
Progress in Catalytic Asymmetric Reactions with 7-Azaindoline as the Directing Group

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
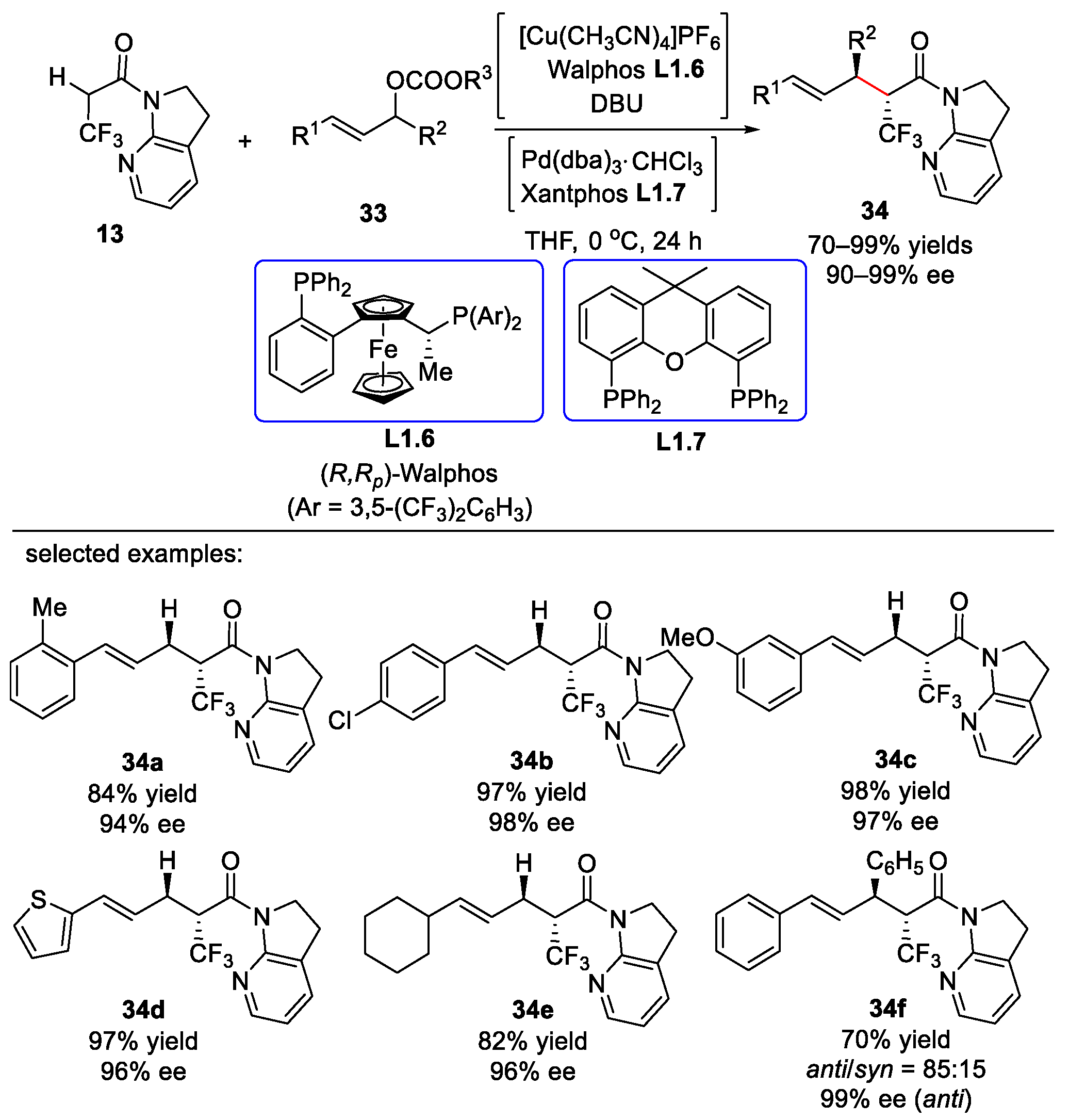{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
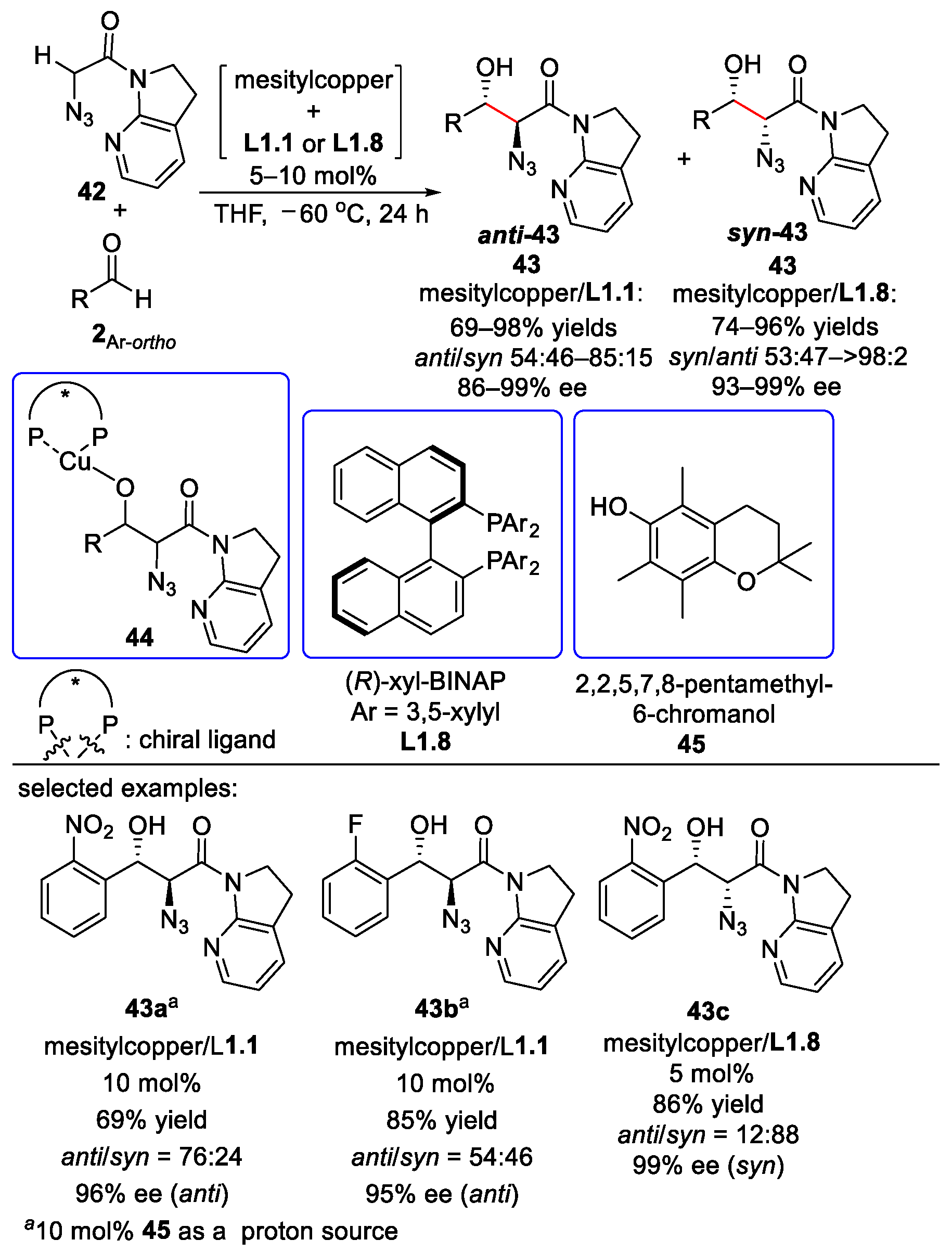{kind=link}
{kind=link}
{kind=link}
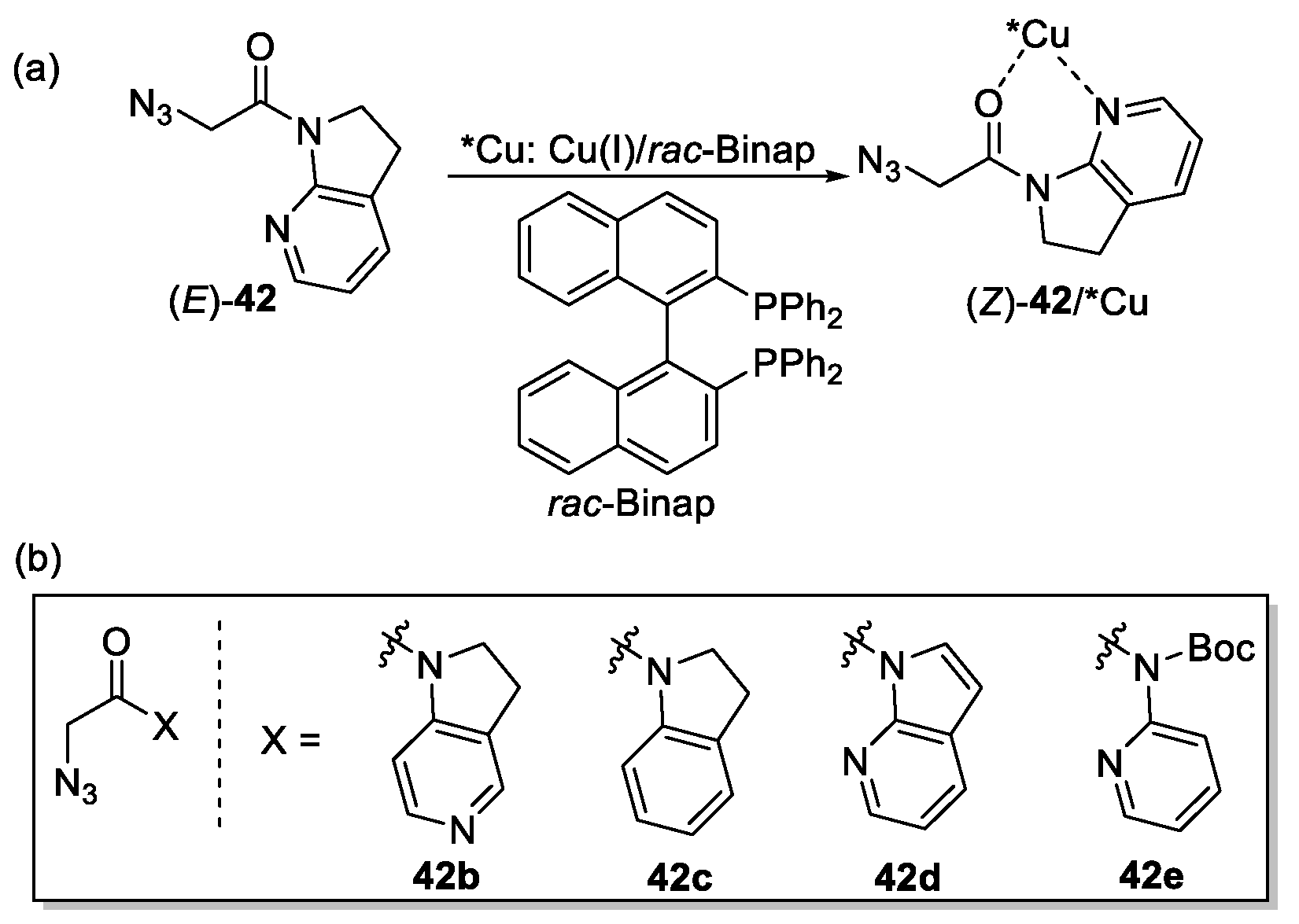{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
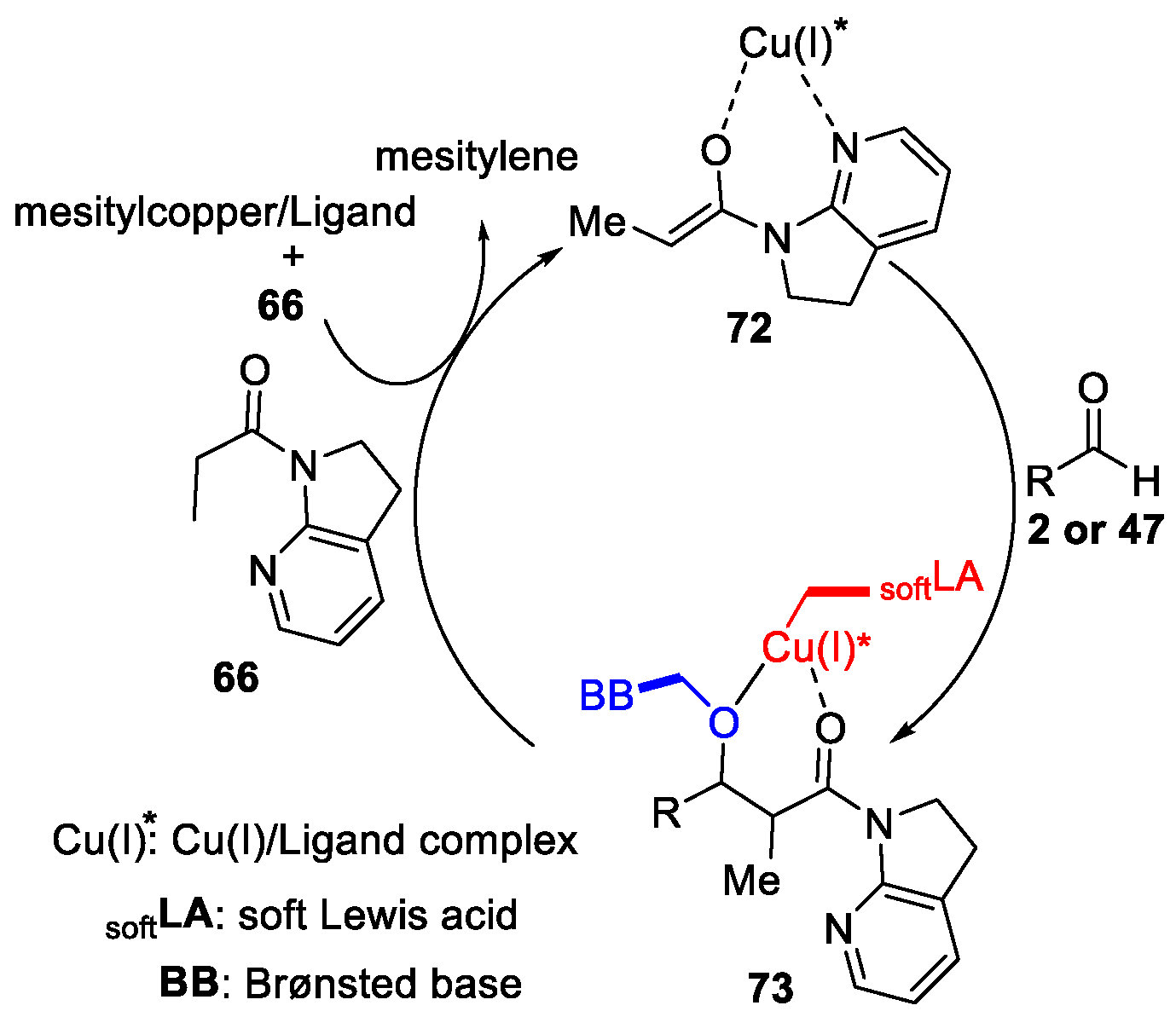{kind=link}
{kind=link}
{kind=link}
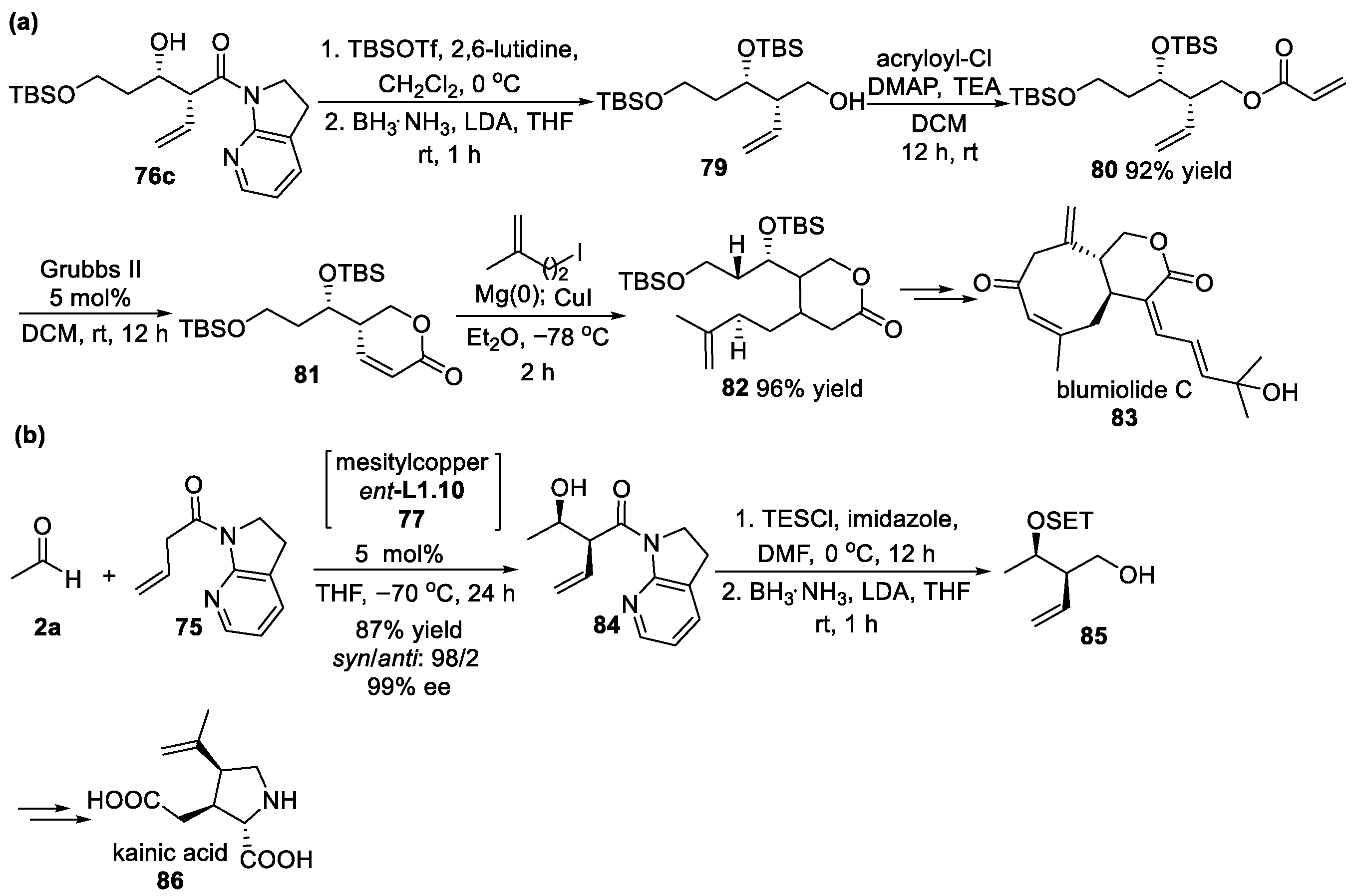{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
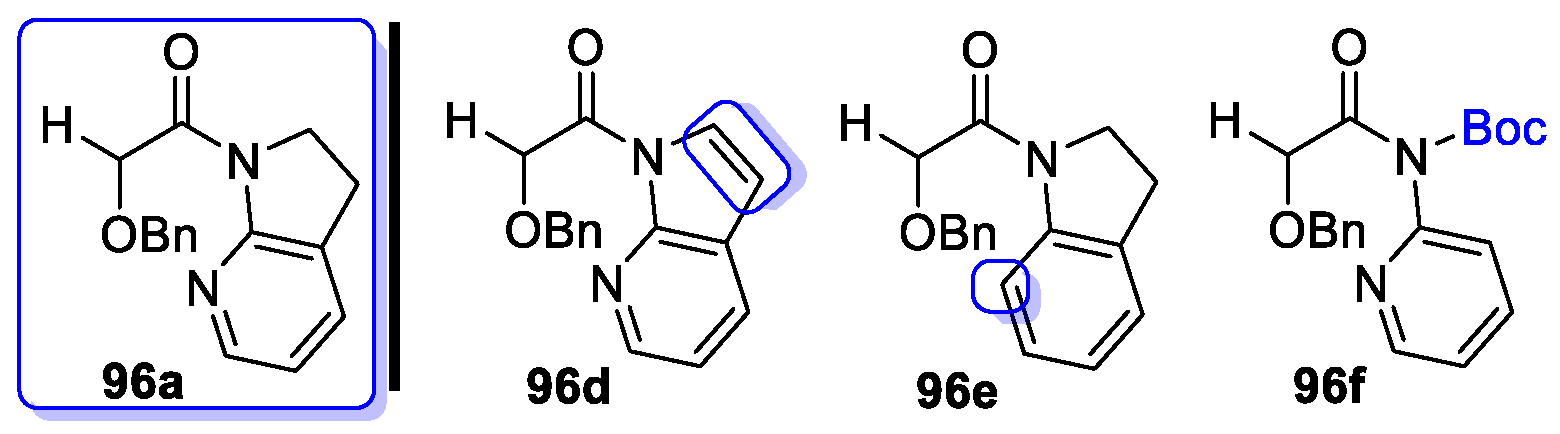{kind=link}
{kind=link}
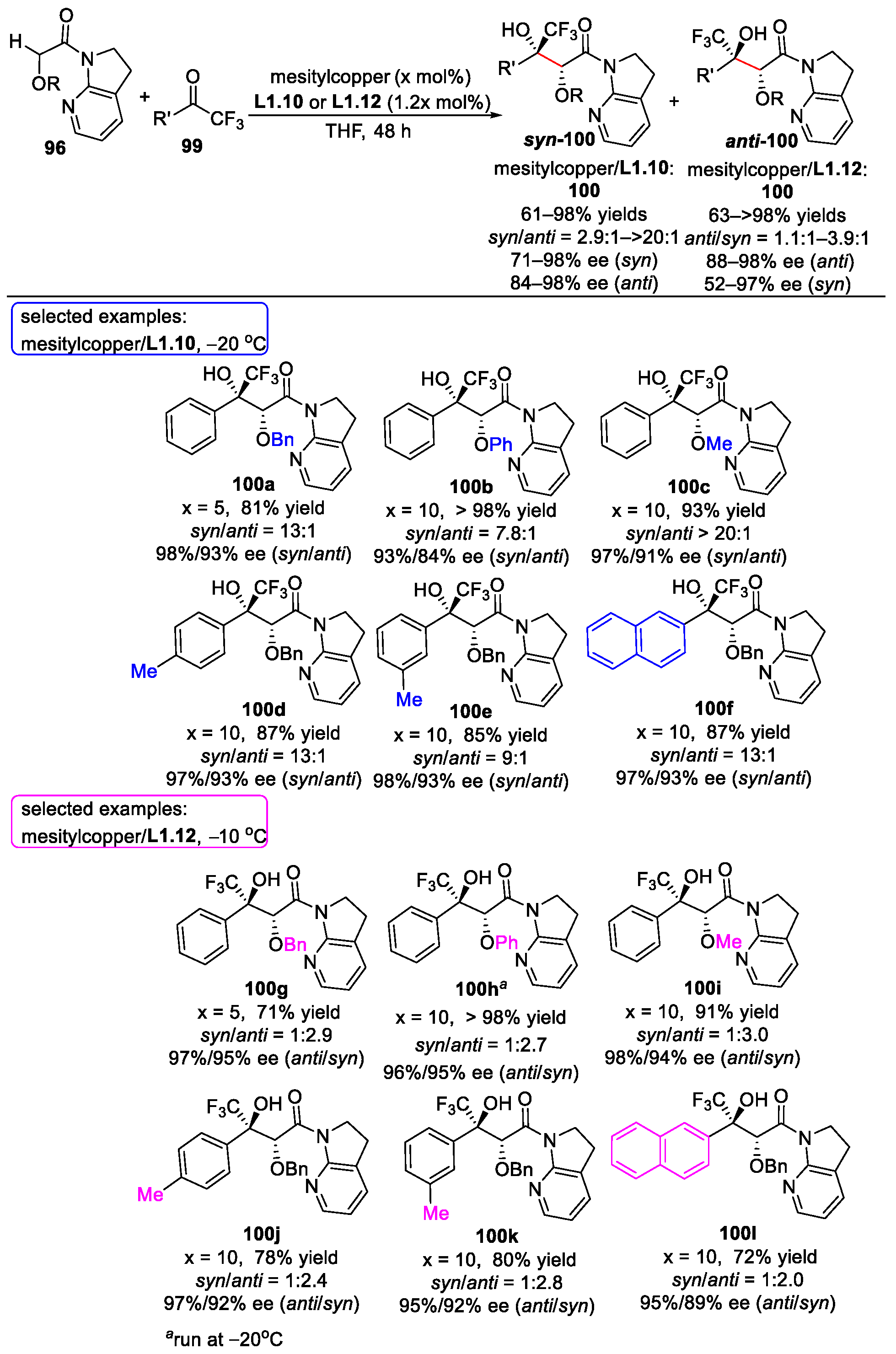{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
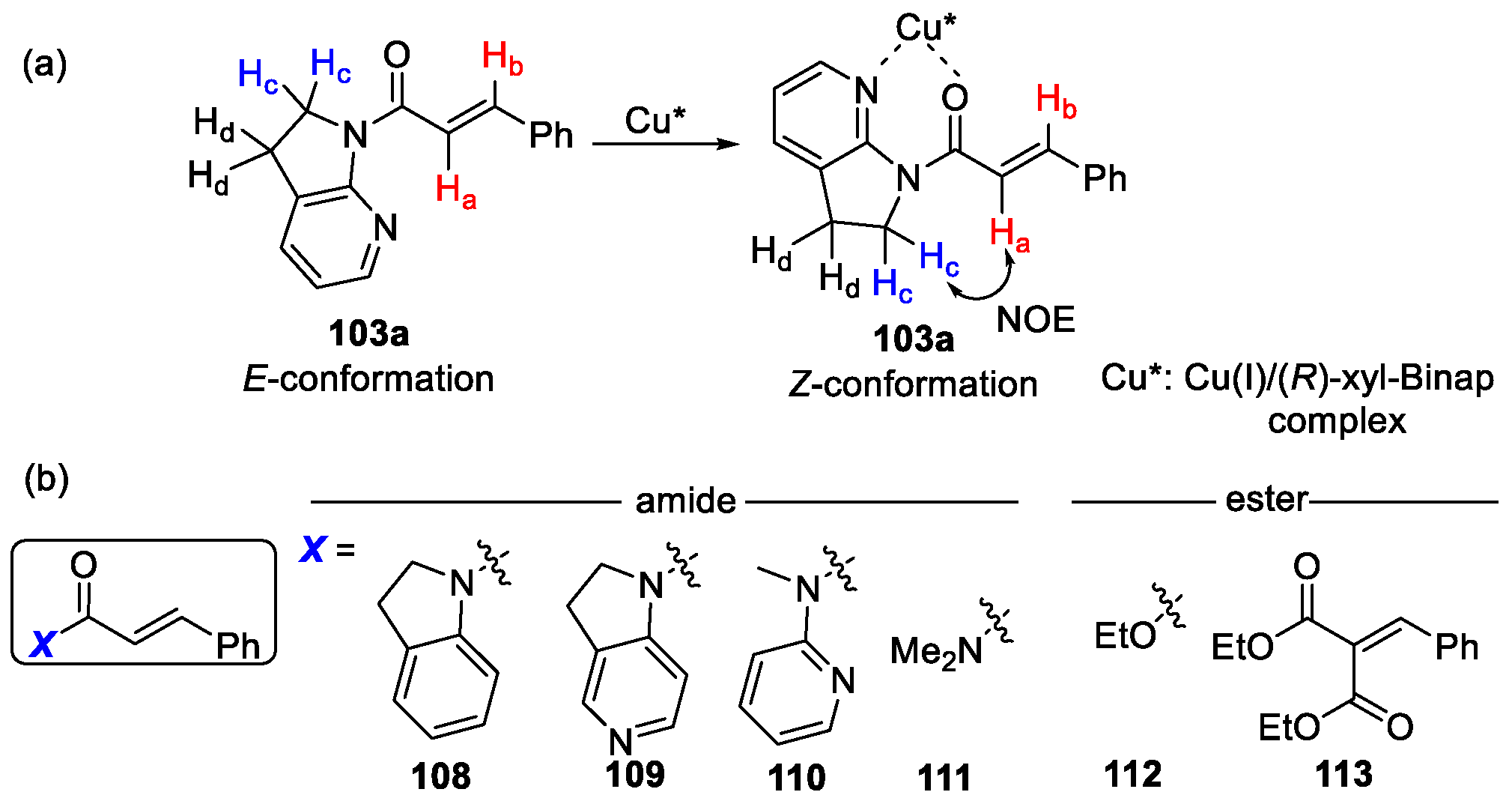{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
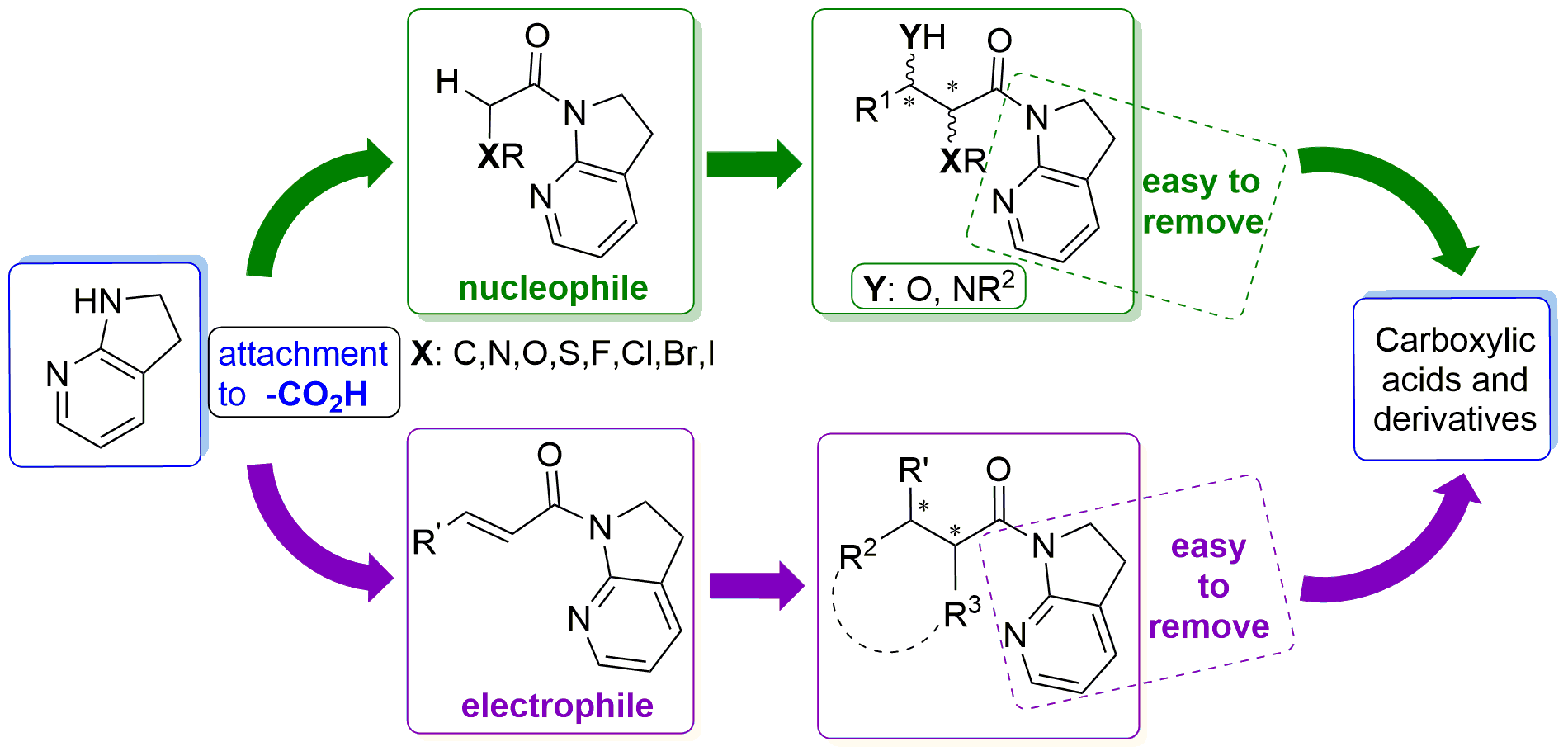
1. Introduction
2. α-Substituted-7-Azaindoline Amide as a Nucleophile
2.1. α-Sulfanyl 7-Azaindoline Amide as an Aldol Donor
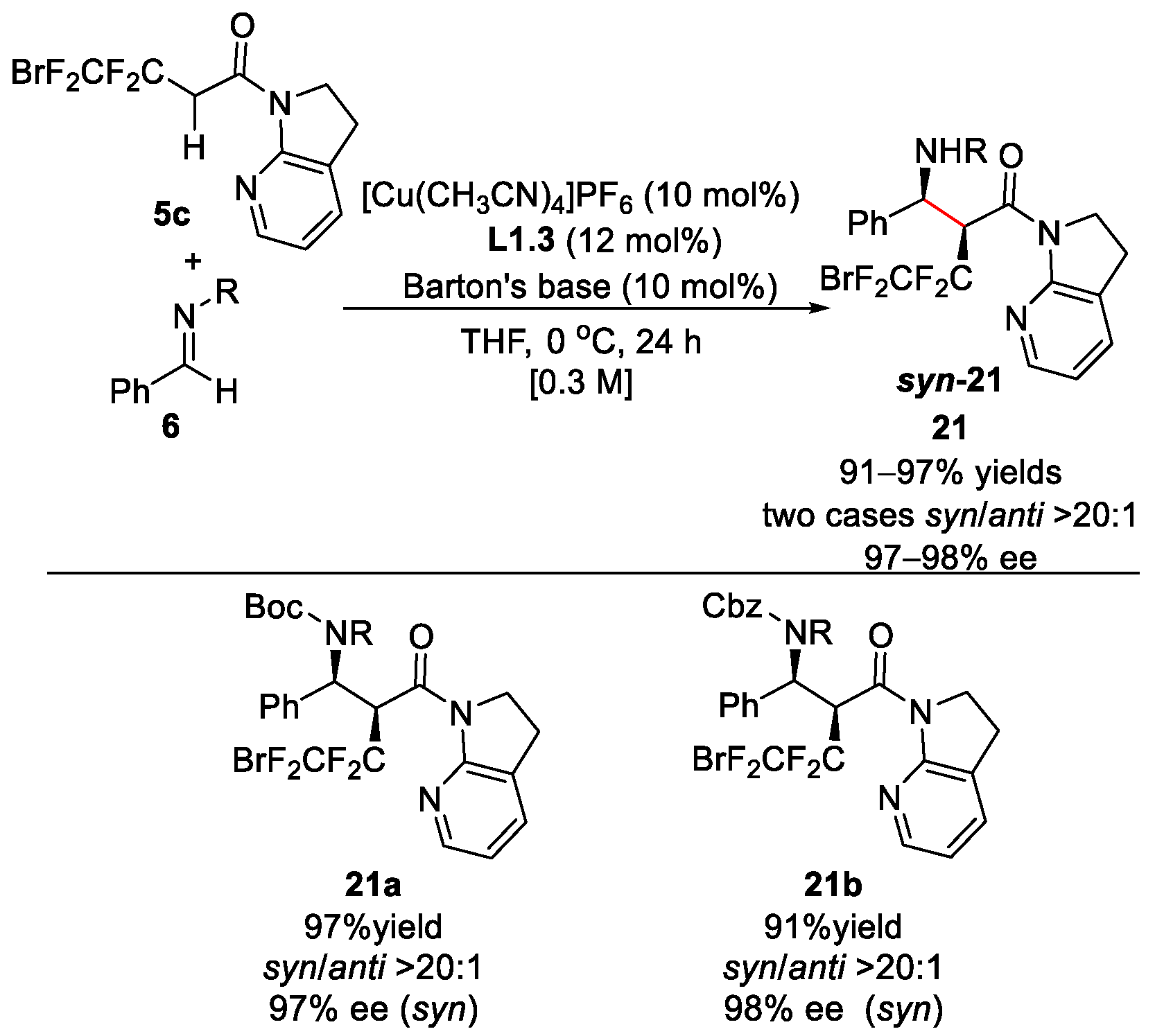
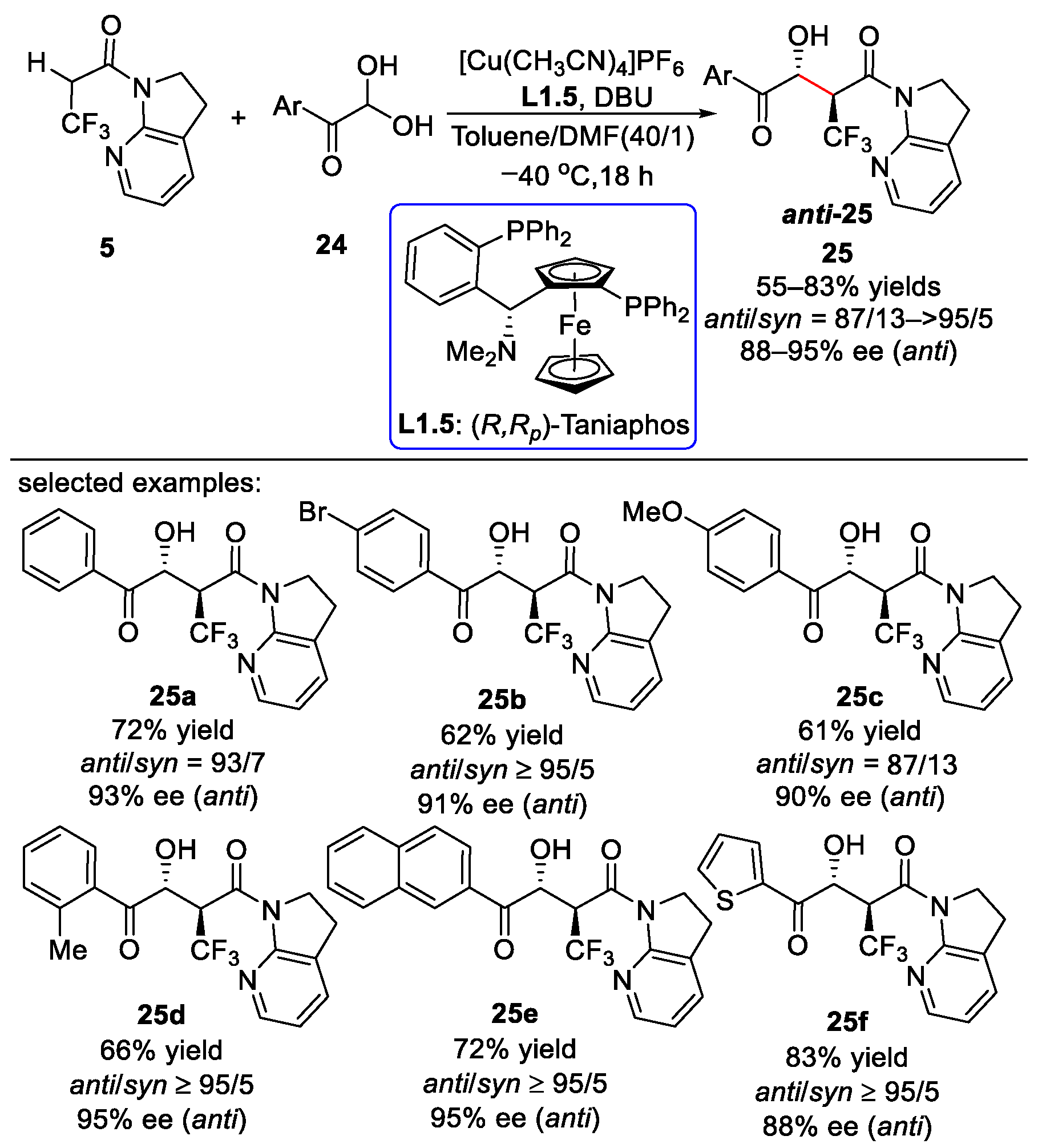
2.2. Fluoroalkyl 7-Azaindoline Amides as Mannich Donors or Aldol Donors
2.3. α-Azido 7-Azaindoline Amide as a Nucleophile
2.4. α-Alkyl Substituted 7-Azaindoline Amide as a Nucleophile
2.5. α-Halo Substituted 7-Azaindoline Amide as Nucleophiles
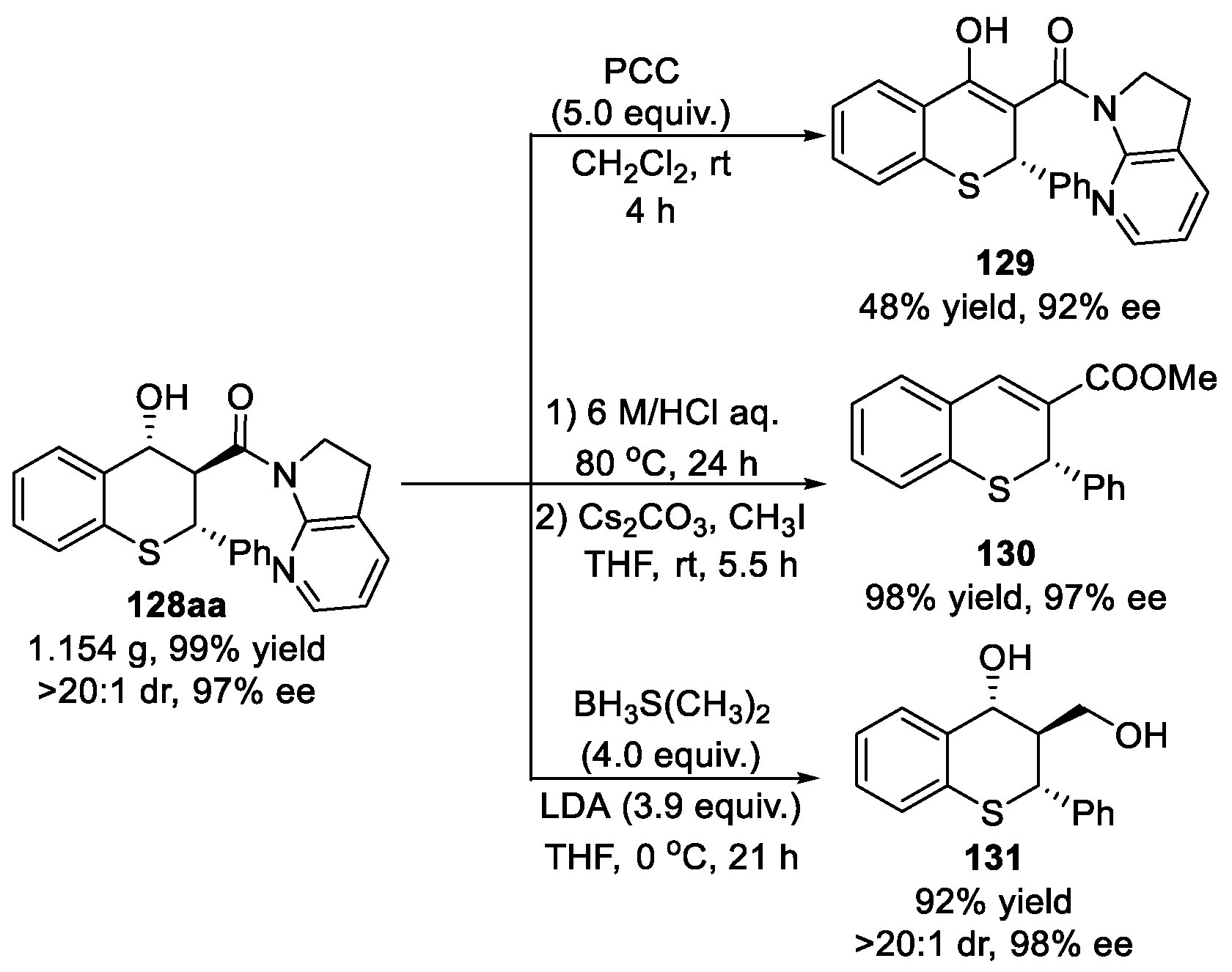
2.6. α-Oxygen-Substituted 7-Azaindoline Amide as Nucleophiles
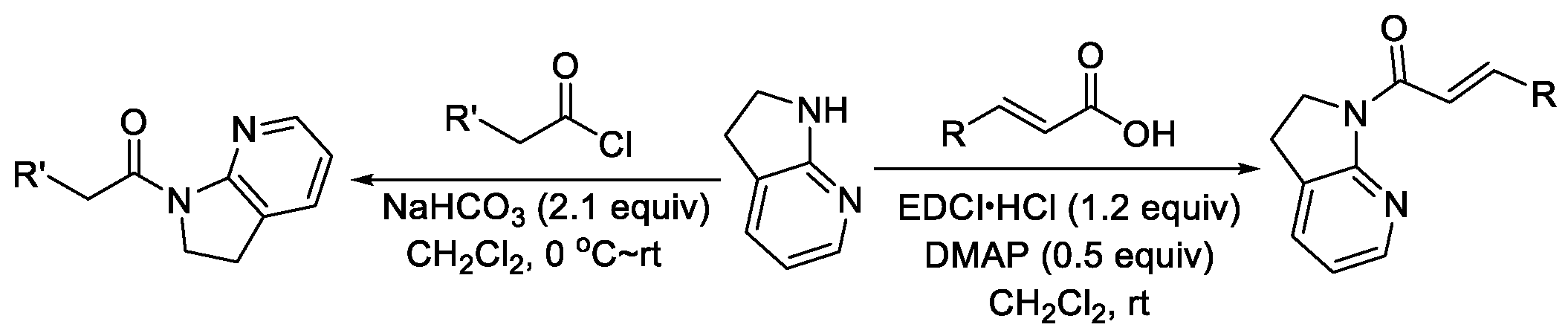
3. α,β-Unsaturated 7-Azaindoline Amides Act as Electrophiles
3.1. α,β-Unsaturated 7-Azaindoline Amides as Electrophiles in Vinylogous Conjugate Addition
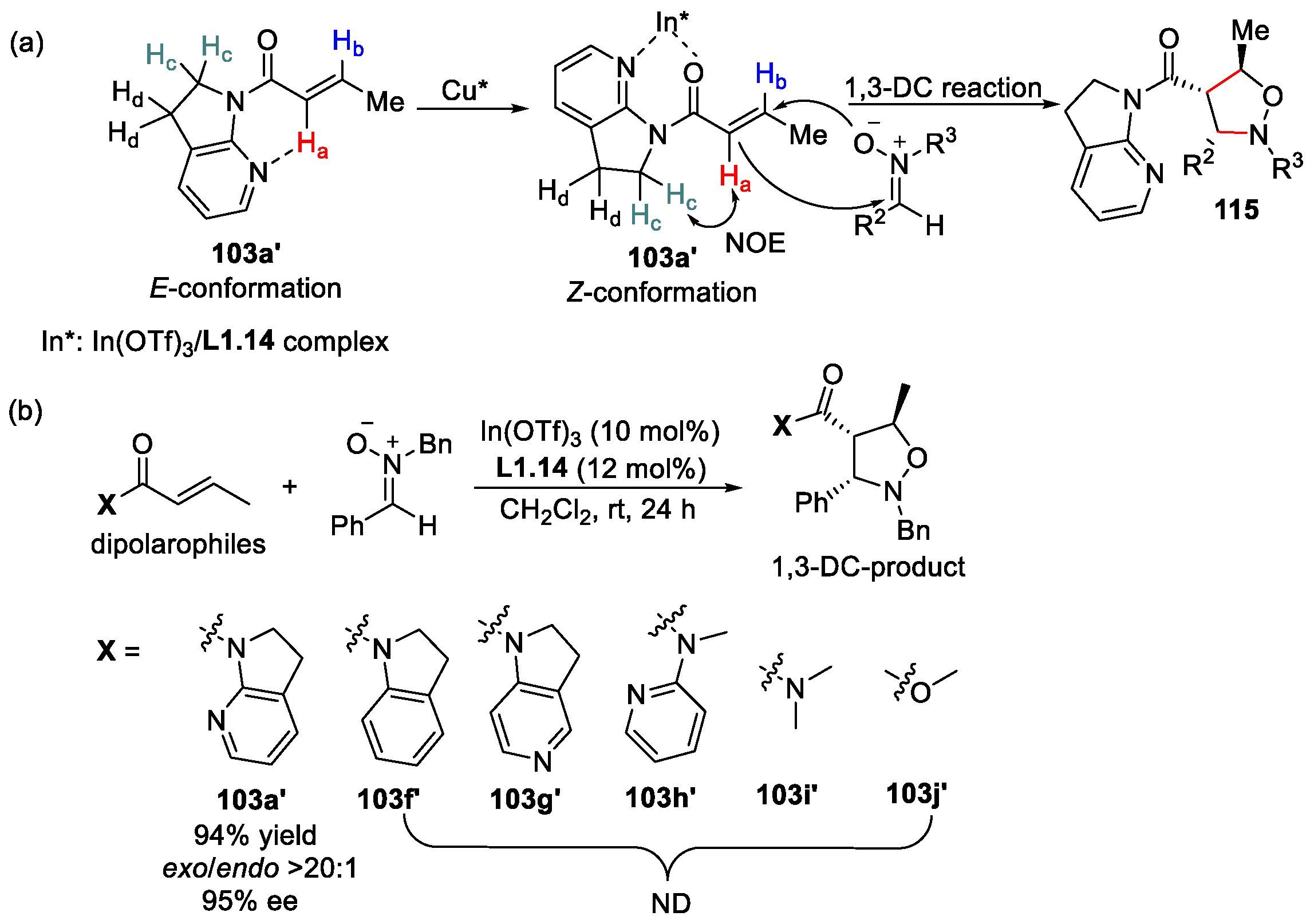
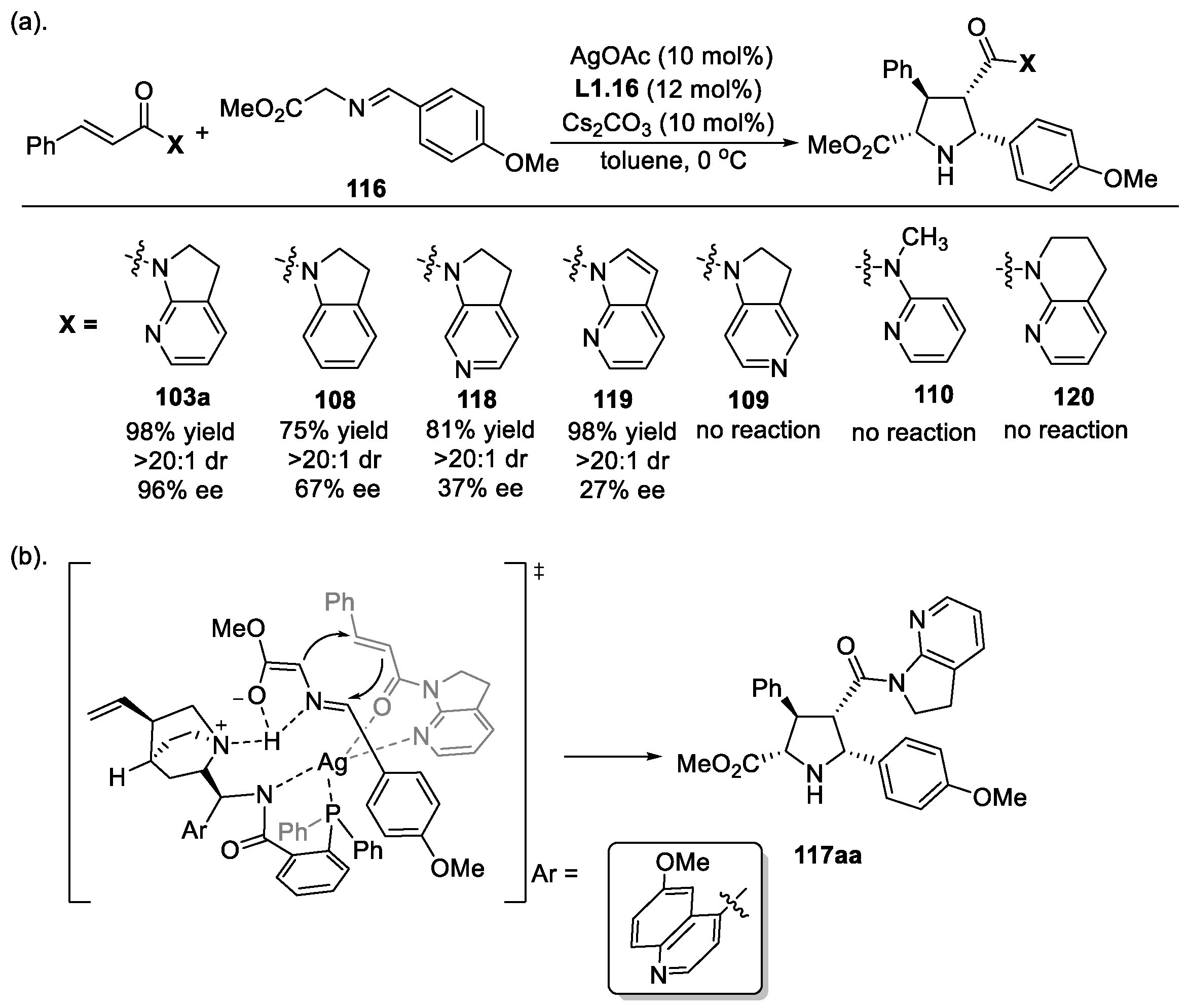
3.2. α,β-Unsaturated 7-Azaindoline Amides as Electrophiles in 1,3-Dipolar Cycloaddition
3.3. α,β-Unsaturated 7-Azaindoline Amides Act as Electrophiles in Michael/Aldol Cascade Reaction
3.4. α,β-Unsaturated 7-Azaindoline Amides Act as Electrophiles in Aminomethylation
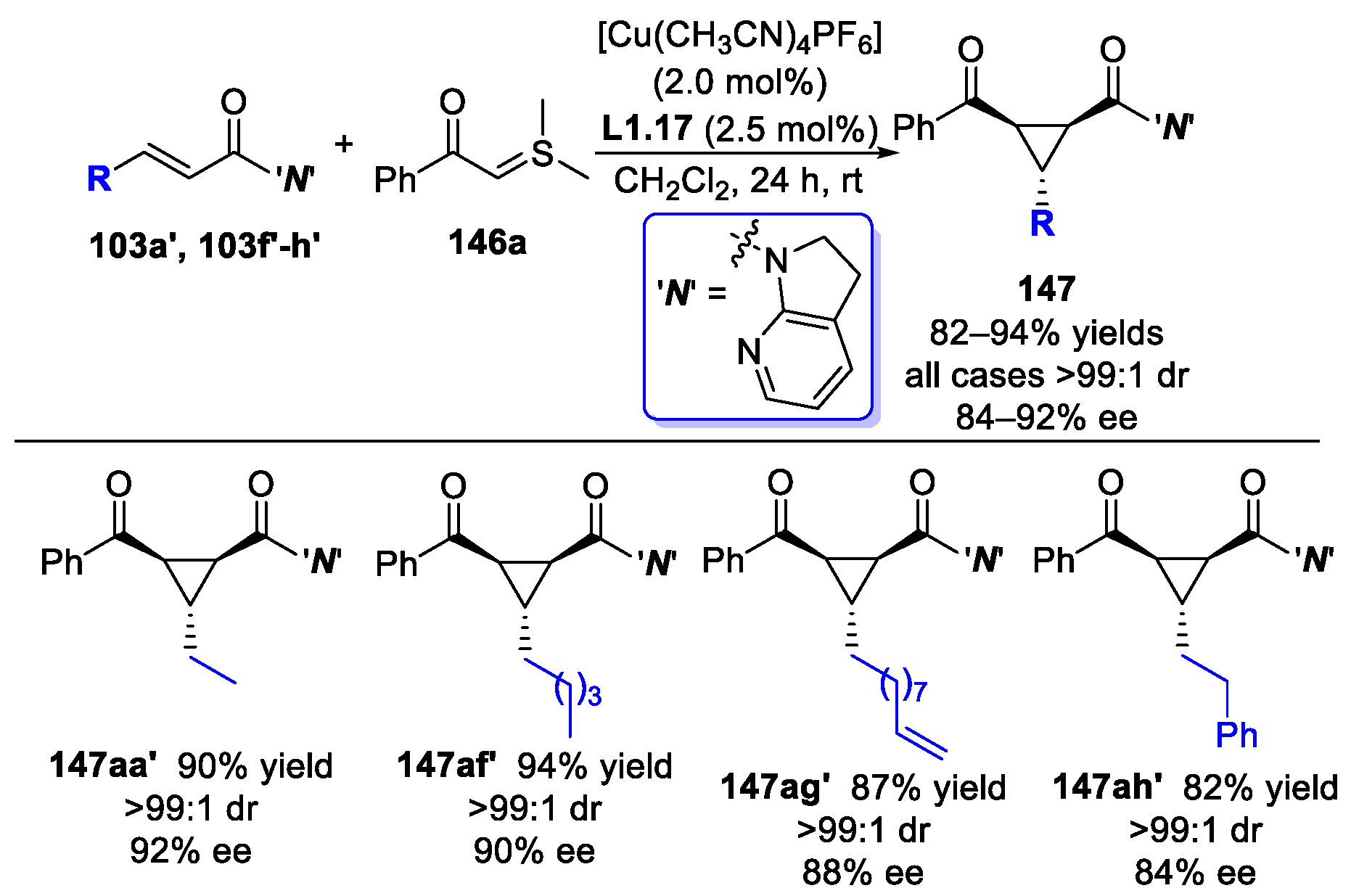
3.5. α,β-Unsaturated 7-Azaindoline Amides Act as Electrophiles in Michael Addition-Initiated Ring-Closure Reaction
4. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stella, V.; Borchardt, R.; Hageman, M.; Oliyai, R.; Maag, H.; Tilley, J. (Eds.) Prodrugs: Challenges and Rewards, Part III; Springer Science & Business Media: New York, NY, USA, 2007; pp. 3–29. [Google Scholar]
- Lamberth, C.; Dinges, J. Different Roles of Carboxylic Functions in Pharmaceuticals and Agrochemicals. In Bioactive Carboxylic Compound Classes: Pharmaceuticals and Agrochemicals; John Wiley & Sons: Hoboken, NJ, USA, 2016; pp. 1–11. [Google Scholar]
- Seibert, J.B.; Bautista-Silva, J.P.; Amparo, T.R.; Petit, A.; Pervier, P.; dos Santos Almeida, J.C.; Azevedoa, M.C.; Silveiraa, B.M.; Brandãoa, G.C.; De Souzaa, G.H.B.; et al. Development of propolis nanoemulsion with antioxidant and antimicrobial activity for use as a potential natural preservative. Food Chem. 2019, 287, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Marchev, A.S.; Vasileva, L.V.; Amirova, K.M.; Savova, M.S.; Koycheva, I.K.; Balcheva-Sivenova, Z.P.; Vasileva, S.M.; Georgiev, M.I. Rosmarinic acid—From bench to valuable applications in food industry. Trends Food Sci. Technol. 2021, 117, 182–193. [Google Scholar] [CrossRef]
- Beg, D.; Kumar, A.; Shende, D.; Wasewar, K. Liquid-liquid extraction of lactic acid using non-toxic solvents. Chem. Data Collect. 2022, 38, 100823. [Google Scholar] [CrossRef]
