
Conformations of Steroid Hormones: Infrared and Vibrational Circular Dichroism Spectroscopy
 and
and {kind=link}
{kind=link}
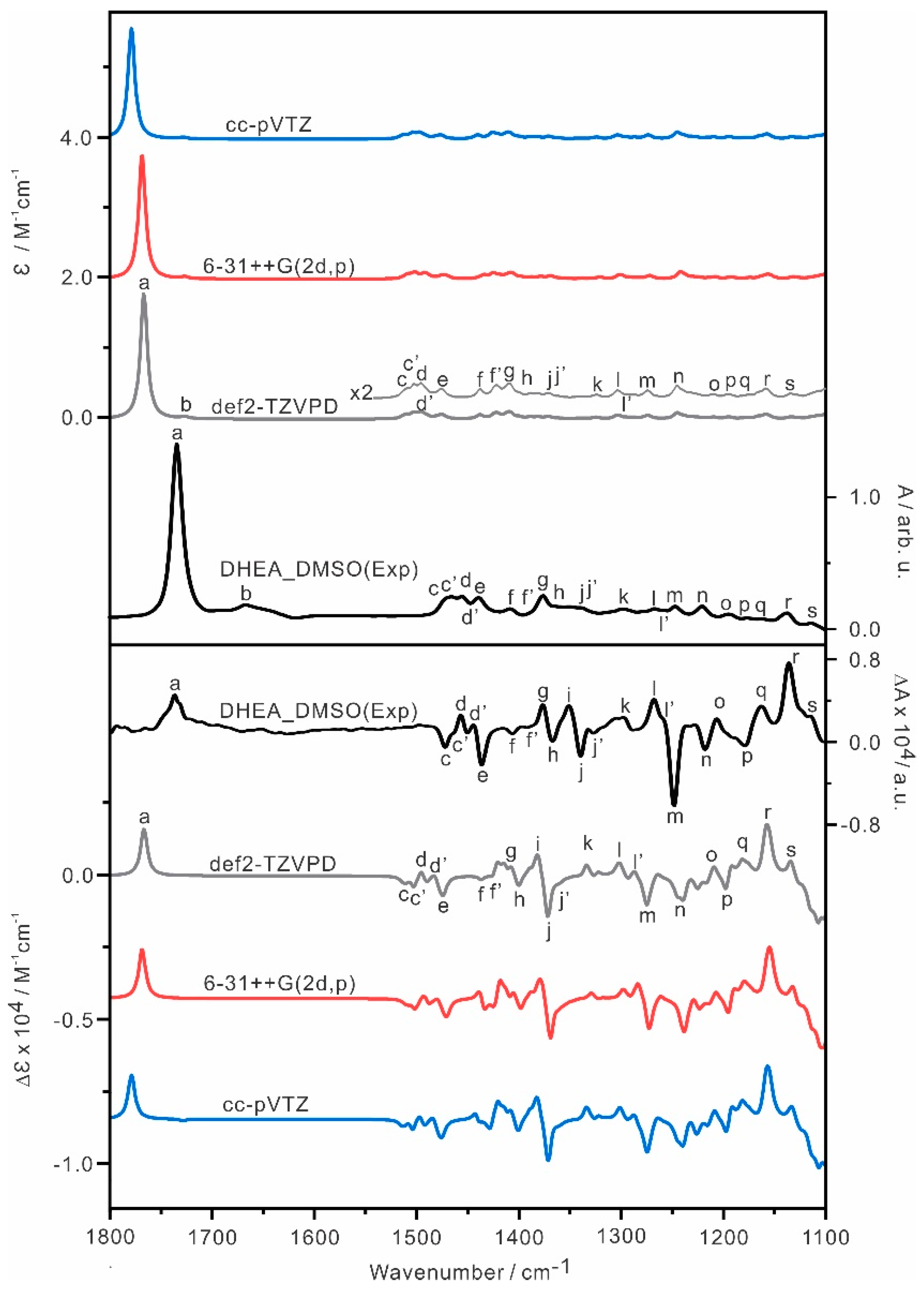{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Using Experimental IR and VCD Spectra to Identify a Mislabeled Sample
2.2. Experimental IR and VCD Spectra of the Four Steroids in DMSO
2.3. Conformational Searches and Identification of Low Energy Minima of the Four Steroids
2.4. Experimental and Simulated IR and VCD Spectra of DHEA, MTTT and Epoxy-P4
2.5. Experimental and Simulated IR and VCD Spectra of AcO-DHEA
2.6. Brief Comments on the Absolute Configurations of the Four Steroids
3. Materials and Methods
3.1. Experimental
3.2. Theoretical
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hu, J.; Zhang, Z.; Shen, W.-J.; Azhar, S. Cellular cholesterol delivery, intracellular processing and utilization for biosynthesis of steroid hormones. Nutr. Metab. 2010, 7, 47. [Google Scholar] [CrossRef] [Green Version]
- Lopez, L.M.; Grimes, D.A.; Schulz, K.F. Steroidal contraceptives: Effect on carbohydrate metabolism in women without diabetes. Cochrane Database Syst. Rev. 2014, 4, CD006133. [Google Scholar] [CrossRef]
- Moss, G.P. Nomenclature of steroids (Recommendations 1989). Pure Appl. Chem. 1989, 61, 1783–1822. [Google Scholar] [CrossRef]
- Li, W.; Covey, D.F.; Alakoskela, J.M.; Kinnunen, P.K.J.; Steinbach, J.H. Enantiomers of neuroactive steroids support a specific interaction with the GABA-C receptor as the mechanism of steroid action. Mol. Pharmacol. 2006, 69, 1779–1782. [Google Scholar] [CrossRef] [Green Version]
- Flack, H.D.; Bernardinelli, G. The use of X-ray crystallography to determine absolute configuration. Chirality 2008, 20, 681–690. [Google Scholar] [CrossRef]
- Parsons, S. Determination of absolute configuration using X-ray diffraction. Tetrahedron Asymmetry 2017, 28, 1304–1313. [Google Scholar] [CrossRef] [Green Version]
- Pitzer, M.; Kunitski, M.; Johnson, A.S.; Jahnke, T.; Sann, H.; Sturm, F.; Schmidt, L.P.H.; Schmidt-Böcking, H.; Dörner, R.; Stohner, J.; et al. Direct determination of absolute molecular stereochemistry in gas phase by coulomb explosion imaging. Science 2013, 341, 1096–1100. [Google Scholar] [CrossRef]
- Fehre, K.; Eckart, S.; Kunitski, M.; Pitzer, M.; Zeller, S.; Janke, C.; Trabert, D.; Rist, J.; Weller, M.; Hartung, A.; et al. Enantioselective fragmentation of an achiral molecule in a strong laser field. Sci. Adv. 2019, 5, eaau7923. [Google Scholar] [CrossRef] [Green Version]
