
Development of a Novel LC-MS/MS Multi-Method for the Determination of Regulated and Emerging Food Contaminants Including Tenuazonic Acid, a Chromatographically Challenging Alternaria Toxin


Abstract
:1. Introduction
2. Results and Discussion
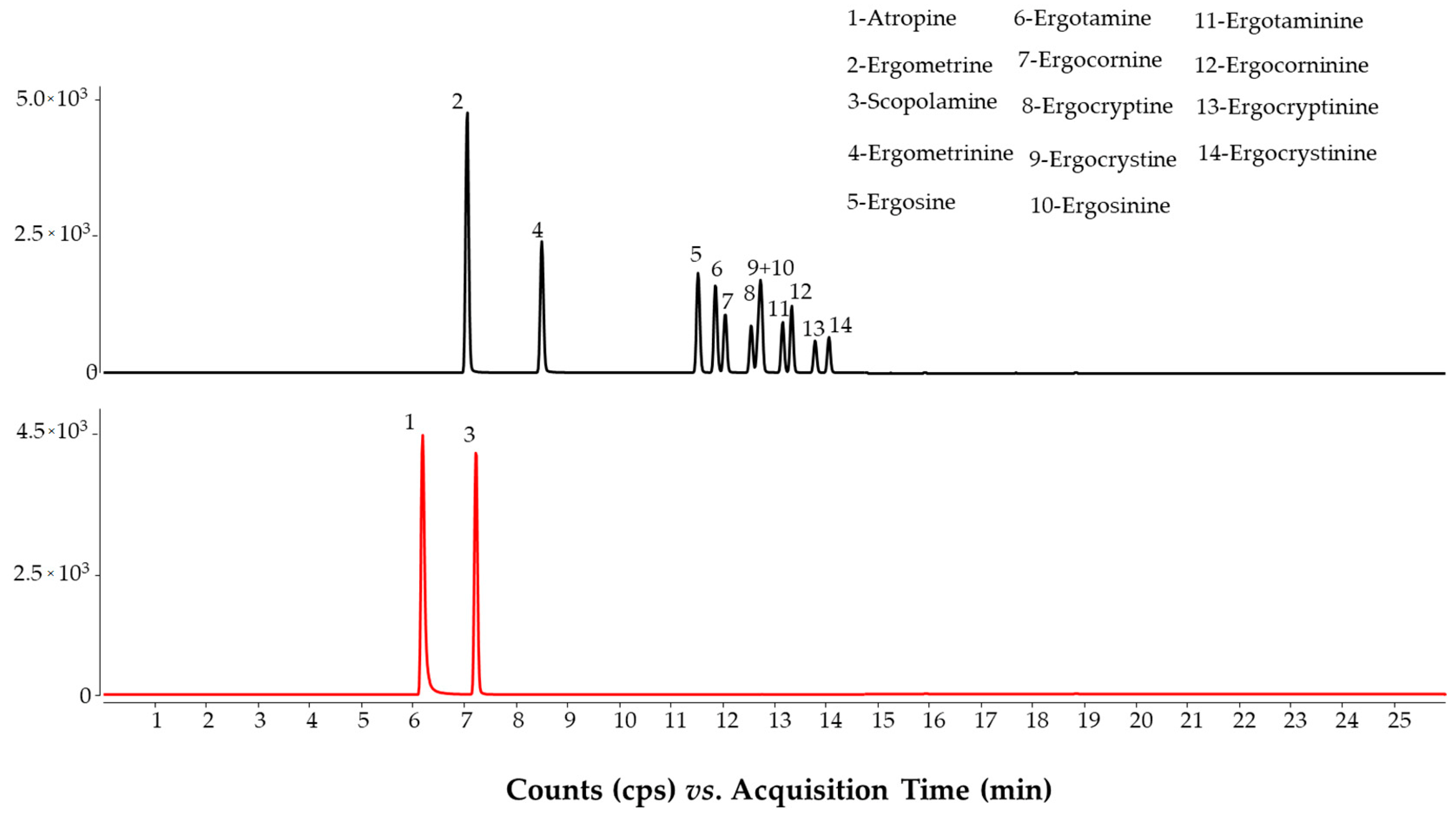
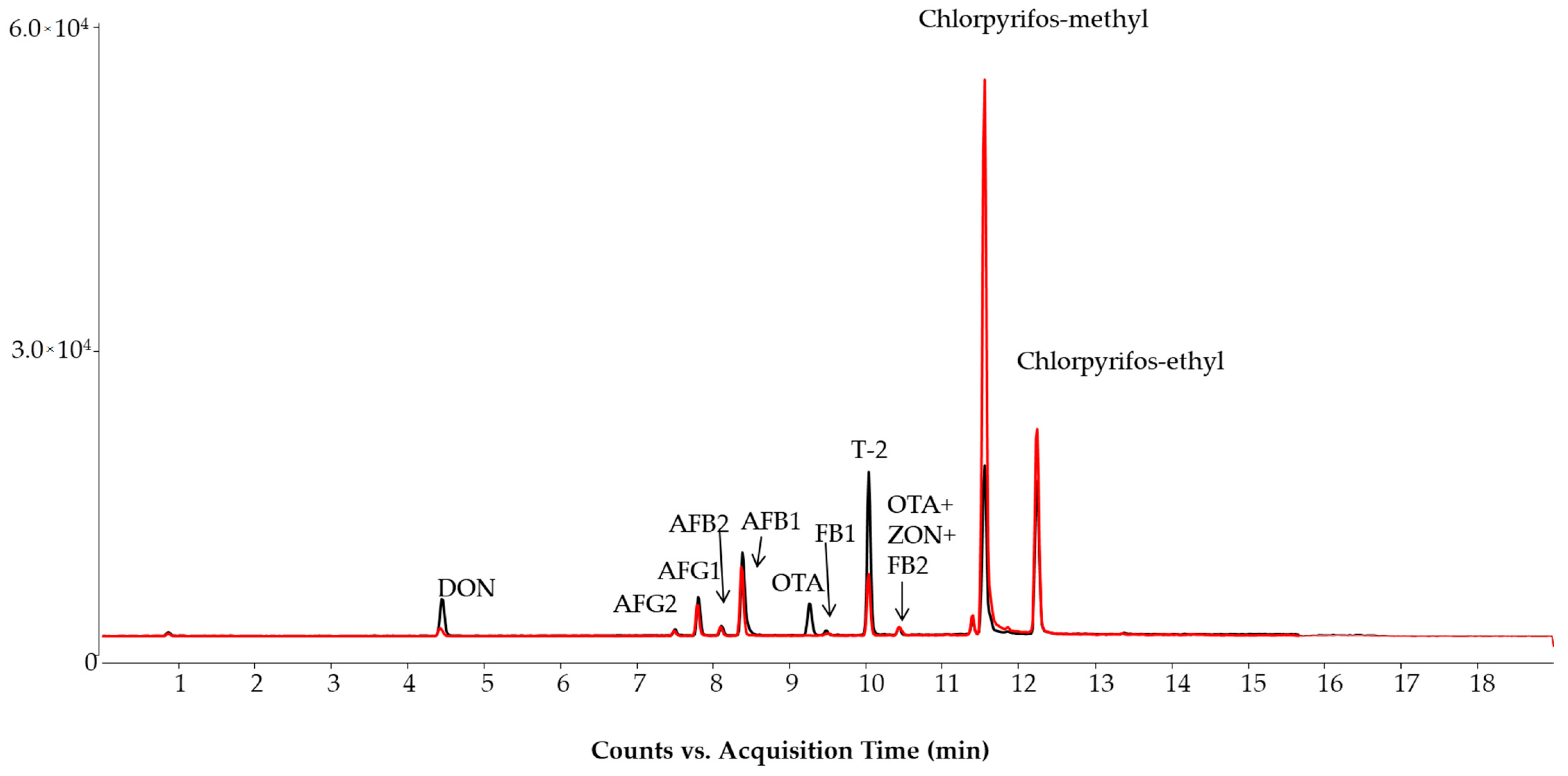
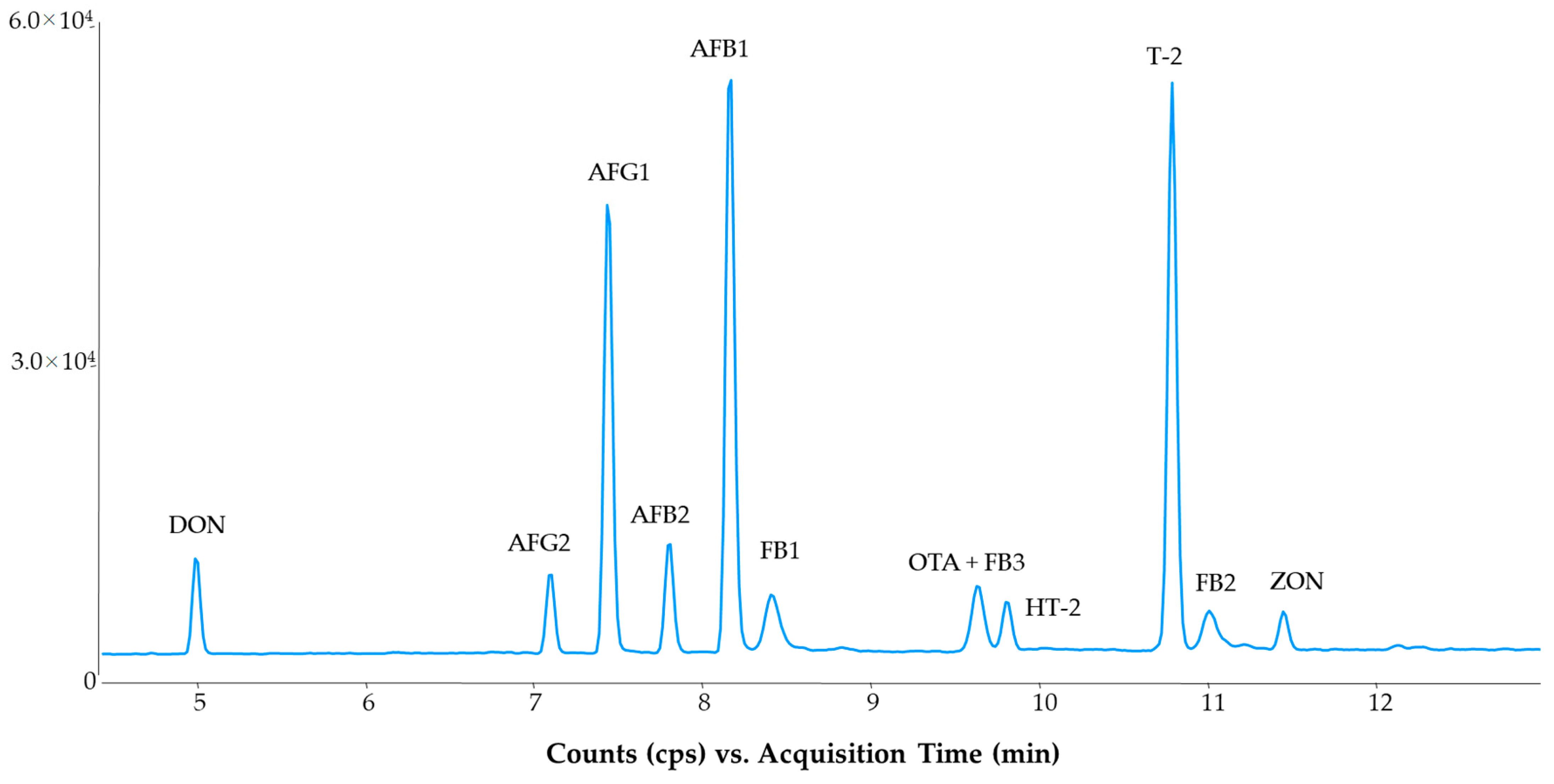
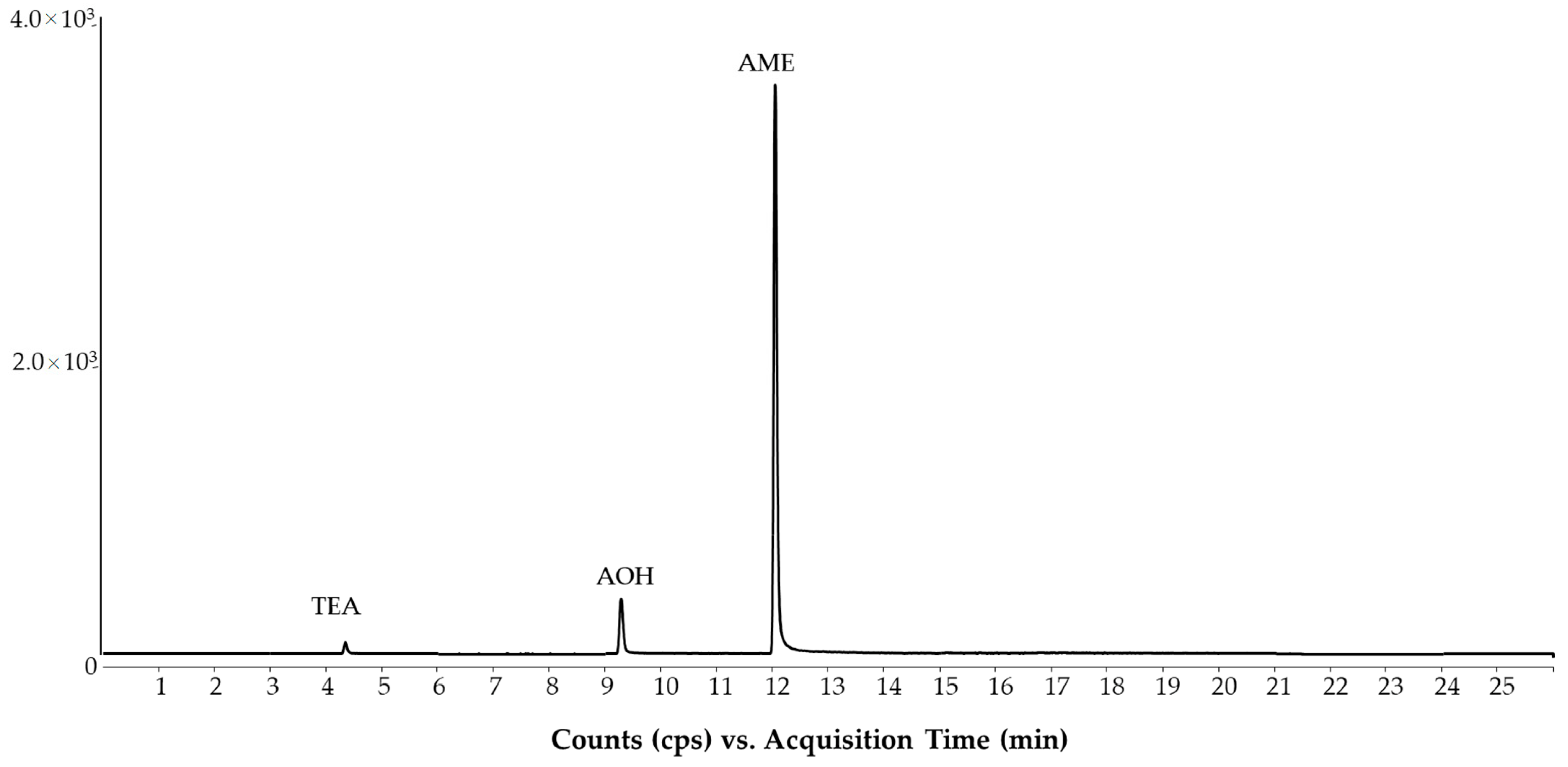
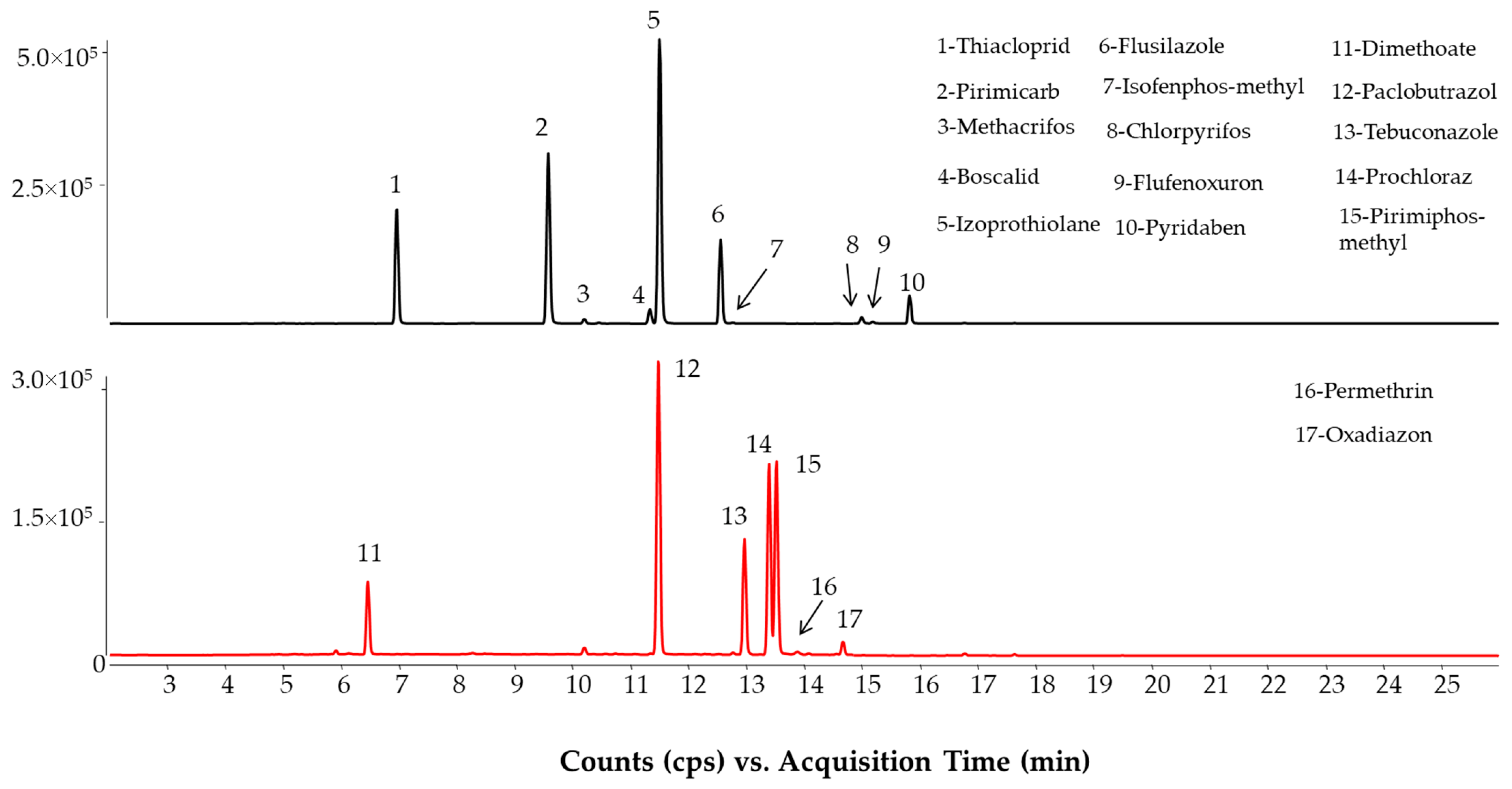
2.1. LC-MS/MS Method
2.2. Method Evaluation
3. Materials and Methods
3.1. Reagents and Samples
3.2. Instrumentation
3.3. Sample Preparation
3.4. LC-MS/MS Separation
3.5. Quantification
3.6. Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Commission Regulation (EU) 2021/1399 of 24 August 2021 Amending Regulation (EC) No 1881/2006 as Regards Maximum Levels of Ergot Sclerotia and Ergot Alkaloids in Certain Foodstuffs. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32021R1399&rid=2 (accessed on 16 October 2022).