- Wang, L.; Pan, X.Q.; Jiang, L.S.; Chu, Y.; Gao, S.; Jiang, X.Y.; Zhang, Y.H.; Chen, Y.; Luo, S.J.; Peng, C. The biological activity mechanism of chlorogenic acid and its applications in food industry: A review. Front. Nutr. 2022, 9, 943911. [Google Scholar] [CrossRef] [PubMed]
- Tavares, L.; Smaoui, S.; Lima, P.S.; de Oliveira, M.M.; Santos, L. Propolis: Encapsulation and application in the food and pharmaceutical industries. Trends Food Sci. Technol. 2022, 127, 169–180. [Google Scholar] [CrossRef]
- El-Sakhawy, M.; Salama, A.; Mohamed, S.A. Propolis applications in food industries and packaging. Biomass Convers. Bior. 2023, 1–16. [Google Scholar] [CrossRef]
- Korth, H.G.; Sustman, R. Carboxylic Acids and Carboxylic Acid Derivatives, 4th ed.; Thieme: Stuttgart, Germany, 1985; pp. 193–469. [Google Scholar]
- Goossen, L.J.; Rodriguez, N.; Gooßen, K. Carboxylic acids as substrates in homogeneous catalysis. Carboxylic acids as substrates in homogeneous catalysis. Angew. Chem. Int. Ed. 2008, 47, 3100–3120. [Google Scholar] [CrossRef]
- Hartman, G.D.; Egbertson, M.S.; Halczenko, W.; Laswell, W.L.; Duggan, M.E.; Smith, R.L.; Naylor, A.M.; Manno, P.D.; Lynch, R.J. Non-peptide fibrinogen receptor antagonists. 1. Discovery and design of exosite inhibitors. J. Med. Chem. 1992, 35, 4640–4642. [Google Scholar] [CrossRef]
- Vranova, V.; Rejsek, K.; Formanek, P. Aliphatic, cyclic, and aromatic organic acids, vitamins, and carbohydrates in soil: A review. Sci. World J. 2013, 2013, 524239. [Google Scholar] [CrossRef]
- Das, J.; Mal, D.K.; Maji, S.; Maiti, D. Recent advances in external-directing-group-free C-H functionalization of carboxylic acids without decarboxylation. ACS Catal. 2021, 11, 4205–4229. [Google Scholar] [CrossRef]
- Majumdar, N. Carboxylic acids as building blocks in catalytic asymmetric reactions. ACS Catal. 2022, 12, 8291–8324. [Google Scholar] [CrossRef]
- Sakuma, S.; Miyaura, N. Rhodium(I)-catalyzed asymmetric 1,4-addition of arylboronic acids to α,β-unsaturated amides. J. Org. Chem. 2001, 66, 8944–8946. [Google Scholar] [CrossRef] [PubMed]
- Oi, S.; Taira, A.; Honma, Y.; Sato, T.; Inoue, Y. Asymmetric 1, 4-addition of aryltrialkoxysilanes to α, β-unsaturated esters and amides catalyzed by a chiral rhodium complex. Tetrahedron Asymmetry 2006, 17, 598–602. [Google Scholar] [CrossRef]
- Li, Y.-B.; Tian, H.; Yin, L. Copper(I)-catalyzed asymmetric 1,4-conjugate hydrophosphination of α,β-unsaturated amides. J. Am. Chem. Soc. 2020, 142, 20098–20106. [Google Scholar] [CrossRef]
- Deng, Y.; Sun, S.; Wang, Y.; Jia, P.; Li, W.; Wang, K.; Yan, W. Asymmetric synthesis of chiral α-CF2H spiro[indoline-3,3′-thiophene] via phase-transfer catalyzed sulfa-Michael/Michael Domino reaction. Adv. Synth. Catal. 2022, 364, 811–830. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, H.; Peng, T.; Xu, Y.; Hou, Y.; Li, S.; Pang, S.; Zhang, H.; Yang, D. A tandem asymmetric oxidation-oxa-Michael sequence for dearomatization of β-naphthols. Chin. Chem. Lett. 2022, 33, 4273–4276. [Google Scholar] [CrossRef]
- Hidasová, D.; Pohl, R.; Císařová, I.; Jahna, U. A diastereoselective catalytic approach to pentasubstituted pyrrolidines by Tandem Anionic-Radical Cross-Over reactions. Adv. Synth. Catal. 2022, 364, 671–678. [Google Scholar] [CrossRef]
- He, C.; Tang, X.; He, X.; Zhou, Y.; Liu, X.; Feng, X. Regio- and enantioselective conjugate addition of β-nitro α,β-unsaturated carbonyls to construct 3-alkenyl disubstituted oxindoles. Chin. Chem. Lett. 2023, 34, 107487. [Google Scholar] [CrossRef]
