitrić, A.; Aoki, S. Amphiphilic Cationic Triscyclometalated Iridium(III) Complex–Peptide Hybrids Induce Paraptosis-like Cell Death of Cancer Cells via an Intracellular Ca2+-Dependent Pathway. ACS Omega 2020, 5, 6983–7001. [Google Scholar] [CrossRef] [Green Version]
- Sperandio, S.; de Belle, I.; Bredesen, D.E. An alternative, nonapoptotic form of programmed cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 14376–14381. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.; Chen, Y.; Zeng, L.; Rees, T.W.; Jin, C.; Huang, J.; Chen, Z.-S.; Ji, L.; Chao, H. Oncosis-inducing cyclometalated iridium(iii) complexes. Chem. Sci. 2018, 9, 5183–5190. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Han, Z.; Chen, Y.; Qi, F.; Fang, H.; Guo, Z.; Zhang, S.; He, W. Ferroptosis Photoinduced by New Cyclometalated Iridium(III) Complexes and Its Synergism with Apoptosis in Tumor Cell Inhibition. Angew. Chem. Int. Ed. 2021, 60, 8174–8181. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tan, C.-P.; Zhang, W.; He, L.; Ji, L.-N.; Mao, Z.-W. Phosphorescent iridium(III)-bis-N-heterocyclic carbene complexes as mitochondria-targeted theranostic and photodynamic anticancer agents. Biomaterials 2015, 39, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Diez-Barra, E.; De la Hoz, A.; Sanchez-Migallon, A.; Tejeda, J. Phase-Transfer Catalysis without Solvent. Synthesis of Bisazolylalkanes. Heterocycles 1992, 34, 1365–1373. [Google Scholar] [CrossRef]
- Cure, J.; Poteau, R.; Gerber, I.; Gornitzka, H.; Hemmert, C. Dimeric Gold Bis(carbene) Complexes by Transmetalation in Water. Organometallics 2012, 31, 619–626. [Google Scholar] [CrossRef]
- King, K.A.; Watts, R.J. Dual Emission from an Ortho-Metalated Ir(III) Complex. J. Am. Chem. Soc. 1987, 109, 1589–1590. [Google Scholar] [CrossRef]
- Paloque, L.; Hemmert, C.; Valentin, A.; Gornitzka, H. Synthesis, characterization, and antileishmanial activities of gold(I) complexes involving quinoline functionalized N-heterocyclic carbenes. Eur. J. Med. Chem. 2015, 94, 22–29. [Google Scholar] [CrossRef]
- Gothe, Y.; Romero-Canelón, I.; Marzo, T.; Sadler, P.J.; Messori, L.; Metzler-Nolte, N. Synthesis and Mode of Action Studies on Iridium(I)-NHC Anticancer Drug Candidates. Eur. J. Inorg. Chem. 2018, 2018, 2461–2470. [Google Scholar] [CrossRef]
- Leo, A.; Hansch, C.; Elkins, D. Partition coefficients and their uses. Chem. Rev. 1971, 71, 525–616. [Google Scholar] [CrossRef]
- Monti, F.; Kessler, F.; Delgado, M.; Frey, J.; Bazzanini, F.; Accorsi, G.; Armaroli, N.; Bolink, H.; Ortí, E.; Scopelliti, R.; et al. Charged Bis-Cyclometalated Iridium(III) Complexes with Carbene-Based Ancillary Ligands. Inorg. Chem. 2013, 52, 10292–10305. [Google Scholar] [CrossRef]
- Yang, C.-H.; Beltran, J.; Lemaur, V.; Cornil, J.; Hartmann, D.; Sarfert, W.; Fröhlich, R.; Bizzarri, C.; De Cola, L. Iridium Metal Complexes Containing N-Heterocyclic Carbene Ligands for Blue-Light-Emitting Electrochemical Cells. Inorg. Chem. 2010, 49, 9891–9901. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.D.; Monti, F.; Accorsi, G.; Barbieri, A.; Bolink, H.; Ortí, E.; Armaroli, N. Photophysical Properties of Charged Cyclometalated Ir(III) Complexes: A Joint Theoretical and Experimental Study. Inorg. Chem. 2011, 50, 7229–7238. [Google Scholar] [CrossRef] [PubMed]
