

Exploration of the Linkages between Lignin and Carbohydrates in Kraft Pulp from Wheat Straw Using a 13C/2H Isotopic Tracer



Abstract
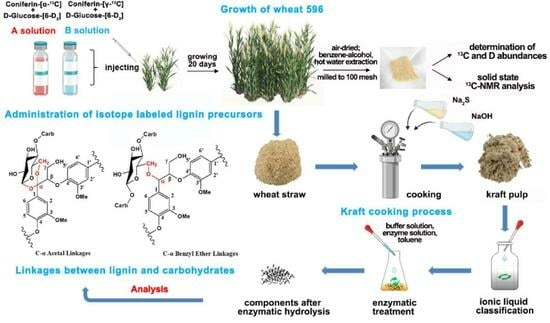
:
1. Introduction
2. Results and Discussion
2.1. Treatment of Wheat Stalks with Lignin and Polysaccharide Precursors
2.2. Analysis of 13C/2H Abundance in Wheat Internode Tissues
2.3. CP/MAS 13C-NMR Analysis of Wheat Straw Powder
2.4. XRD Characterization of KP-CLC and KP
2.5. Chemical Structure Analysis of Ac-En-KP-CLC
2.5.1. 13C-NMR Characterization of Ac-En-KP-CLC
2.5.2. 1H-NMR Characterization of Ac-En-KP-CLC
2.6. Chemical Structure Analysis of En-KP-XLC
13C-NMR characterization of En-KP-XLC
3. Experiment
3.1. Materials
3.2. Methods
3.2.1. Synthesis of Isotope-Labeled Lignin Precursors
3.2.2. Administration of Wheat Stalks with 13C and D Double-Isotope-Labeled Precursors
3.2.3. Preparation of Wheat Straw Powder
3.2.4. Determination of 13C and D Abundances
3.2.5. Determination of Cross-Polarization/Magic Angle Spinning (CP/MAS) 13C-NMR Spectrum of Wheat Internode Tissue Powder
3.2.6. Preparation of Kraft Pulp (KP) Cellulose–Lignin Complex (KP-CLC) and KP Xylan–Lignin Complex (KP-XLC) from KP
Kraft Cooking Process
Determination of Klason Lignin Content of KP
Classification of KP Using Ionic Liquid
3.2.7. Determination of KP and KP-CLC Using an X-ray Diffractometer (XRD)
3.2.8. Enzymatic Treatment of KP-CLC and KP-XLC
3.2.9. Acetylation of the En-KP-CLC Sample
3.2.10. Determination of NMR Spectra of Ac-En-KP-CLC and En-KP-XLC Samples
4. Conclusions
- (1)
- The abundances of 13C and D in the experimental groups were substantially higher than those in the control group. These results indicate that the injected exogenous coniferin-[α-13C], coniferin-[γ-13C], and d-glucose-[6-D2] were effectively absorbed and metabolized during the growth of wheat internode tissues. Therefore, the lignin in the cell wall of wheat straw was labeled with 13C and the polysaccharides were labeled with D.
- (2)
- CP/MAS 13C-NMR determination and analysis of the differential spectroscopy of wheat straw powder in the 13Cα-labeled experimental group, 13Cγ-labeled experimental group, and unlabeled control group showed that after the labeling of lignin side chain 13Cα and 13Cγ, the signals in the CP/MAS 13C-NMR spectra were substantially enhanced compared with that of the control group. The chemical linkages between lignin and lignin were mainly β-aryl ether, β-5, β-1, and β-β. Lignin was primarily linked to polysaccharides via acetal, benzyl ether, and benzyl ester bonds.
- (3)
- After kraft cooking of the wheat straw, a large amount of lignin and xylan was dissolved, but most of the CLC and XLC was retained. The obtained KP was fractionated with the ionic liquid DMSO/TBAH to obtain cellulose–lignin and xylan–lignin complexes. The XRD results of the KP and KP-CLC showed that the KP-CLC was mainly converted from cellulose type I to cellulose type II in the crystalline region after ionic liquid classification.