- Commission Regulation (EC) No 1881/2006 of 19 December 2006 Setting Maximum Levels for Certain Contaminants in Foodstuffs. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32006R1881&from=HU (accessed on 16 October 2022).
- Commission Regulation (EU) 2022/553 of 5 April 2022 on Monitoring the Presence of Alternaria Toxins in Food. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32022H0553&from=EN (accessed on 16 October 2022).
- Commission Regulation of 27 March 2013 on the Presence of T-2 and HT-2 Toxin in Cereals and Cereal Products. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2013:091:0012:0015:EN:PDF (accessed on 16 October 2022).
- Commission Regulation (EU) 2021/1408 of 27 August 2021 Amending Regulation (EC) No 1881/2006 as Regards Maximum Levels of Tropane Alkaloids in Certain Foodstuffs. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32021R1408&from=EN (accessed on 16 October 2022).
- Regulation (EC) No 396/2005 of the European Parliament and of the Council of 23 February 2005 on Maximum Residue Levels of Pesticides in or on Food and Feed of Plant and Animal Origin and Amending Council Directive 91/414/EEC. Available online: http://data.europa.eu/eli/reg/2005/396/2016-05-13 (accessed on 18 September 2022).
- Pesticide Evaluations: Overview and Procedure. Available online: https://www.efsa.europa.eu/en/applications/pesticides (accessed on 18 September 2022).
- Pesticide Residue Monitoring Program, Fiscal Year 2020 Pesticide Report, U.S. Food and Drug Administration. Available online: https://www.fda.gov/media/160464/download (accessed on 18 September 2022).
- De Girolamo, A.; Lippolis, V.; Pascale, M. Overview of Recent Liquid Chromatography Mass Spectrometry-Based Methods for Natural Toxins Detection in Food Products. Toxins 2022, 14, 328. [Google Scholar] [CrossRef] [PubMed]
- Sulyok, M.; Stadler, D.; Steiner, D.; Krska, R. Validation of an LC-MS/MS-based dilute-and-shoot approach for the quantification of > 500 mycotoxins and other secondary metabolites in food crops: Challenges and solutions. Anal. Bioanal. Chem. 2020, 412, 2607–2620. [Google Scholar] [CrossRef] [PubMed]
- Sulyok, M.; Berthiller, F.; Krska, R.; Schuhmacher, R. Development and validation of a liquid chromatography/ tandem mass spectrometric method for the determination of 39 mycotoxins in wheat and maize. Rapid Commun. Mass Spectrom. 2006, 20, 2649–2659. [Google Scholar] [CrossRef] [PubMed]
- EN 17194:2019. Animal Feeding Stuffs: Methods of Sampling and Analysis. Determination of Deoxynivalenol, Aflatoxin B1, Fumonisin B1 & B2, T-2 & HT-2 Toxins, Zearalenone and Ochratoxin A in Feed Materials and Compound Feed by LC-MS/MS. Available online: https://www.en-standard.eu/search/?q=EN+17194%3A2019 (accessed on 16 October 2022).