- Domingos, S.R.; Pérez, C.; Schnell, M. Sensing chirality with rotational spectroscopy. Annu. Rev. Phys. Chem. 2018, 69, 499–519. [Google Scholar] [CrossRef] [Green Version]
- Pate, B.H.; Evangelisti, L.; Caminati, W.; Xu, Y.; Thomas, J.; Patterson, D.; Perez, C.; Schnell, M. Quantitative chiral analysis by molecular rotational spectroscopy. In Chiral Analysis, Advances in Spectroscopy, Chromatography and Emerging Methods, 2nd ed.; Polavarapu, P.L., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 679–729. [Google Scholar]
- Xie, F.; Seifert, N.A.; Jäger, W.; Xu, Y. Conformational panorama and chirality controlled structure—Energy relationship in a chiral carboxylic acid dimer. Angew. Chem. Int. Ed. 2020, 59, 15703–15710. [Google Scholar] [CrossRef]
- Decleva, P. Photoelectron circular dichroism as a probe of chiral hydrocarbons. Chemistry 2022, 4, 31–41. [Google Scholar] [CrossRef]
- Krüger, P.; Both, J.H.; Linne, U.; Chirot, F.; Weitzel, K.-M. Photoelectron circular dichroism of electrosprayed gramicidin anions. J. Phys. Chem. Lett. 2022, 13, 6110–6116. [Google Scholar] [CrossRef] [PubMed]
- Polavarapu, P.L. Optical rotation: Recent advances in determining the absolute configuration. Chirality 2002, 14, 768–781. [Google Scholar] [CrossRef] [PubMed]
- Padula, D.; Pescitelli, G. How and how much molecular conformation affects electronic circular dichroism: The case of 1,1-diarylcarbinols. Molecules 2018, 23, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aharon, T.; Lemler, P.; Vaccaro, P.H.; Caricato, M. Comparison of measured and predicted specific optical rotation in gas and solution phases: A test for the polarizable continuum model of solvation. Chirality 2018, 30, 383–395. [Google Scholar] [CrossRef]
- Lahiri, P.; Wiberg, K.B.; Vaccaro, P.H.; Caricato, M.; Crawford, T.D. Large solvation effect in the optical rotatory dispersion of norbornenone. Angew. Chem. Int. Ed. 2014, 53, 1386–1389. [Google Scholar] [CrossRef]
- Nafie, L.A.; Keiderling, T.A.; Stephens, P.J. Vibrational circular dichroism. J. Am. Chem. Soc. 1976, 98, 2715–2723. [Google Scholar] [CrossRef]
- Freedman, T.B.; Cao, X.; Dukor, R.K.; Nafie, L.A. Absolute configuration determination of chiral molecules in the solution state using vibrational circular dichroism. Chirality 2003, 15, 743–758. [Google Scholar] [CrossRef]
- Yang, G.; Xu, Y. Vibrational circular dichroism spectroscopy of chiral molecules. Top. Curr. Chem. 2011, 298, 189–236. [Google Scholar]