- Drobnik, J.; Yeargers, E. On the use of quinine sulfate as a fluorescence standard. J. Mol. Spectrosc. 1966, 19, 454–455. [Google Scholar] [CrossRef]
- Srinivasan, A.; Li, F.; Wong, A.; Kodandapani, L.; Smidt, R.; Krebs, J.F.; Fritz, L.C.; Wu, J.C.; Tomaselli, K.J. Bcl-xL Functions Downstream of Caspase-8 to Inhibit Fas- and Tumor Necrosis Factor Receptor 1-induced Apoptosis of MCF7 Breast Carcinoma Cells. J. Biol. Chem. 1998, 273, 4523–4529. [Google Scholar] [CrossRef] [Green Version]
- Cuvillier, O.; Nava, V.E.; Murthy, S.K.; Edsall, L.C.; Levade, T.; Milstien, S.; Spiegel, S. Sphingosine generation, cytochrome c release, and activation of caspase-7 in doxorubicin-induced apoptosis of MCF7 breast adenocarcinoma cells. Cell Death Differ. 2001, 8, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Medema, J.P.; Scaffidi, C.; Krammer, P.H.; Peter, M.E. Bcl-xL Acts Downstream of Caspase-8 Activation by the CD95 Death-inducing Signaling Complex. J. Biol. Chem. 1998, 273, 3388–3393. [Google Scholar] [CrossRef] [Green Version]
- Scaffidi, C.; Fulda, S.; Srinivasan, A.; Friesen, C.; Li, F.; Tomaselli, K.J.; Debatin, K.M.; Krammer, P.H.; Peter, M.E. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998, 17, 1675–1687. [Google Scholar] [CrossRef] [Green Version]
- Kalkavan, H.; Green, D. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef]
- Kaufmann, T.; Schlipf, S.; Sanz, J.; Neubert, K.; Stein, R.; Borner, C. Characterization of the signal that directs Bcl-xL, but not Bcl-2, to the mitochondrial outer membrane. J. Cell Biol. 2003, 160, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, D.W.; Ali, A.; Thornberry, N.A.; Vaillancourt, J.P.; Ding, C.K.; Gallant, M.; Gareau, Y.; Griffin, P.R.; Labelle, M.; Lazebnik, Y.A.; et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 1995, 376, 37–43. [Google Scholar] [CrossRef]
- Jänicke, R.U.; Sprengart, M.L.; Wati, M.R.; Porter, A.G. Caspase-3 Is Required for DNA Fragmentation and Morphological Changes Associated with Apoptosis. J. Biol. Chem. 1998, 273, 9357–9360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamaraeva, M.; Sabirov, R.Z.; Maeno, E.; Ando-Akatsuka, Y.; Bessonova, S.V.; Okada, Y. Cells die with increased cytosolic ATP during apoptosis: A bioluminescence study with intracellular luciferase. Cell Death Differ. 2005, 12, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Vit, J.-P.; Guillouf, C.; Rosselli, F. Futile Caspase-8 Activation during the Apoptotic Cell Death Induced by DNA Damaging Agents in Human B-Lymphoblasts. Exp. Cell Res. 2001, 269, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Phase annealing in SHELX-90: Direct methods for larger structures. Acta Crystallogr. Sect. A Found. Crystallogr. 1990, 46, 467–473. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, S.; Flack, H.D.; Wagner, T. Use of intensity quotients and differences in absolute structure refinement. Acta Crystallogr. Sect. B Struct. Sci. 2013, 69, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Crosby, G.A.; Demas, J.N. Measurement of photoluminescence quantum yields. Review. J. Phys. Chem. 1971, 75, 991–1024. [Google Scholar] [CrossRef]