- Chaulagain, M.R.; Aron, Z.D. A diastereoselective three-component coupling approach to highly substituted pyrrolidines. J. Org. Chem. 2010, 75, 8271–8274. [Google Scholar] [CrossRef]
- Honga, H.R.; Wang, H.Y.; Zheng, C.W.; Zhao, G.; Shang, Y.J. Organophosphine bearing multiple hydrogen-bond donors for asymmetric Michael addition reaction of 1-oxoindane-2-carboxylic acid ester via dual-reagent catalysis. Chin. Chem. Lett. 2021, 32, 708–712. [Google Scholar] [CrossRef]
- Ma, Z.W.; Wang, C.C.; Liu, X.F.; Chen, X.P.; Tao, J.C.; Lv, Q.J. Enantioselective synthesis of coumarins catalyzed by an isosteviol-derived tertiary amine-squaramide catalyst. Chirality 2022, 34, 325–332. [Google Scholar] [PubMed]
- Ma, Z.W.; Wang, C.C.; Chen, X.P.; Li, A.Q.; Tao, J.C.; Lv, Q.J. Highly enantioselective Michael addition of cyclic diketones to β,γ-unsaturated α-keto esters catalyzed by squaramide organocatalyst. Synthesis 2022, 54, 1785–1792. [Google Scholar] [CrossRef]
- Wang, X.B.; Tian, Y.; Zhou, L.; Xie, M.S.; Qu, G.R.; Guo, H.M. Rational design of chiral tridentate ligands: Bifunctional cobalt (II) complex/hydrogen bond for enantioselective Michael reactions. Org. Lett. 2022, 24, 3861–3866. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.S.; Li, K.L.; Sun, X.; Zha, Z.G.; Wang, Z.Y. Copper-catalyzed stereoselective [4+2] cycloaddition of β,γ-unsaturated α-keto esters and 2-uinylpyrroles in water. Org. Lett. 2022, 24, 4224–4228. [Google Scholar] [CrossRef]
- Rousseau, G.; Breit, B. Removable directing groups in organic synthesis and catalysis. Angew. Chem. Int. Ed. 2011, 50, 2450–2494. [Google Scholar] [CrossRef]
- Cheng, E.W.C.; Mandalia, R.T.; Motevalli, M.; Mothia, B.; Wyatt, P.B. Stereoselective diels–alder reactions of 3-phosphonopropenoyl derivatives of 1,3-oxazolidin-2-ones. Tetrahedron 2006, 62, 12398–12407. [Google Scholar] [CrossRef]
- Sibi, M.P.; Soeta, T.; Jasperse, C.P. Nitrile ylides: Diastereoselective cycloadditions using chiral oxzolidinones without lewis acid. Org. Lett. 2009, 11, 5366–5369. [Google Scholar] [CrossRef]
- Lam, Y.H.; Cheong, H.Y.; Mata, J.; Stanway, S.J.; Houk, K.N. Diels-alder exo selectivity in terminal-substituted dienes and dienophiles: Experimental discoveries and computational explanations. J. Am. Chem. Soc. 2009, 131, 1947–1957. [Google Scholar] [CrossRef]
- Martinet, S.; Méou, A.; Brun, P. Access to enantiopure 2,5-diaryltetrahydrofurans—Application to the synthesis of (-)-virgatusin and (+)-urinaligran. Eur. J. Org. Chem. 2009, 2009, 2306–2311. [Google Scholar] [CrossRef]
- Dentel, H.; Chataigner, I.; Lohier, J.F.; Gulea, M. Asymmetric diastereoselective thia-hetero-Diels-Alder reactions of dithioesters. Tetrahedron 2012, 68, 2326–2335. [Google Scholar] [CrossRef]
- Zhang, W.; Tan, D.; Lee, R.; Tong, G.H.; Chen, W.C.; Qi, B.J.; Huang, K.W.; Tan, C.H.; Jiang, Z.Y. Highly enantio- and diastereoselective reactions of γ-substituted butenolides through direct vinylogous conjugate additions. Angew. Chem. Int. Ed. 2012, 51, 10069–10073. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Kamma, S.; Gundla, R.; Adepally, U.; Kuncha, S.; Thirnathi, S.; Prasad, U.V. A reagent based dos strategy via evans chiral auxiliary: Highly stereoselective michael reaction towards optically active quinolizidinones, piperidinones and pyrrolidinones. RSC Adv. 2013, 3, 2404–2411. [Google Scholar] [CrossRef]
- Leitch, J.A.; Wilson, P.B.; McMullin, C.L.; Mahon, M.F.; Bhonoah, Y.; Williams, I.H.; Frost, C.G. Ruthenium(II) catalyzed C-H functionalization using the oxazolidinone heterocycle as a weakly coordinating directing group: Experimental and computational insights. ACS Catal. 2016, 6, 5520–5529. [Google Scholar] [CrossRef]