- Polavarapu, P.L.; Santoro, E. Vibrational optical activity for structural characterization of natural products. Nat. Prod. Rep. 2020, 37, 1661–1699. [Google Scholar] [CrossRef]
- Yokomichi, M.A.S.; Silva, H.R.L.; Brandao, L.E.V.N.; Vicente, E.F.; Batista, J.M., Jr. Conformational preferences induced by cyclization in orbitides: A vibrational CD study. Org. Biomol. Chem. 2022, 20, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Poopari, M.R.; Cai, X.; Savin, A.; Dezhahang, Z.; Cheramy, J.; Xu, Y. IR and vibrational circular dichroism spectroscopy of matrine- and artemisinin-type herbal products: Stereochemical characterization and solvent effects. J. Nat. Prod. 2016, 79, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Merten, C.; Li, F.; Bravo-Rodriguez, K.; Sanchez-Garcia, E.; Xu, Y.; Sander, W. Solvent-induced conformational changes in cyclic peptides: A vibrational circular dichroism study. Phys. Chem. Chem. Phys. 2014, 16, 5627–5633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessler, J.; Andrushchenko, V.; Kapitán, J.; Bouř, P. Insight into vibrational circular dichroism of proteins by density functional modeling. Phys. Chem. Chem. Phys. 2018, 20, 4926–4935. [Google Scholar] [CrossRef] [Green Version]
- Dezhahang, Z.; Poopari, M.R.; Cheramy, J.; Xu, Y. Conservation of helicity in a chiral pyrrol-2-yl Schiff-base ligand and its transition metal complexes. Inorg. Chem. 2015, 54, 4539–4549. [Google Scholar] [CrossRef]
- Merten, C.; Hiller, K.; Xu, Y. Effects of electron configuration and coordination number on the vibrational circular dichroism spectra of metal complexes of trans-1,2-diaminocyclohexane. Phys. Chem. Chem. Phys. 2012, 14, 12884–12891. [Google Scholar] [CrossRef]
- Yang, Y.; Cheramy, J.; Brehm, M.; Xu, Y. Raman optical activity of N-acetyl-L-cysteine in water and in methanol: The “clusters-in-a-liquid” model and ab initio molecular dynamics simulations. ChemPhysChem 2022, 23, e202200161. [Google Scholar] [CrossRef]
- Le Barbu-Debus, K.; Bowles, J.; Jähnigen, S.; Clavaguéra, C.; Calvo, F.; Vuilleumier, R.; Zehnacker, A. Assessing cluster models of solvation for the description of vibrational circular dichroism spectra: Synergy between static and dynamic approaches. Phys. Chem. Chem. Phys. 2020, 22, 26047–26068. [Google Scholar] [CrossRef]
- Perera, A.S.; Cheramy, J.; Merten, C.; Thomas, J.; Xu, Y. IR, Raman, and vibrational optical activity spectra of methyl glycidate in chloroform and water: The clusters-in-a-liquid solvation model. ChemPhysChem 2018, 19, 2234–2242. [Google Scholar] [CrossRef]