- EN 15662:2018. Foods of Plant Origin—Multimethod for the Determination of Pesticide Residues Using GC- and LC-Based Analysis Following Acetonitrile Extraction/Partitioning and Clean-Up by Dispersive SPE—Modular QuEChERS-Method. Available online: https://www.en-standard.eu/bs-en-15662-2018-foods-of-plant-origin-multimethod-for-the-determination-of-pesticide-residues-using-gc-and-lc-based-analysis-following-acetonitrile-extraction-partitioning-and-clean-up-by-dispersive-spe-modular-quechers-method/ (accessed on 16 October 2022).
- Lee, S.-H.; Kwak, S.-Y.; Sarker, A.; Moon, J.-K.; Kim, J.-E. Optimization of a Multi-Residue Analytical Method during Determination of Pesticides in Meat Products by GC-MS/MS. Foods 2022, 11, 2930. [Google Scholar] [CrossRef]
- Maragou, N.C.; Balayiannis, G.; Karanasios, E.; Markellou, E.; Liapis, K. Targeted Multiresidue Method for the Analysis of Different Classes of Pesticides in Agro-Food Industrial Sludge by Liquid Chromatography Tandem Mass Spectrometry. Molecules 2021, 26, 6888. [Google Scholar] [CrossRef]
- González-Gómez, L.; Morante-Zarcero, S.; Pérez-Quintanilla, D.; Sierra, I. Occurrence and Chemistry of Tropane Alkaloids in Foods, with a Focus on Sample Analysis Methods: A Review on Recent Trends and Technological Advances. Foods 2022, 11, 407. [Google Scholar] [CrossRef]
- Martinello, M.; Borin, A.; Stella, R.; Bovo, D.; Biancotto, G.; Gallina, A.; Mutinelli, F. Development and validation of a QuEChERS method coupled to liquid chromatography and high resolution mass spectrometry to determine pyrrolizidine and tropane alkaloids in honey. Food Chem. 2017, 234, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Rausch, A.-K.; Brockmeyer, R.; Schwerdtle, T. Development and Validation of a QuEChERS-Based Liquid Chromatography Tandem Mass Spectrometry Multi-Method for the Determination of 38 Native and Modified Mycotoxins in Cereals. J. Agric. Food Chem. 2020, 68, 4657–4669. [Google Scholar] [CrossRef]
- EN 17425:2021. Foodstuffs. Determination of Ergot Alkaloids in Cereals and Cereal Products by dSPE Clean-Up and HPLC-MS/MS. Available online: https://www.en-standard.eu/search/?q=EN+17425%3A2021 (accessed on 16 October 2022).
- Carbonell-Rozas, L.; Gámiz-Gracia, L.; Lara, F.J.; García-Campaña, A.M. Determination of the Main Ergot Alkaloids and Their Epimers in Oat-Based Functional Foods by Ultra-High Performance Liquid Chromatography Tandem Mass Spectrometry. Molecules 2021, 26, 3717. [Google Scholar] [CrossRef]
- González-Gómez, L.; Morante-Zarcero, S.; Pereira, J.A.M.; Câmara, J.S.; Sierra, I. Improved Analytical Approach for Determination of Tropane Alkaloids in Leafy Vegetables Based on µ-QuEChERS Combined with HPLC-MS/MS. Toxins 2022, 14, 650. [Google Scholar] [CrossRef]
- Mujahid, C.; Savoy, M.-C.; Baslé, Q.; Woo, P.M.; Ee, E.C.Y.; Mottier, P.; Bessaire, T. Levels of Alternaria Toxins in Selected Food Commodities Including Green Coffee. Toxins 2020, 12, 595. [Google Scholar] [CrossRef] [PubMed]
- Bessaire, T.; Mujahid, C.; Mottier, P.; Desmarchelier, A. Multiple Mycotoxins Determination in Food by LC-MS/MS: An International Collaborative Study. Toxins 2019, 11, 658. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.; Lee, D.; Cho, H.-K.; Choi, S.-D. Review of the QuEChERS method for the analysis of organic pollutants: Persistent organic pollutants, polycyclic aromatic hydrocarbons, and pharmaceuticals. Trends Environ. Anal. Chem. 2019, 22, e00063. [Google Scholar] [CrossRef]
- EN 17256:2019. Animal Feeding Stuffs: Methods of Sampling and Analysis. Determination of Ergot Alkaloids and Tropane Alkaloids in Feed Materials and Compound Feeds by LC-MS/MS. Available online: https://www.en-standard.eu/search/?q=EN+17256%3A2019 (accessed on 16 October 2022).
- EN 17521:2021. Foodstuffs. Determination of Alternaria Toxins in Tomato, Wheat and Sunflower Seeds by SPE Clean-Up and HPLC-MS/MS. Available online: https://www.en-standard.eu/search/?q=EN+17521%3A2021 (accessed on 16 October 2022).