- Murre, A.; Erkman, K.; Kaabel, S.; Jrving, I.; Kanger, T. Diastereoselective [2,3]-sigmatropic rearrangement of N-allyl ammonium ylides. Synthesis 2019, 51, 4183–4197. [Google Scholar] [CrossRef]
- Nazari, A.; Heravi, M.M.; Zadsirjan, V. Oxazolidinones as chiral auxiliaries in asymmetric aldol reaction applied to natural products total synthesis. J. Organomet. Chem. 2021, 932, 121629. [Google Scholar] [CrossRef]
- Bhamboo, P.; Bera, S.; Mondal, D. TiCl4-promoted asymmetric aldol reaction of oxazolidinones and its sulphur-congeners for natural product synthesis. Asian J. Org. Chem. 2021, 10, 2763–2819. [Google Scholar] [CrossRef]
- Lauzon, S.; Schouwey, L.; Ollevier, T. C2-symmetric 2,2′-bipyridine-α,α′-1-adamantyl-diol ligand: Bulky iron complexes in asymmetric catalysis. Org. Lett. 2022, 24, 1116–1120. [Google Scholar] [CrossRef]
- Jensen, K.B.; Gothelf, K.V.; Hazell, R.G.; Jørgensen, K.A. Improvement of TADDOLate-TiCl2-catalyzed 1,3-dipolar nitrone cycloaddition reactions by substitution of the oxazolidinone auxiliary of the alkene with succinimide. J. Org. Chem. 1997, 62, 2471–2477. [Google Scholar] [CrossRef]
- Minakata, S.; Ezoe, T.; Nakamura, K.; Ryu, I.; Komatsu, M. Yb(OTf)3-catalyzed 1,3-dipolar cycloaddition of nitrone with alkene; switch in diastereoselectivity by solvent and bidentate auxiliary. Tetrahedron Lett. 1998, 39, 5205–5208. [Google Scholar] [CrossRef]
- Zhao, B.-L.; Du, D.-M. Enantioselective synthesis of enol lactones from tandem Michael addition/lactonization catalyzed by a chiral squaramide catalyst. Tetrahedron Asymmetry 2014, 25, 310–317. [Google Scholar] [CrossRef]
- Itoh, K.; Hasegawa, M.; Tanaka, J.; Kanemasa, S. Enantioselective enol lactone synthesis under double catalytic conditions. Org. Lett. 2005, 7, 979–981. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.B.; Yang, W.; Yang, W.K.; Liu, X.H.; Feng, X.M. Asymmetric catalytic [2,3]-stevens and Sommelet-Hauser rearrangements of α-diazo pyrazoleamides with sulfides. Angew. Chem. Int. Ed. 2019, 58, 13492–13498. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Z.; Wang, Y.C.; Wang, S.Z.; Ma, Y.Y.; Zhao, D.G.; Zhan, R.T.; Huang, H.C. Asymmetric construction of cyclobutanes via direct vinylogous Michael addition/cyclization of β, γ-unsaturated amides. Org. Lett. 2020, 22, 7135–7140. [Google Scholar] [CrossRef]
- Lin, Q.C.; Hu, B.W.; Xu, X.; Dong, S.X.; Liu, X.H.; Feng, X.M. Chiral N,N′-dioxide/Mg(OTf)2 complex-catalyzed asymmetric [2,3]-rearrangement of in situ generated ammonium salts. Chem. Sci. 2020, 11, 3068–3073. [Google Scholar] [CrossRef]
- Zhang, H.-J.; Zhong, F.; Xie, Y.-C.; Yin, L. Catalytic asymmetric Mannich-type reaction enabled by efficient dienolization of α,β-unsaturated pyrazoleamides. Chin. J. Chem. 2021, 39, 55–61. [Google Scholar] [CrossRef]
- Biswas, R.G.; Ray, S.K.; Kannaujiya, V.K.; Unhalea, R.A.; Singh, V.K. Cu(I)-catalyzed asymmetric exo-selective synthesis of substituted pyrrolidines via a 1,3-dipolar cycloaddition reaction. Org. Biomol. Chem. 2021, 19, 4685–4690. [Google Scholar] [CrossRef]
- Grell, Y.; Xie, X.L.; Ivlev, S.I.; Meggers, E. Enantioselective α-fluorination and α-chlorination of N-acyl pyrazoles catalyzed by a non-C2-symmetric chiral-at-Rhodium catalyst. ACS Catal. 2021, 11, 11396–11406. [Google Scholar] [CrossRef]