- (4)
- The 13C-NMR and 1H-NMR spectra of Ac-En-KP-CLC showed that the signal intensity between lignin–lignin and lignin–cellulose was substantially enhanced after the lignin side chain was labeled with 13Cα and 13Cγ. The 13C-NMR and 1H-NMR spectra of AC-En-KP-CLC showed that the CLC structure obtained after enzymatic hydrolysis by cellulase was mainly chemically bonded by acetal and benzyl ether bonds. The 13C-NMR spectrum of En-KP-XLC showed that the XLC structure obtained after enzymatic hydrolysis of xylanase contained some lignin side chain xylan linked to Cα by acetal and ether bonds.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, M.X.; Wang, Y.T.; Shi, L. Environmental performance of straw-based pulp making: A life cycle perspective. Sci. Total. Environ. 2018, 616, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Sharma, D.; Nagpal, R.; Kaur, A.; Bhardwaj, N.; Mahajan, R. Valorisation of wheat straw into paper with improved quality characteristics using ultrafiltered xylano-pectinolytic pulping approach. Biotech 2023, 13, 106. [Google Scholar]
- Volynets, B.; Dahman, Y. Assessment of pretreatments and enzymatic hydrolysis of wheat straw as a sugar source for bioprocess industry. Int. J. Energy Environ. 2011, 2, 427–446. [Google Scholar]
- Deshpande, R.; Giummarella, N.; Henriksson, G.; Germgard, U.; Sundvall, L.; Grundberg, H.; Lawoko, M. The reactivity of lignin carbohydrate complex (LCC) during manufacture of dissolving sulfite pulp from softwood. Ind. Crop. Prod. 2018, 115, 315–322. [Google Scholar] [CrossRef]
- Santos, R.B.; Jameel, H.; Chang, H.M.; Hart, P.W. Impact of Lignin and Carbohydrate Chemical Structures on Degradation Reactions during Hardwood Kraft Pulping Processes. Bioresources 2013, 8, 158–171. [Google Scholar] [CrossRef]
- Santos, R.B.; Jameel, H.; Chang, H.M.; Hart, P.W. Impact of Lignin and Carbohydrate Chemical Structures on Kraft Pulping Process and Biofuel Production. Tappi J. 2013, 12, 23. [Google Scholar] [CrossRef]
- You, T.T.; Zhang, L.M.; Zhou, S.K.; Xu, F. Structural elucidation of lignin–Carbohydrate complex (LCC) preparations and lignin from Arundo donax Linn. Ind. Crop. Prod. 2015, 71, 65–74. [Google Scholar] [CrossRef]
- Lundquist, K.; Simonson, R.; Tingsvik, K. Studies on Lignin Carbohydrate Linkages in Milled Wood Lignin Preparations. Svensk Papperstidn. 1980, 83, 452–454. [Google Scholar]
- Johnson, K.G.; Overend, R.P. Lignin-Carbohydrate Complexes from Populus deltoides. I. Purification and Characterization. Holzforschung 1991, 45, 469–475. [Google Scholar] [CrossRef]
- Scalbert, A.; Monties, B.; Guittet, E.; Lallemand, J.Y. Comparison of Wheat Straw Lignin Preparations—I. Chemical and Spectroscopic Characterizations. Holzforschung 1986, 40, 119–127. [Google Scholar] [CrossRef]
- Iversen, T.; Wannstrom, S. Lignin-Carbohydrate Bonds in a Residual Lignin Isolated from Pine Kraft Pulp. Holzforschung 1986, 40, 19–22. [Google Scholar] [CrossRef]
- Balakshin, M.Y.; Capanema, E.A.; Chen, C.L.; Gracz, H.S. Elucidation of the Structures of Residual and Dissolved Pine Kraft Lignins Using an HMQC NMR Technique. J. Agric. Food Chem. 2003, 51, 6116–6127. [Google Scholar] [CrossRef] [PubMed]
- Pinto, P.C.; Evtuguin, D.V.; Neto, C.P.; Silvestre, A.J.D.; Amado, F.M.L. Behavior of Eucalyptus globulus lignin during kraft pulping. II. Analysis by NMR, ESI/MS, and GPC. J. Wood Chem. Technol. 2002, 22, 109–125. [Google Scholar] [CrossRef]
- Corbett, W.M.; Kenner, J.; Richards, G.N. The degradation of carbohydrate by alkali. Part X. Acetal derivatives. J. Chem. Soc. 1955, 1709–1711. [Google Scholar] [CrossRef]
- Minor, J.L. Chemical Linkage of Polysaccharides to Residual Lignin in Loblolly Pine Kraft Pulps. J. Wood Chem. Technol. 1986, 6, 185–201. [Google Scholar] [CrossRef]