- Tölgyesi, Á.; Berky, R.; Békési, K.; Fekete, S.; Fekete, J.; Sharma, V.K. Analysis of sulfonamide residue in real honey samples using liquid chromatography with fluorescence and tandem mass spectrometry detection. J. Liq. Chromatogr Relat. Technol. 2013, 36, 1105–1125. [Google Scholar] [CrossRef]
- Kresse, M.; Drinda, H.; Romanotto, A.; Speer, K. Simultaneous determination of pesticides, mycotoxins, and metabolites as well as other contaminants in cereals by LC-LC-MS/MS. J. Chromatogr. B 2019, 1117, 86–102. [Google Scholar] [CrossRef]
- Reichert, B.; de Kok, A.; Pizzutti, I.R.; Scholten, J.; Cardoso, C.D.; Spanjer, M. Simultaneous determination of 117 pesticides and 30 mycotoxins in raw coffee, without clean-up, by LC-ESI-MS/MS analysis. Anal. Chim. Acta 2018, 1004, 40–50. [Google Scholar] [CrossRef]
- Na, T.W.; Seo, H.-J.; Jang, S.-N.; Kim, H.; Yun, H.; Kim, H.; Ahn, J.; Cho, H.; Hong, S.-H.; Kim, H.J.; et al. Multi-residue analytical method for detecting pesticides, veterinary drugs, and mycotoxins in feed using liquid- and gas chromatography coupled with mass spectrometry. J. Chromatogr. A 2022, 1676, 463257. [Google Scholar] [CrossRef]
- Eyring, P.; Tienstra, M.; Mol, H.; Herrmann, S.S.; Rasmussen, P.H.; Frandsen, H.L.; Poulsen, M.E. Development of a new generic extraction method for the analysis of pesticides, mycotoxins, and polycyclic aromatic hydrocarbons in representative animal feed and food samples. Food Chem. 2021, 356, 129653. [Google Scholar] [CrossRef]
- Asam, S.; Liu, Y.; Konitzer, K.; Rychlik, M. Development of a stable isotope dilution assay for tenuazonic acid. J. Agric. Food Chem. 2011, 59, 2980–2987. [Google Scholar] [CrossRef]
- Siegel, D.; Rasenko, T.; Koch, M.; Nehls, I. Determination of the Alternaria mycotoxin tenuazonic acid. in cereals by high-performance liquid chromatography–electrospray ionization iontrap multistage mass spectrometry after derivatization with 2,4-dinitrophenylhydrazine. J. Chromatogr. A 2009, 1216, 4582–4588. [Google Scholar] [CrossRef]
- Tölgyesi, Á.; Stroka, J.; Tamosiunas, V.; Zwickel, T. Simultaneous analysis of Alternaria toxins and citrinin in tomato: An optimised method using liquid chromatography-tandem mass spectrometry. Food Addit. Contam. 2015, 32, 1512–1522. [Google Scholar] [CrossRef]
- Tölgyesi, Á.; Kozma, L.K.; Sharma, V. Determination of Alternaria Toxins in Sunflower Oil by Liquid Chromatography Isotope Dilution Tandem Mass Spectrometry. Molecules 2020, 25, 1685. [Google Scholar] [CrossRef] [PubMed]
- Tölgyesi, Á.; Farkas, T.; Bálint, M.; McDonald, T.J.; Sharma, V.K. A Dilute and Shoot Strategy for Determining Alternaria Toxins in Tomato-Based Samples and in Different Flours Using LC-IDMS Separation. Molecules 2021, 26, 1017. [Google Scholar] [CrossRef] [PubMed]
- Tóth, E.; Tölgyesi, Á.; Simon, A.; Bálint, M.; Ma, X.; Sharma, V.K. An Alternative Strategy for Screening and Confirmation of 330 Pesticides in Ground- and Surface Water Using Liquid Chromatography Tandem Mass Spectrometry. Molecules 2022, 27, 1872. [Google Scholar] [CrossRef] [PubMed]
- Lauwers, M.; De Baere, S.; Letor, B.; Rychlik, M.; Croubels, S.; Devreese, M. Multi LC-MS/MS and LC-HRMS Methods for Determination of 24 Mycotoxins including Major Phase I and II Biomarker Metabolites in Biological Matrices from Pigs and Broiler Chickens. Toxins 2019, 11, 171. [Google Scholar] [CrossRef] [PubMed]
- Tölgyesi, Á.; Tóth Kovács, B.; Tóth, E.; Simon, A.; Bálint, M.; Sharma, V.K. Unexpected sensitivity enhancement in analysing alfatoxin M1 using LC-IDMS. Microchem. J. 2022, 179, 107469. [Google Scholar] [CrossRef]
- Gonçalves, C.; Tölgyesi, Á.; Bouten, K.; Cordeiro, F.; Stroka, J. Determination of Alternaria Toxins in Food by SPE and LC-IDMS: Development and In-House Validation of a Candidate Method for Standardisation. Separations 2022, 9, 70. [Google Scholar] [CrossRef]
- Main Changes Introduced in Document Nº SANTE/11312/2021 with Respect to the Previous Version (Document Nº SANTE 12682/2019). Available online: https://food.ec.europa.eu/system/files/2022-02/pesticides_mrl_guidelines_wrkdoc_2021-11312.pdf (accessed on 16 October 2022).
- CEN/TR 16059:2010. Food Analysis—Performance Criteria for Single Laboratory Validated Methods of Analysis for the Determination of Mycotoxins. Available online: https://www.nbn.be/shop/en/standard/preview/6283/en/ (accessed on 16 October 2022).