- Li, Z.J.; Xu, N.; Guo, N.; Zhou, Y.Q.; Lin, L.L.; Feng, X.M. Asymmetric catalytic synthesis of hexahydropyrroloisoquinolines via three-component 1,3-dipolar-cycloaddition. Chem. Eur. J. 2021, 27, 14841–14845. [Google Scholar] [CrossRef]
- Xiong, W.; Jiang, X.; Zhang, M.M.; Xiao, W.J.; Lu, L.Q. A Cooperative Pd/Co catalysis system for the asymmetric (4+2) cycloaddition of vinyl benzoxazinones with N-acylpyrazoles. Chem. Commun. 2021, 57, 13566–13569. [Google Scholar] [CrossRef]
- Chen, L.; Pu, M.P.; Li, S.Y.; Sang, X.P.; Liu, X.H.; Wu, Y.-D.; Feng, X.M. Enantioselective synthesis of nitriles containing a quaternary carbon center by Michael reactions of silyl ketene imines with 1-acrylpyrazoles. J. Am. Chem. Soc. 2021, 143, 19091–19098. [Google Scholar] [CrossRef]
- Zhong, F.; Yue, W.J.; Yin, X.H.; Zhang, H.M.; Yin, L. Copper(I)-catalyzed asymmetric synthesis of α-allenylamines and β-lactams through regioselective Mannich-type reactions. ACS Catal. 2022, 12, 9181–9189. [Google Scholar] [CrossRef]
- He, F.S.; Zhang, C.; Jiang, M.H.; Lou, L.J.; Wu, J.; Ye, S.Q. Access to chiral β-sulfonyl carbonyl compounds via photoinduced organocatalytic asymmetric radical sulfonylation with sulfur dioxide. Chem. Sci. 2022, 13, 8834–8839. [Google Scholar] [CrossRef]
- Nishimura, K.; Wang, Y.Z.; Ogura, Y.; Kumagai, J.; Ishihara, K. A π-Cu(II)-π complex as an extremely active catalyst for enantioselective α-halogenation of N-acyl-3,5-dimethylpyrazoles. ACS Catal. 2022, 12, 1012–1017. [Google Scholar] [CrossRef]
- Liu, Y.J.; Liu, Y.H.; Zhang, Z.Z.; Yan, S.Y.; Chen, K.; Shi, B.F. Divergent and stereoselective synthesis of β-silyl-α-amino acids through Palladium-catalyzed intermolecular silylation of unactivated primary and secondary C-H bonds. Angew. Chem. Int. Ed. 2016, 55, 13859–13862. [Google Scholar] [CrossRef]
- Sattar, M.; Shareef, M.; Patidar, K.; Kumar, S. Copper-catalyzed 8-aminoquinoline assisted aryl chalcogenation of ferroceneamide with aryl disulfides, diselenides, and ditellurides. J. Org. Chem. 2018, 83, 8241–8249. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bai, Z.B.; Jiao, T.Q.; Deng, Z.Q.; Tong, H.R.; He, G.; Peng, Q.; Chen, G. Palladium-catalyzed amide-directed enantioselective hydrocarbofunctionalization of unactivated alkenes using a chiral monodentate oxazoline ligand. J. Am. Chem. Soc. 2018, 140, 3542–3546. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.G.; Wei, J.L.; Lin, J.B.; Wang, G.J.; Zhou, J.; Chen, K.; Fan, C.A.; Zhang, S.Y. Asymmetric organocatalytic synthesis of 2,3-allenamides from hydrogen-bond-stabilized enynamides. Org. Lett. 2019, 21, 2468–2472. [Google Scholar] [CrossRef]
- Shen, H.C.; Zhang, L.; Chen, S.S.; Feng, J.J.; Zhang, B.W.; Zhang, Y.; Zhang, X.H.; Wu, Y.D.; Gong, L.Z. Enantioselective addition of cyclic ketones to unactivated alkenes enabled by amine/Pd(II) cooperative catalysis. ACS Catal. 2019, 9, 791–797. [Google Scholar] [CrossRef]
- Tong, H.R.; Zheng, W.R.; Lv, X.Y.; He, G.; Liu, P.; Chen, G. Asymmetric synthesis of β-lactam via Palladium-catalyzed enantioselective intramolecular C(sp3)-H amidation. ACS Catal. 2020, 10, 114–120. [Google Scholar] [CrossRef]
- Yuan, W.K.; Shi, B.F. Synthesis of chiral spirolactams via sequential C-H olefination/asymmetric [4+1] spirocyclization under a simple CoII/chiral spiro phosphoric acid binary system. Angew. Chem. Int. Ed. 2021, 60, 23187–23192. [Google Scholar] [CrossRef]
- Kim, Y.B.; Won, J.; Lee, J.; Kim, J.; Zhou, B.W.; Park, J.W.; Baik, M.H.; Chang, S. Ni-catalyzed intermolecular C(sp3)-H amidation tuned by bidentate directing groups. ACS Catal. 2021, 11, 3067–3072. [Google Scholar] [CrossRef]