- Galvao, J.; Davis, B.; Tilley, M.; Normando, E.; Duchen, M.R.; Cordeiro, M.F. Unexpected low-dose toxicity of the universal solvent DMSO. FASEB J. 2014, 28, 1317–1330. [Google Scholar] [CrossRef]
- Zhang, C.; Fortin, P.; Barnoin, G.; Qin, X.; Wang, X.; Alvarez, A.F.; Bijani, C.; Maddelein, M.; Hemmert, C.; Cuvillier, O.; et al. An Artemisinin-Derivative–(NHC)Gold(I) Hybrid with Enhanced Cytotoxicity through Inhibition of NRF2 Transcriptional Activity. Angew. Chem. Int. Ed. 2020, 59, 12062–12068. [Google Scholar] [CrossRef]
- Bonhoure, E.; Lauret, A.; Barnes, D.J.; Martin, C.; Malavaud, B.; Kohama, T.; Melo, J.V.; Cuvillier, O. Sphingosine kinase-1 is a downstream regulator of imatinib-induced apoptosis in chronic myeloid leukemia cells. Leukemia 2008, 22, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Hemmert, C.; Fabié, A.; Fabre, A.; Benoit-Vical, F.; Gornitzka, H. Synthesis, structures, and antimalarial activities of some silver(I), gold(I) and gold(III) complexes involving N-heterocyclic carbene ligands. Eur. J. Med. Chem. 2013, 60, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Hemmert, C.; Ramadani, A.P.; Boselli, L.; Álvarez, Á.F.; Paloque, L.; Augereau, J.-M.; Gornitzka, H.; Benoit-Vical, F. Antiplasmodial activities of gold(I) complexes involving functionalized N-heterocyclic carbenes. Bioorg. Med. Chem. 2016, 24, 3075–3082. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | λem (nm) a | ΦPL(%) b | τ1/ns |
|---|---|---|---|
| 4a | 469, 505 | 1.2 | 191 |
| 4b | 470, 503 | 1.6 | 166 |
| 4c | 469, 499 | 1.6 | 180 |
| 4d | 471, 506 | 1.1 | 167 |
| 4e | 480, 510 | 0.2 | - |
| 4f | 485, 510 | 0.2 | - |
| 4g | 425, 535 | 0.2 | - |
| 4a | 4b | 4c | 4d | Precursor | ||
|---|---|---|---|---|---|---|
| NON TUMORAL CELLS | ||||||
| NIH-3T3 | Dark | 3.67 | 1.95 [a] | 2.73 | 2.33 | 40 |
| Fibroblasts | Blue | 2.79 | 1.52 [a] | 2.67 | 2.78 | 14 |
| p value | 0.1117 (NS) | 0.1658 (NS) | 0.8524 (NS) | 0.3882 (NS) | ||
| PI [b] | 1.3 | 1.3 | 1.0 | 0.8 | 2.9 | |
| MC3T3 | Dark | 2.46 | 0.979 | 1.16 | 2.08 | |
| Osteoblasts | Blue | 1.74 | 0.704 | 1.33 | 1.80 | |
| p value | 0.1008 (NS) | 0.2273 (NS) | 0.4313 (NS) | 0.1856 (NS) | ||
| PI | 1.3 | 1.3 | 1.0 | 0.8 | ||
| TUMORAL CELLS | ||||||
| T24 | Dark | 0.288 | 0.259 | 0.358 | 0.501 | 26 |
| Bladder | SI [c] vs NIH | 12.7 | 7.5 | 7.6 | 4.7 | 1.5 |
| SI [c] vs MC3T3 | 8.5 | 3.8 | 3.2 | 4.2 | ||
| Blue | 0.082 | 0.090 | 0.091 | 0.165 | 22 | |
| SI vs NIH | 34.0 | 16.7 | 29.3 | 16.8 | 0.7 | |
| SI vs MC3T3 | 21.2 | 7.7 | 14.6 | 10.9 | ||
| p value | 0.0080 (**) | 0.0027 (**) | 0.0027 (**) | 0.0007 (***) | ||
| PI | 3.5 | 2.8 | 3.9 | 3.0 | 1.2 | |
| PC-3 | Dark | 0.233 | 0.257 | 0.336 | 0.509 | 29 |
| Prostate | SI vs NIH | 15.8 | 7.6 | 8.1 | 4.6 | 1.4 |