- Tang, J.; Chen, L.-R.; Chen, K.-H. The Utilization of dehydroepiandrosterone as a sexual hormone precursor in premenopausal and postmenopausal women: An overview. Pharmaceuticals 2022, 15, 46. [Google Scholar] [CrossRef]
- Huang, X.; Shen, Q.-K.; Zhang, H.-J.; Li, J.-L.; Tian, Y.-S.; Quan, Z.-S. Design and synthesis of novel dehydroepiandrosterone analogues as potent antiproliferative agents. Molecules 2018, 23, 2243. [Google Scholar] [CrossRef] [PubMed]
- Lou, S.-J.; Li, X.-H.; Zhou, X.-L.; Fang, D.-M.; Gao, F. Palladium-catalyzed synthesis and anticancer activity of paclitaxel−dehydroepiandrosterone hybrids. ACS Omega 2020, 5, 5589–5600. [Google Scholar] [CrossRef] [PubMed]
- Olesti, E.; Boccard, J.; Visconti, G.; González-Ruiz, V.; Rudaz, S. From a single steroid to the steroidome: Trends and analytical challenges. J. Steroid Biochem. Mol. Biol. 2021, 206, 105797. [Google Scholar] [CrossRef] [PubMed]
- King, T.L.; Brucker, M.C. Pharmacology for Women’s Health; Jones & Bartlett Publishers: Sudbury, MA, USA, 2010; pp. 372–373. [Google Scholar]
- Giannoni, E.; Guignard, L.; Reymond, M.K.; Perreau, M.; Roth-Kleiner, M.; Calandra, T.; Roger, T. Estradiol and progesterone strongly inhibit the innate immune response of mononuclear cells in newborns. Infect. Immun. 2011, 79, 2690–2698. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.X.; Guan, Y.X.; Wang, H.Q.; Yao, S.J. 11α-Hydroxylation of 16α,17-epoxyprogesterone by Rhizopus nigricans in a biphasic ionic liquid aqueous system. Bioresour. Technol. 2011, 102, 9368–9373. [Google Scholar] [CrossRef]
- Thanasupsin, S.P.; Chheang, L.; Math, C. Ecological risk of 17α-methyltestosterone contaminated water discharged from a full water recirculating earthen masculinization pond. Hum. Ecol. Risk Assess. Int. J. 2021, 27, 1696–1714. [Google Scholar] [CrossRef]
- Carlson, C.D.; Seifert, N.A.; Heger, M.; Xie, F.; Thomas, J.; Xu, Y. Conformational dynamics of 1-phenyl-2,2,2-trifluoroethanol by rotational spectroscopy and ab initio calculations. J. Mol. Spectrosc. 2018, 351, 62–67. [Google Scholar] [CrossRef]
- Carlson, C.D.; Hazrah, A.S.; Mason, D.; Yang, Q.; Seifert, N.A.; Xu, Y. Alternating 1-phenyl-2,2,2-trifluroethanol conformation landscape with the addition of one water: Conformations and large amplitude motions. J. Phys. Chem. A 2022, 126, 7250–7260. [Google Scholar] [CrossRef]
- 17α-Methyltestosterone. Available online: https://webbook.nist.gov/cgi/cbook.cgi?ID=C58184&Mask=80#Refs (accessed on 23 October 2015).