- Wu, J.M.; Zhang, J.L.; Jiao, Y.J.; Deng, G.T.; Li, Y.M.; Zhang, Z.Y.; Jiang, Y.J. Palladium-catalyzed decarbonylation of amino acid derivatives via C-C bond and C-N bond dual activations. J. Org. Chem. 2021, 86, 17462–17470. [Google Scholar] [CrossRef] [PubMed]
- Weidner, K.; Kumagai, N.; Shibasaki, M. A Designed amide as an aldol donor in the direct catalytic asymmetric aldol reaction. Angew. Chem. Int. Ed. 2014, 53, 6150–6154. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Brewitz, L.; Kumagai, N.; Shibasaki, M. Catalytic generation of α-CF3 enolate: Direct catalytic asymmetric Mannich-type reaction of α-CF3 amide. J. Am. Chem. Soc. 2014, 136, 17958–17961. [Google Scholar] [CrossRef]
- Brewitz, L.; Arteaga, F.A.; Yin, L.; Alagiri, K.; Kumagai, N.; Shibasaki, M. Direct catalytic asymmetric Mannich-type reaction of α- and β-fluorinated amides. J. Am. Chem. Soc. 2015, 137, 15929–15939. [Google Scholar] [CrossRef]
- Cooper, J.A.; Olivares, C.M.; Sandford, G. Nucleophilic substitution and Claisen rearrangement reactions of model fluoroalkenes of general Structure R-CF=CF-CF3. J. Org. Chem. 2001, 66, 4887–4891. [Google Scholar] [CrossRef] [PubMed]
- Ogu, K.-i.; Akazome, M.; Ogura, K. A novel reaction of allylic alcohols with hexafluoropropene-diethylamine adduct (PPDA) to form 2-fluoro-2-trifluoromethyl-4-alkenamide. J. Fluorine Chem. 2003, 124, 69–80. [Google Scholar] [CrossRef]
- Loska, R.; Makosza, M. New synthesis of 2-heteroarylperfluoropropionic acids derivatives by reaction of azine N-Oxides with hexafluoropropene. Chem.-Eur. J. 2008, 14, 2577–2589. [Google Scholar] [CrossRef]
- Hafner, A.; Feuerstein, T.J.; Brase, S. Silver-mediated methoxycarbonyltetrafluoroethylation of arenes. Org. Lett. 2013, 15, 3468–3471. [Google Scholar] [CrossRef]
- Li, L.; Chen, Q.-Y.; Guo, Y. Synthesis of α-trifluoromethyl ketones via the Cu-catalyzed trifluoromethylation of silyl enol ethers using an electrophilic trifluoromethylating agent. J. Org. Chem. 2014, 79, 5145–5152. [Google Scholar] [CrossRef]
- Brewitz, L.; Noda, H.; Kumagai, N.; Shibasaki, M. (2R,3S)-3,4,4,4-tetrafluorovaline: A fluorinated bioisostere of isoleucine. Eur. J. Org. Chem. 2020, 2020, 1745–1752. [Google Scholar] [CrossRef]
- Brewitz, L.; Kumagai, N.; Shibasaki, M. Catalytic asymmetric synthesis of 2,3,3,3-tetrafluoro-2-methyl-1-arylpropan-1-amines as useful building blocks for SAR-studies. J. Fluor. Chem. 2017, 194, 1–7. [Google Scholar] [CrossRef]
- Matsuzawa, A.; Noda, H.; Kumagai, N.; Shibasaki, M. Direct catalytic asymmetric aldol addition of an α-CF3 amide to arylglyoxal hydrates. J. Org. Chem. 2017, 82, 8304–8308. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Kumagai, N.; Shibasaki, M. Cu/Pd synergistic dual catalysis: Asymmetric α-allylation of an α-CF3 amide. Angew. Chem. Int. Ed. 2017, 56, 5551–5555. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Sun, B.; Kumagai, N.; Shibasaki, M. Direct catalytic asymmetric 1,6-conjugate addition of amides to p-quinone methides. Org. Lett. 2018, 20, 3070–3073. [Google Scholar] [CrossRef]
- Yu, J.S.; Noda, H.; Kumagai, N.; Shibasaki, M. Direct catalytic asymmetric Mannich-type reaction of an α-CF3 amide to isatin imines. Synlett 2019, 30, 488–492. [Google Scholar] [CrossRef]
- Weidner, K.; Sun, Z.; Kumagai, N.; Shibasaki, M. Direct catalytic asymmetric aldol reaction of an α-azido amide. Angew. Chem. Int. Ed. 2015, 54, 6236–6240. [Google Scholar] [CrossRef]