- EU Pesticides Database. Available online: https://ec.europa.eu/food/plants/pesticides/eu-pesticides-database_hu (accessed on 16 October 2022).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Components | Repeatability RSD% (n = 5) | Reproducibility RSD% (n = 15) | Recovery% (n = 15) | LOQ(µg/kg) | Linearity | ||||
|---|---|---|---|---|---|---|---|---|---|
| Level 1 | Level 2 | Level 1 | Level 2 | Level 1 | Level 2 | Equation | R2 | ||
| AFB1 | 4.79 | 4.21 | 8.30 | 6.24 | 88.9 | 85.8 | 0.20 | 0.8850x − 0.0985 | 0.9992 |
| AFB2 | 11.1 | 11.0 | 23.8 | 19.9 | 111 | 89.3 | 0.05 | 0.6217x − 0.0876 | 0.9999 |
| AFG1 | 11.9 | 4.48 | 13.7 | 16.7 | 88.7 | 94.3 | 0.20 | 0.6388x + 0.0125 | 0.9998 |
| AFG2 | 19.1 | 13.6 | 26.5 | 10.4 | 119 | 101 | 0.05 | 0.4611x − 0.00985 | 0.9988 |
| AME | 5.31 | 6.05 | 5.32 | 4.82 | 87.4 | 86.3 | 0.20 | 0.0745x − 0.0253 | 0.9992 |
| AOH | 21.3 | 14.6 | 23.0 | 14.6 | 108 | 103 | 0.20 | 0.1455x − 0.0325 | 0.9999 |
| Atropine | 4.02 | 2.18 | 4.11 | 2.18 | 78.6 | 83.9 | 0.20 | 0.6052x + 0.0897 | 0.9992 |
| Chlorpyrifos-ethyl | 11.3 | 3.86 | 11.3 | 15.6 | 97.1 | 72.7 | 0.20 | 0.0407x + 0.0014 | 0.9994 |
| DON | 21.1 | 6.71 | 20.5 | 8.86 | 86.0 | 82.9 | 10.0 | 0.04475x − 0.0014 | 0.9995 |
| FB1 | 10.1 | 5.7 | 12.3 | 9.7 | 83.1 | 73.9 | 10.0 | 0.1118x − 0.00547 | 0.9979 |
| FB2 | 9.9 | 5.2 | 17.2 | 9.8 | 91.4 | 81.5 | 10.0 | 0.0954x − 0.0145 | 0.9999 |
| FB3 | 7.7 | 6.4 | 13.0 | 10.1 | 95.3 | 90.5 | 10.0 | 0.0954x − 0.0145 | 0.9999 |
| HT-2 | 17.7 | 7.89 | 26.8 | 12.8 | 102.6 | 92 | 5.0 | 0.0051x + 0.0011 | 0.9998 |
| OTA | 21.7 | 4.25 | 21.7 | 10.5 | 101 | 70.6 | 1.00 | 0.1045x + 0.0745 | 0.9983 |
| Scopolamine | 3.49 | 2.80 | 5.57 | 2.80 | 74.9 | 76.6 | 0.20 | 0.3750x−0.0455 | 0.9975 |
| T-2 | 8.86 | 3.44 | 11.9 | 13.6 | 96.1 | 89.2 | 1.00 | 0.04459x + 0.0084 | 0.9994 |
| TEA | 12.1 | 11.8 | 24.2 | 28.2 | 100 | 67.1 | 200 | 0.0153x + 0.00632 | 0.9988 |
| ZON | 13.8 | 9.87 | 14.6 | 13.3 | 95.4 | 88.2 | 1.00 | 0.0397x − 0.00754 | 0.9998 |
| Sample Code | Matrix | Detected Compounds | Detected Concentrations (µg/kg) | Calculated Z-Score | Evaluation | Detected Concentrations with Standard Method (Reference Value, µg/kg) | Evaluation |
|---|---|---|---|---|---|---|---|
| GAFTA PT 2022-M2 | Maize | AFB1 AFB2 AFG1 AFG2 Total Aflatoxins | 1.77 0.507 2.05 0.69 5.05 | 0.49 0.0 1.67 1.90 1.61 | Satisfactory | 1.79 (1.60) 0.524 (0.50) 1.86 (1.50) 0.552 (0.50) 4.73 (3.73) | Satisfactory |
| GAFTA PT 2022-M1 | Wheat | HT-2 T-2 | 7.8 30.1 | 0.31 −1.42 | Satisfactory | 9.2 (7.3) 38.1 (43.8) | Satisfactory |
| Romer PT M22411 AF | Maize | AFB1 AFB2 AFG1 AFG2 Total Aflatoxins FB1 FB2 FB3 Total Fumonisins | 8.35 0.576 0.706 − 9.73 1136 296 121 1553 | −0.22 −0.38 1.96 − 0.0 −1.56 −1.21 −1.31 −1.55 | Satisfactory | 9.44 (8.79) 0.450 (0.63) 0.554 (0.49) − 10.4 (9.72) 1414 (1425) 402 (387) 168 (168) 1984 (1911) | Satisfactory |
| Romer PT M22161 DZO | Wheat | DON OTA ZON | 2032 30.3 702 | 0.78 0.79 1.81 | Satisfactory | 1694 (1826) 27.5 (25.9) 519 (545) | Satisfactory |
| Romer QC M21161DZO | Wheat | DON OTA ZON | 2597 25.0 200 | −0.96 −0.84 0.62 | Satisfactory | 2802 (2841) 28.7 (30.7) 195 (177) | Satisfactory |
| EURL QC 2016 O161 | Oat | HT-2 T-2 | 161 63.8 | 0.33 −0.41 | Satisfactory | 98 (150) 58.8 (70.3) | Satisfactory |
| EURL QC 2017 A004 | Wheat | DON | 434 | −0.97 | Satisfactory | 388 (551) | Satisfactory |
| EURL QC 2016 C257 | Maize | AFB1 DON FB1 FB2 ZON | 10.9 553 501 237 210 | 0.13 −0.43 −1.58 0.27 1.33 | Satisfactory | 9.10 (10.6) 454 (618) 653 (768) 246 (224) 147 (162) | Satisfactory |
| Romer QC DZO10006460 | Wheat | DON OTA ZON | 618 7.4 34.6 | −1.67 −1.24 −0.05 | Satisfactory | 859 (825) 7.2 (10) 34.4 (34.9) | Satisfactory |