- Kasal, A.; Budesinsky, M.; Griffiths, W.J. Spectroscopic methods of steroid analysis. In Steroid Analysis; Makin, H.L.J., Gower, D.B., Eds.; Springer: Dordrecht, The Netherlands, 2010; p. 56. [Google Scholar] [CrossRef] [Green Version]
- Pracht, P.; Bohle, F.; Grimme, S. Automated exploration of the low-energy chemical space with fast quantum chemical methods. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. [Google Scholar] [CrossRef]
- Xie, F.; Seifert, N.A.; Heger, M.; Thomas, J.; Jäger, W.; Xu, Y. The rich conformational landscape of perillyl alcohol revealed by broadband rotational spectroscopy and theoretical modelling. Phys. Chem. Chem. Phys. 2019, 21, 15408–15416. [Google Scholar] [CrossRef]
- Oswald, S.; Seifert, N.A.; Bohle, F.; Gawrilow, M.; Grimme, S.; Jäger, W.; Xu, Y.; Suhm, M.A. The chiral trimer and a metastable chiral dimer of achiral hexafluoroisopropanol: A multi-messenger study. Angew. Chem. Int. Ed. 2019, 58, 5080–5084. [Google Scholar] [CrossRef] [PubMed]
- Marshall, M.D.; Leung, H.O.; Domingos, S.R.; Krin, A.; Schnell, M.; Seifert, N.A.; Xu, Y.; Jäger, W. Examining the gas-phase homodimers of 3,3,3-trifluoro-1,2-epoxypropane using quantum chemistry and microwave spectroscopy. Phys. Chem. Chem. Phys. 2022, 24, 28495–28505. [Google Scholar] [CrossRef]
- Wang, H.; Heger, M.; Al-Jabiri, M.H.; Xu, Y. Vibrational spectroscopy of homo- and heterochiral amino acid dimers: Conformational landscapes. Molecules 2022, 27, 38. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Fusè, M.; Hazrah, A.S.; Jäger, W.; Barone, V.; Xu, Y. Discovering the elusive global minimum in a ternary chiral cluster: Rotational spectra of propylene oxide trimer. Angew. Chem. Int. Ed. 2020, 59, 22427–22430. [Google Scholar] [CrossRef] [PubMed]
- Becke, A. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796. [Google Scholar] [CrossRef] [Green Version]
- Mennucci, B.; Tomasi, J.; Cammi, R.; Cheeseman, J.R.; Frisch, M.J.; Devlin, F.J.; Gabriel, S.; Stephens, P.J. Polarizable continuum model (PCM) calculations of solvent effects on optical rotations of chiral molecules. J. Phys. Chem. A 2002, 106, 6102–6113. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.G.A.; Burns, L.A.; Patkowski, K.; Sherrill, C.D. Revised damping parameters for the D3 dispersion correction to density functional theory. J. Phys. Chem. Lett. 2016, 7, 2197–2203. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D.; Johnson, A.D. A density-functional model of the dispersion interaction. J. Chem. Phys. 2005, 123, 154101. [Google Scholar] [CrossRef] [PubMed]
- Jensen, F.R.; Bushweller, C.H. Conformational preferences and interconversion barriers in cyclohexene and derivatives. J. Am. Chem. Soc. 1969, 91, 5774–5782. [Google Scholar] [CrossRef]
- Scharpen, L.H.; Wollrab, J.E.; Ames, D.P. Microwave spectrum, structure, and dipole moment of cyclohexene. J. Chem. Phys. 1968, 49, 2368–2372. [Google Scholar] [CrossRef]
- Lin, W.; Brooks, A.H.; Minei, A.J.; Novick, S.E.; Pringle, W.C. Microwave spectra and structure of the Argon−cyclopentanone and neon−cyclopentanone van der waals complexes. J. Phys. Chem. A 2014, 118, 856–861. [Google Scholar] [CrossRef]
- Ikeda, T.; Lord, R.C. Far-Infrared Spectra of Ring Molecules. Far-infrared spectrum and hindered pseudorotation in cyclopentanone. J. Chem. Phys. 1972, 56, 4450–4466. [Google Scholar] [CrossRef]
- Dragojlovic, V. Conformational analysis of cycloalkanes. ChemTexts 2015, 1, 14. [Google Scholar] [CrossRef]
- Saebø, S.; Cordell, F.R.; Boggs, J.E. Structures and conformations of cyclopentane, cyclopentene, and cyclopentadiene. J. Mol. Struct. THEOCHEM 1983, 104, 221–232. [Google Scholar] [CrossRef]
- Johnson, W.S.; Bauer, V.J.; Margrave, J.L.; Frisch, M.A.; Dreger, L.H.; Hubbard, W.N. The energy difference between the chair and boat forms of cyclohexane. The twist conformation of cyclohexane. J. Am. Chem. Soc. 1961, 83, 606–614. [Google Scholar] [CrossRef]