- Lawoko, M.; Berggren, R.; Berthold, F.; Henriksson, G.; Gellerstedt, G. Changes in the lignin-carbohydrate complex in softwood kraft pulp during kraft and oxygen delignification. Holzforschung 2004, 58, 603–610. [Google Scholar] [CrossRef]
- Lu, D.S.; Zhang, K.; Chen, X.D.; Xie, Y.M. Study on the Structure of Residual Lignin in Unbleached Kraft Pulp by 13C-2H Dual Isotope Tracer. China Pulp Pap. 2022, 41, 8–15. [Google Scholar]
- Balakshin, M.Y.; Capanema, E.A.; Chang, H.M. MWL Fraction with a High Concentration of Lignin-Carbohydrate Linkages: Isolation and 2D NMR Spectroscopic Analysis. Holzforschung 2007, 61, 1–7. [Google Scholar] [CrossRef]
- Du, X.Y.; Pérez-Boada, M.M.; Fernández, C.; Rencoret, J.; del Río, J.C.; Jiménez-Barbero, J.; Li, J.; Gutiérrez, A.; Martínez, A.T. Analysis of Lignin-Carbohydrate and Lignin-Lignin Linkages after Hydrolase Treatment of Xylan-Lignin, Glucomannan-Lignin and Glucan-Lignin Complexes from Spruce Wood. Planta 2014, 239, 1079–1090. [Google Scholar] [CrossRef]
- Tugarinov, V.; Kanelis, V.; Kay, L. Isotope labeling strategies for the study of high-molecular-weight proteins by solution NMR spectroscopy. Nat. Protoc. 2006, 1, 749–754. [Google Scholar] [CrossRef]
- Xie, Y.; Terashima, N. Selective carbon 13-enrichment of side chain carbons of ginkgo lignin traced by carbon 13 nuclear magnetic resonance. Mokuzai Gakkaishi 1991, 37, 935–941. [Google Scholar]
- Hafrén, J.; Westermark, U.; Lennholm, H.; Terashima, N. Formation of 13C-enriched cell-wall DHP using isolated soft xylem from Picea abies. Holzforschung 2002, 56, 585–591. [Google Scholar] [CrossRef]
- Xie, Y.; Yasuda, S.; Wu, H.; Liu, H.B. Analysis of the structure of lignin-carbohydrate complexes by the specific 13C tracer method. J. Wood Sci. 2000, 46, 130–136. [Google Scholar] [CrossRef]
- He, L.; Terashima, N. Formation and structure of lignin in monocotyledons I. Selective labeling of the structural units of lignin in rice plant (Oryza sativa) with 3H and visualization of their distribution in the tissue by microautoradiography. Mokuzai Gakkaishi 1989, 35, 116–122. [Google Scholar]
- He, L.; Terashima, N. Formation and structure of lignin in monocotyledons. II. Deposition and distribution of phenololic acids and their association with cell wall polymers s in rice plants (Oryzasativa). Mokuzai Gakkaishi 1989, 35, 123–129. [Google Scholar]
- He, L.F.; Terashima, N. Formation and Structure of Lignin in Monocotyledons. III. Heterogeneity of Sugarcane (Saccharum officinarum L.) Lignin with Respect to the Composition of Structural Units in Different Morphological Regions. J. Wood Chem. Technol. 1990, 10, 435–459. [Google Scholar] [CrossRef]
- Balakshin, M.; Capanema, E.; Berlin, A. Isolation and Analysis of Lignin-Carbohydrate Complexes Preparations with Traditional and Advanced Methods. Stud. Nat. Prod. Chem. 2014, 42, 83–115. [Google Scholar]
- Xie, Y.; Terashima, N. Selective carbon 13-enrichment of side chain carbons of rice stalk lignin traced by carbon 13 nuclear magnetic resonance. Mokuzai Gakkaishi 1993, 39, 91–97. [Google Scholar]
- Terashima, N.; Seguchi, Y. Selective 13C Enrichment of Side Chain Carbons of Guaiacyl Lignin in Pine. Holzforschung 1991, 45, 35–39. [Google Scholar] [CrossRef]
- Zhang, K.; Liu, Y.C.; Cui, S.; Xie, Y. Elucidation of the structure of lignin–carbohydrate complexes in ginkgo CW-DHP by 13C-2H dual isotope tracer. Molecules 2021, 26, 5740. [Google Scholar] [CrossRef]
- Nishida, Y.; Ohrui, H.; Meguro, H. 1H-NMR studies of (6r)-and (6s)-deuterated d-hexoses: Assignment of the preferred rotamers about C5-C6 bond of D-glucose and D-galactose derivatives in solutions. Tetrahedron Lett. 1984, 25, 1575–1578. [Google Scholar] [CrossRef]
- Hikichi, K.; Kakuta, Y.; Katoh, T. 1H NMR study on substituent distribution of cellulose diacetate. Polym. J. 1995, 27, 659–663. [Google Scholar] [CrossRef]