- Sun, Z.; Weidner, K.; Kumagai, N.; Shibasaki, M. Direct catalytic asymmetric Mannich-type reaction of α-N3 amide. Chem. Eur. J. 2015, 21, 17574–17577. [Google Scholar] [CrossRef]
- Noda, H.; Amemiya, F.; Weidner, K.; Kumagai, N.; Shibasaki, M. Catalytic asymmetric synthesis of CF3-substituted tertiary propargylic alcohols via direct aldol reaction of α-N3 amide. Chem. Sci. 2017, 8, 3260–3269. [Google Scholar] [CrossRef]
- Arteaga, F.A.; Liu, Z.; Brewitz, L.; Chen, J.; Sun, B.; Kumagai, N.; Shibasaki, M. Direct catalytic asymmetric Mannich-type reaction of alkylamides. Org. Lett. 2016, 18, 2391–2394. [Google Scholar] [CrossRef]
- Liu, Z.; Takeuchi, T.; Pluta, R.; Arteaga, F.A.; Kumagai, N.; Shibasaki, M. Direct catalytic asymmetric aldol reaction of α-alkylamides. Org. Lett. 2017, 19, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Kumagai, N.; Shibasaki, M. Direct catalytic asymmetric aldol reaction of α-vinyl acetamide. J. Org. Chem. 2018, 83, 5851–5858. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Li, Z.; Kumagai, N.; Shibasaki, M. Z-Enolate geometry in the thioamide aldol reaction illuminated by the 7-azaindoline auxiliary. Org. Lett. 2020, 22, 791–794. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Balaji, P.V.; Kumagai, N.; Shibasaki, M. α-Halo amides as competent latent enolates: Direct catalytic asymmetric Mannich-type reaction. J. Am. Chem. Soc. 2017, 139, 8295–8301. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Pluta, R.; Kumagai, N.; Shibasaki, M. Direct catalytic asymmetric Mannich-type reaction en route to α-hydroxy-β-amino acid derivatives. Org. Lett. 2018, 20, 526–529. [Google Scholar] [CrossRef]
- Pluta, R.; Kumagai, N.; Shibasaki, M. Direct catalytic asymmetric aldol reaction of α-alkoxyamides to α-fluorinated ketones. Angew. Chem. Int. Ed. 2019, 58, 2459–2485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kumagai, N.; Shibasaki, M. Electrophilic activation of α,β-unsaturated amides: Catalytic asymmetric vinylogous conjugate addition of unsaturated γ-butyrolactones. Chem. Eur. J. 2016, 22, 5525–5529. [Google Scholar] [CrossRef]
- Zhang, M.; Kumagai, N.; Shibasaki, M. α,β-Unsaturated amides as dipolarophiles: Catalytic asymmetric exo-selective 1,3-dipolarcycloaddition with nitrones. Chem. Eur. J. 2017, 23, 12450–12455. [Google Scholar] [CrossRef]
- Zhang, Y.P.; You, Y.; Zhao, J.Q.; Zhou, X.J.; Zhang, X.M.; Xu, X.Y.; Yuan, W.C. A AgOAc/quinine-derived aminophosphine complex as an efficient catalyst for diastereoand enantioselective 1,3-dipolar cycloaddition of α,β-unsaturated 7-azaindoline amides and azomethine ylides. Org. Chem. Front. 2019, 6, 1879–1884. [Google Scholar] [CrossRef]
- Li, Z.; Kumagai, N.; Shibasaki, M. Catalytic asymmetric 1,3-dipolar cycloaddition of α,β-unsaturated amide and azomethine imine. Chem. Pharm. Bull. 2020, 68, 552–554. [Google Scholar] [CrossRef]
- Zhang, Y.P.; You, Y.; Zhao, J.Q.; Zhang, X.M.; Xu, X.Y.; Yuan, W.C. Chiral bifunctional amine-squaramide-catalyzed highly diastereoand enantioselective Michael/aldol cascade reaction of 2-mercaptobenzaldehyde and α,β-unsaturated 7-azaindoline amides. J. Org. Chem. 2019, 84, 7984–7994. [Google Scholar] [CrossRef] [PubMed]
- Zu, L.; Wang, J.; Li, H.; Xie, H.; Jiang, W.; Wang, W. Cascade Michael−aldol reactions promoted by hydrogen bonding mediated catalysis. J. Am. Chem. Soc. 2007, 129, 1036–1037. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.-Q.; Fang, X.; Tao, H.-Y.; Zhou, X.; Wang, C.-J. Organocatalytic asymmetric domino sulfa-Michael–aldol reactions of 2-mercaptobenzaldehyde with α,β-unsaturated N-acylpyrazoles for the construction of thiochromane. Chem. Commun. 2012, 48, 7238–7240. [Google Scholar] [CrossRef]