- Brutcher, F.V., Jr.; Roberts, T.; Barr, S.J.; Pearson, N. The conformations of substituted cyclopentanes. I. The infrared analysis and structure of the α-halocamphors, the α-halo-2-indanones and the α-halocyclopentanones. J. Am. Chem. Soc. 1959, 81, 4915–4920. [Google Scholar] [CrossRef]
- Heshmat, M.; Baerends, E.J.; Polavarapu, P.L.; Nicu, V.P. The importance of large-amplitude motions for the interpretation of mid-infrared vibrational absorption and circular dichroism spectra: 6,6′-dibromo-[1,1′-binaphthalene]-2,2′-diol in dimethyl sulfoxide. J. Phys. Chem. A 2014, 118, 4766–4777. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Peña, I.; Carlson, C.D.; Yang, Y.; Jäger, W.; Xu, Y. Structural and Dynamical Features of the 2,2,2-Trifluoroethanol···Ammonia Complex. Phys. Chem. Chem. Phys. 2020, 22, 23019–23027. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Carlson, C.D.; Jäger, W.; Xu, Y. Conformational landscape of the hydrogen-bonded 1-phenyl-2,2,2-trilfuoroethanol···1,4-dioxane complex: Dispersion interactions and conformational conversion. J. Phys. Chem. A 2022, 126, 2942–2949. [Google Scholar] [CrossRef]
- Barone, V.; Ceselin, G.; Fusè, M.; Tasinato, N. Accuracy meets interpretability for computational spectroscopy by means of hybrid and double-hybrid functionals. Front. Chem. 2020, 8, 584203. [Google Scholar] [CrossRef]
- Jarota, A.; Drwal, D.; Pięta, J.; Pastorczak, E. Wide-range IR spectra of diarylethene derivatives and their simulation using the density functional theory. Sci. Rep. 2022, 12, 16834. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian Development Version; Gaussian Inc.: Wallingford, CT, USA, 2022. [Google Scholar]
- Coriani, S.; Thorvaldsen, A.J.; Kristensen, K.; Jorgensen, P. Variational response-function formulation of vibrational circular dichroism. Phys. Chem. Chem. Phys. 2011, 13, 4224–4229. [Google Scholar] [CrossRef]
- Losada, M.; Tran, H.; Xu, Y. Lactic acid in solution: Investigations of lactic acid self-aggregation and hydrogen bonding interactions with water and methanol using VA and VCD spectroscopy. J. Chem. Phys. 2008, 128, 014508. [Google Scholar] [CrossRef]
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended tight-binding quantum chemistry methods. WIREs Comput. Mol. Sci. 2021, 11, e1493. [Google Scholar] [CrossRef]
- Grimme, S.; Bannwarth, C.; Shushkov, P. A robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent interactions of large molecular systems parametrized for all spd-block elements (Z = 1–86). J. Chem. Theory Comput. 2017, 13, 1989–2009. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.-J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M. Molpro: A general-purpose quantum chemistry program package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 242–253. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.03; Gaussian Inc.: Wallingford, CT, USA, 2019; Available online: https://gaussian.com/ (accessed on 24 December 2022).










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Krin, A.; Cai, X.; Poopari, M.R.; Zhang, Y.; Cheeseman, J.R.; Xu, Y. Conformations of Steroid Hormones: Infrared and Vibrational Circular Dichroism Spectroscopy. Molecules 2023, 28, 771. https://doi.org/10.3390/molecules28020771
Yang Y, Krin A, Cai X, Poopari MR, Zhang Y, Cheeseman JR, Xu Y. Conformations of Steroid Hormones: Infrared and Vibrational Circular Dichroism Spectroscopy. Molecules. 2023; 28(2):771. https://doi.org/10.3390/molecules28020771
Chicago/Turabian StyleYang, Yanqing, Anna Krin, Xiaoli Cai, Mohammad Reza Poopari, Yuefei Zhang, James R. Cheeseman, and Yunjie Xu. 2023. "Conformations of Steroid Hormones: Infrared and Vibrational Circular Dichroism Spectroscopy" Molecules 28, no. 2: 771. https://doi.org/10.3390/molecules28020771
APA StyleYang, Y., Krin, A., Cai, X., Poopari, M. R., Zhang, Y., Cheeseman, J. R., & Xu, Y. (2023). Conformations of Steroid Hormones: Infrared and Vibrational Circular Dichroism Spectroscopy. Molecules, 28(2), 771. https://doi.org/10.3390/molecules28020771