- Tanbda, H.; Nakano, J.; Hosoya, S.; Chang, H.-M. Stability of α-Ether type Model Compounds During Chemical Pulping Processes. J. Wood. Chem. Technol. 1987, 7, 485–497. [Google Scholar] [CrossRef]
- Besombes, S.; Robert, D.; Utille, J.P.; Taravel, F.R.; Mazeau, K. Molecular modeling of syringyl and p-hydroxyphenyl β-O-4 dimers. Comparative study of the computed and experimental conformational properties of lignin β-O-4 model compounds. J. Agric. Food Chem. 2003, 51, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Slater, W.G.; Beevers, H. Utilization of D-Glucuronate by Corn Coleoptiles. Plant Physiol. 1958, 33, 146–151. [Google Scholar] [CrossRef]
- Bailey, R.W.; Hassid, W.Z. Xylan synthesis from uridine-diphosphate-d-xylose by particulate preparations from immature corncobs. Proc. Natl. Acad. Sci. USA 1966, 56, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, A.S.; Tyshkunova, I.V.; Poshina, D.N.; Guryanova, A.A.; Chukhchin, D.G.; Sinelnikov, I.G.; Terentyev, K.Y.; Skorik, Y.A.; Novozhilov, E.V.; Synitsyn, A.P. Biocatalysis of Industrial Kraft Pulps: Similarities and Differences between Hardwood and Softwood Pulps in Hydrolysis by Enzyme Complex of Penicillium verruculosum. Catalysts 2020, 10, 536. [Google Scholar] [CrossRef]
- Spies, J.R. Determination of tryptophan in proteins. Anal. Chem. 1967, 39, 1412–1416. [Google Scholar] [CrossRef]
- Olley, J.; Lovern, J.A. Phospholipid hydrolysis in cod flesh stored at various temperatures. J. Sci. Food Agric. 1960, 11, 644–652. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| δ13C (‰) | 13C/12C (%) | 13Cα/12Cα (%) | 13Cγ/12Cγ (%) | |
|---|---|---|---|---|
| Control | −27.56 | 1.076 | 1.076 | 1.076 |
| A | −1.28 | 1.104 | 2.604 | - |
| B | 7.92 | 1.114 | - | 3.089 |
| δD (‰) | D/H (%) | D6/H6 (%) | |
|---|---|---|---|
| Control | 58.692 | 0.016 | 0.016 |
| A | 1356.568 | 0.037 | 0.280 |
| B | 1911.743 | 0.045 | 0.370 |
| Signal | 13C (ppm) | Assignments |
|---|---|---|
| 1 | 109.0 | Cα in lignin linked to carbohydrate by acetal bond |
| 2 | 82–88 | Cα in lignin linked to carbohydrate by benzyl ether bond, Cα in lignin β-5, β-β structure |
| 3 | 76.4 | Cα in lignin linked to carbohydrates by benzyl ester bond |
| 4 | 74.6 | Cα in β-O-4 structure |
| 1′ | 167.3 | Cγ of ferulic acid derivatives |
| 2′ | 72.6 | Cγ in β-β structure of lignin |
| 3′ | 64.4 | Cγ in lignin β-5 |
| 4′ | 62.5 | Cγ in β-O-4 and β-1 structure of lignin |
| Coniferin-[α-13C] | Coniferin-[γ-13C] | d-Glucose-[6-D2] | 4CL Inhibitor | |
|---|---|---|---|---|
| Control | - | - | - | - |
| Group A | 5 mg/mL | - | 3.33 mg/mL | 4 mg/mL |
| Group B | - | 5 mg/mL | 3.33 mg/mL | 4 mg/mL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, H.; Chen, X.; Zhao, Y.; Zhou, J.; Xie, Y. Exploration of the Linkages between Lignin and Carbohydrates in Kraft Pulp from Wheat Straw Using a 13C/2H Isotopic Tracer. Molecules 2023, 28, 7493. https://doi.org/10.3390/molecules28227493
Niu H, Chen X, Zhao Y, Zhou J, Xie Y. Exploration of the Linkages between Lignin and Carbohydrates in Kraft Pulp from Wheat Straw Using a 13C/2H Isotopic Tracer. Molecules. 2023; 28(22):7493. https://doi.org/10.3390/molecules28227493
Chicago/Turabian StyleNiu, Hujun, Xudong Chen, Yunbo Zhao, Junyi Zhou, and Yimin Xie. 2023. "Exploration of the Linkages between Lignin and Carbohydrates in Kraft Pulp from Wheat Straw Using a 13C/2H Isotopic Tracer" Molecules 28, no. 22: 7493. https://doi.org/10.3390/molecules28227493
APA StyleNiu, H., Chen, X., Zhao, Y., Zhou, J., & Xie, Y. (2023). Exploration of the Linkages between Lignin and Carbohydrates in Kraft Pulp from Wheat Straw Using a 13C/2H Isotopic Tracer. Molecules, 28(22), 7493. https://doi.org/10.3390/molecules28227493