- Storer, R.I.; Aciro, C.; Jones, L.H. Squaramides: Physical properties, synthesis and applications. Chem. Soc. Rev. 2011, 40, 2330–2346. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, P.; Mahajan, S.; Kaya, U.; Hack, D.; Enders, D. Bifunctional amine-squaramides: Powerful hydrogen-bonding organocatalysts for asymmetric domino/cascade reactions. Adv. Synth. Catal. 2015, 357, 253–281. [Google Scholar] [CrossRef]
- Pagire, S.K.; Kumagai, N.; Shibasaki, M. Introduction of a 7-aza-6-MeO-indoline auxiliary in Lewis-acid/photoredox cooperative catalysis: Highly enantioselective aminomethylation of α,β-unsaturated amides. Chem. Sci. 2020, 11, 5168–5174. [Google Scholar] [CrossRef] [PubMed]
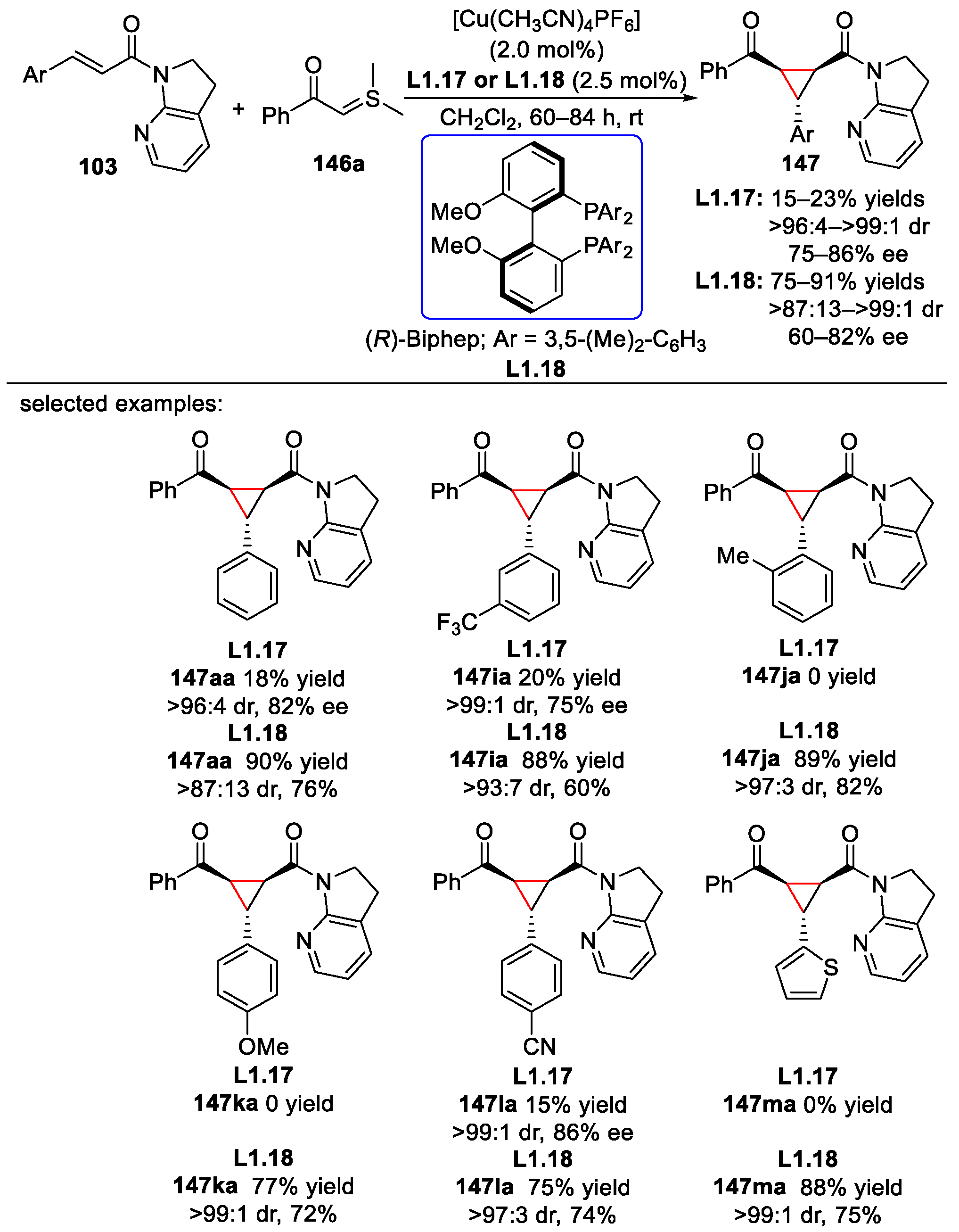
- Pagire, S.K.; Kumagai, N.; Shibasaki, M. Highly enantio- and diastereoselective synthesis of 1,2,3-trisubstituted cyclopropanes from α,β-unsaturated amides and stabilized sulfur ylides catalyzed by a chiral copper(I) complex. ACS Catal. 2021, 11, 11597–11606. [Google Scholar] [CrossRef]
- Yan, H.H.; Sha, F.; Wu, X.Y. Copper-catalyzed enantioselective 1,4-hydroboration of α,β-unsaturated amides. Tetrahedron Lett. 2023, 117, 154364. [Google Scholar] [CrossRef]
- Xiang, S.-H.; Tan, B. Advances in asymmetric organocatalysis over the last 10 years. Nat. Commun. 2020, 11, 3786. [Google Scholar] [CrossRef]
- Cheng, J.K.; Xiang, S.-H.; Tan, B. Organocatalytic enantioselective synthesis of axially chiral molecules: Development of strategies and skeletons. Acc. Chem. Res. 2022, 55, 2920–2937. [Google Scholar] [CrossRef]
- Tan, W.; Zhang, J.-Y.; Gao, C.-H.; Shi, F. Progress in organocatalytic asymmetric (4+3) cycloadditions for the enantioselective construction of seven-membered rings. Sci. China Chem. 2023, 66, 966–992. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Sun, M.; Yu, X.-Y.; Zhang, Y.-C.; Tan, W.; Shi, F. Atroposelective construction of axially chiral alkene-indole scaffolds via catalytic enantioselective addition reaction of 3-alkynyl-2-indolylmethanols. Chin. J. Chem. 2021, 39, 2163–2171. [Google Scholar]
- Sheng, F.-T.; Yang, S.; Wu, S.-F.; Zhang, Y.-C.; Shi, F. Catalytic asymmetric synthesis of axially chiral 3,3′-bisindoles by direct coupling of indole rings. Chin. J. Chem. 2022, 40, 2151–2160. [Google Scholar] [CrossRef]














































































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.-P.; You, Y.; Yin, J.-Q.; Wang, Z.-H.; Zhao, J.-Q.; Yuan, W.-C. Progress in Catalytic Asymmetric Reactions with 7-Azaindoline as the Directing Group. Molecules 2023, 28, 7898. https://doi.org/10.3390/molecules28237898
Zhang Y-P, You Y, Yin J-Q, Wang Z-H, Zhao J-Q, Yuan W-C. Progress in Catalytic Asymmetric Reactions with 7-Azaindoline as the Directing Group. Molecules. 2023; 28(23):7898. https://doi.org/10.3390/molecules28237898
Chicago/Turabian StyleZhang, Yan-Ping, Yong You, Jun-Qing Yin, Zhen-Hua Wang, Jian-Qiang Zhao, and Wei-Cheng Yuan. 2023. "Progress in Catalytic Asymmetric Reactions with 7-Azaindoline as the Directing Group" Molecules 28, no. 23: 7898. https://doi.org/10.3390/molecules28237898
APA StyleZhang, Y. -P., You, Y., Yin, J. -Q., Wang, Z. -H., Zhao, J. -Q., & Yuan, W. -C. (2023). Progress in Catalytic Asymmetric Reactions with 7-Azaindoline as the Directing Group. Molecules, 28(23), 7898. https://doi.org/10.3390/molecules28237898