| SI vs MC3T3 | 10.6 | 3.8 | 3.5 | 4.1 | ||
| Blue | 0.057 | 0.071 | 0.116 | 0.157 | 14 | |
| SI vs NIH | 48.9 | 21.4 | 23.0 | 17.7 | 1.0 | |
| SI vs MC3T3 | 30.5 | 9.9 | 11.5 | 11.5 | ||
| p value | <0.0001 (***) | <0.0001 (***) | 0.0001 (***) | <0.0001 (***) | ||
| PI | 4.1 | 3.6 | 2.9 | 3.2 | 2.1 | |
| MCF7 | Dark | 0.213 | 0.211 | 0.248 | 0.365 | |
| Breast | SI vs NIH | 17.2 | 9.2 | 11.0 | 6.4 | |
| SI vs MC3T3 | 11.5 | 4.6 | 4.7 | 5.7 | ||
| Blue | 0.050 | 0.053 | 0.061 | 0.117 | ||
| SI vs NIH | 55.8 | 28.7 | 43.8 | 23.8 | ||
| SI vs MC3T3 | 34.8 | 13.3 | 21.8 | 15.4 | ||
| p value | 0.0003 (***) | 0.0003 (***) | 0.0029 (**) | <0.0001 (***) | ||
| PI | 4.3 | 4.0 | 4.1 | 3.1 | ||
| A549 | Dark | 0.355 | 0.354 | 0.407 | 1.51 | |
| Lung | SI vs NIH | 10.3 | 5.5 | 6.7 | 1.5 | |
| SI vs MC3T3 | 6.9 | 2.8 | 2.9 | 1.4 | ||
| Blue | 0.107 | 0.084 | 0.171 | 0.617 | ||
| SI vs NIH | 26.1 | 18.1 | 15.6 | 4.5 | ||
| SI vs MC3T3 | 16.3 | 8.4 | 7.8 | 2.9 | ||
| p value | 0.0039 (**) | 0.0043 (**) | 0.0005 (***) | <0.0001 (***) | ||
| PI | 3.3 | 4.2 | 2.4 | 2.4 | ||
| HeLa | Dark | 1.29 | 0.625 | 0.494 | 1.54 | |
| Cervix | SI vs NIH | 2.8 | 3.1 | 5.5 | 1.5 | |
| SI vs MC3T3 | 1.9 | 1.6 | 2.3 | 1.4 | ||
| Blue | 0.525 | 0.294 | 0.275 | 0.817 | ||
| SI vs NIH | 5.3 | 5.2 | 9.7 | 3.4 | ||
| SI vs MC3T3 | 3.3 | 2.4 | 4.8 | 2.2 | ||
| p value | 0.0029 (**) | 0.0009 (***) | 0.0038(**) | 0.0048 (***) | ||
| PI | 2.5 | 2.1 | 1.8 | 1.9 | ||
| AVERAGE SI [d] | ||||||
| Blue light | SI vs NIH | 34.0 | 18.0 | 24.3 | 13.2 | |
| SI vs MC3T3 | 21.2 | 8.3 | 12.1 | 8.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Zhang, C.; Madji, R.; Voros, C.; Mazères, S.; Bijani, C.; Deraeve, C.; Cuvillier, O.; Gornitzka, H.; Maddelein, M.-L.; et al. N-Heterocyclic Carbene-Iridium Complexes as Photosensitizers for In Vitro Photodynamic Therapy to Trigger Non-Apoptotic Cell Death in Cancer Cells. Molecules 2023, 28, 691. https://doi.org/10.3390/molecules28020691
Wang X, Zhang C, Madji R, Voros C, Mazères S, Bijani C, Deraeve C, Cuvillier O, Gornitzka H, Maddelein M-L, et al. N-Heterocyclic Carbene-Iridium Complexes as Photosensitizers for In Vitro Photodynamic Therapy to Trigger Non-Apoptotic Cell Death in Cancer Cells. Molecules. 2023; 28(2):691. https://doi.org/10.3390/molecules28020691
Chicago/Turabian StyleWang, Xing, Chen Zhang, Ryma Madji, Camille Voros, Serge Mazères, Christian Bijani, Céline Deraeve, Olivier Cuvillier, Heinz Gornitzka, Marie-Lise Maddelein, and et al. 2023. "N-Heterocyclic Carbene-Iridium Complexes as Photosensitizers for In Vitro Photodynamic Therapy to Trigger Non-Apoptotic Cell Death in Cancer Cells" Molecules 28, no. 2: 691. https://doi.org/10.3390/molecules28020691
APA StyleWang, X., Zhang, C., Madji, R., Voros, C., Mazères, S., Bijani, C., Deraeve, C., Cuvillier, O., Gornitzka, H., Maddelein, M. -L., & Hemmert, C. (2023). N-Heterocyclic Carbene-Iridium Complexes as Photosensitizers for In Vitro Photodynamic Therapy to Trigger Non-Apoptotic Cell Death in Cancer Cells. Molecules, 28(2), 691. https://doi.org/10.3390/molecules28020691