| Romer QC 10003613 | Maize | AFB1 AFB2 | 8.5 1.93 | −0.59 −0.49 | Satisfactory | 8.6 (9.5) 2.5 (2.1) | Satisfactory |
| Trilogy QC TQC-MMF11-100 | Maize | AFB1 AFB2 Total Aflatoxins DON FB1 FB2 FB3 Total Fumonisins HT-2 T-2 OTA ZON | 20.8 1.32 22.1 1932 1168 442 95 1705 121.9 104.5 17.5 374 | 0.54 0.07 0.50 0.16 −1.55 1.2 −0.2 −0.68 −0.41 1.22 −0.31 0.35 | Satisfactory | 17.6 (18.6) 1.20 (1.30) 18.8 (19.9) 1758 (1900) 1276 (1400) 366 (400) 93 (100) 1735 (1900) 149 (127) 92.5 (94.8) 22.6 (18.5) 373 (360) | Satisfactory |
| FAPAS QC T09133QC | Kidney Beans (Dried) | Boscalid Chlorpyrifos Flufenoxuron Flusilazole Isofenphos-methyl Isoprothiolane Methacrifos Pirimicarb Pyridaben Thiacloprid | 81 143 26 119 25 154 70 96.4 32.3 67.2 | −1.12 1.74 −2.38 −1.09 −3.06 −0.06 −1.88 −0.21 −1.79 −0.95 | Questionable | 94 (107) 68 (103) 38.5 (55) 152 (155) 80 (77) 142 (156) 118 (119) 82 (101) 69 (53) 62 (85) | Satisfactory |
| FAPAS QC T09140QC | Wheat flour | Dimethoate Oxadiazon Paclobutrazol Permethrin Pirimiphos-methyl Prochloraz Tebuconazole | 47.1 101 134 28.1 104 166 79 | 2.10 1.35 1.02 −2.57 0.00 0.40 0.44 | Questionable | 41.1 (32.1) 65.4 (77.8) 112 (98.3) 66.8 (64.7) 99.3 (104) 111 (153) 101 (87.3) | Satisfactory |
| PT, Chlorpyrifos-ethyl | Wheat | Chlorpyrifos-ethyl | 28.0 | −0.68 | Satisfactory | 27.0 (33.0) | Satisfactory |
| PT, Chlorpyrifos-ethyl | Wheat | Chlorpyrifos-ethyl | 17.0 | 0.0 | Satisfactory | 19.6 (16.9) | Satisfactory |
| EURL 2017 QC EA047 | Rye | Ergocornine/inine α-Ergocryptine/inine Ergocrystine/inine Ergometrine/inine Ergosine/inine Ergotamine/inine | 294 304 676 92.1 136 641 | 0.00 +1.60 −0.55 −1.20 −2.65 −0.45 | Questionable | 280 (295) 337 (231) 651 (752) 114 (116) 222 (242) 606 (695) | Satisfactory |
| FAPAS QC 22180 | Rye | Ergocornine Ergocorninine α-Ergocryptinine Ergocrystine Ergocrystinine Ergometrine Ergometrinine Ergosine Ergotamine Ergotaminine Total Ergot Alkaloides | 45.2 12.6 13.9 85.6 20.8 27.1 4.30 16.9 34.5 6.03 338 | 1.79 0.07 −1.30 −0.91 −1.95 0.21 0.11 −1.08 −1.42 −2.29 −1.06 | Questionable | 40.7 (32.4) 15.8 (12.4) 15.7 (19.5) 141 (107) 23.6 (36.4) 32 (25.9) 4.64 (4.2) 19.1 (22.2) 41.6 (50.2) 18.0 (13.6) 353 (419) | Satisfactory |
| EURL QC 2016 C029 | Cereal | Atropine Scopolamine | 0.81 0.111 | −1.37 −1.85 | Questionable | 1.11 (1.16) 0.169 (0.183) | Satisfactory |
| FAPAS QC 22179 | Cereal | Atropine Scopolamine | 6.5 3.6 | −1.53 −0.37 | Satisfactory | 8.82 (9.8) 4.83 (3.88) | Satisfactory |
| EURL QC 2016 E087 | Cereal | Atropine Scopolamine | 6.7 0.63 | −0.45 −1.77 | Satisfactory | 6.29 (7.44) 0.76 (1.03) | Satisfactory |
| QC 2018T15 | Wheat | AME AOH TEA | 4.0 3.3 76.0 | −0.95 −1.63 2.1 | Satisfactory | 4.08 (5.06) 5.96 (5.11) 71.0 (52.0) | Satisfactory |
| QC 2018 B56 | Wheat | AME AOH TEA | 0.78 1.51 180 | −1.53 −1.28 −1.95 | Satisfactory | 0.69 (1.17) 2.51 (2.1) 314 (297) | Satisfactory |
| QC 2018 X06 | Sunflower seed | AME AOH TEA | 1.68 1.57 87 | −0.91 −1.06 −1.84 | Satisfactory | 2.01 (2.1) 1.32 (2.06) 102 (146) | Satisfactory |
| Compounds | Cal 1 (µg/kg) | Cal 2 (µg/kg) | Cal 3 (µg/kg) | Cal 4 (µg/kg) | Cal 5 (µg/kg) | Cal 6 (µg/kg) |
|---|---|---|---|---|---|---|
| AFB1 | 0.2 | 1 | 2 | 10 | 20 | 50 |
| AFB2 | 0.05 | 0.25 | 0.5 | 2.5 | 5 | 12.5 |
| AFG1 | 0.2 | 1 | 2 | 10 | 20 | 50 |
| AFG2 | 0.05 | 0.25 | 0.5 | 2.5 | 5 | 12.5 |
| AME | 0.2 | 1 | 2 | 10 | 20 | 50 |
| AOH | 0.2 | 1 | 2 | 10 | 20 | 50 |
| Atropine/scopolamine | 0.2 | 1 | 2 | 10 | 20 | 50 |
| DON | 10 | 50 | 100 | 500 | 1000 | 2500 |
| Ergot alkaloids | 0.2 | 1 | 2 | 10 | 20 | 50 |
| Fumonisins | 10 | 50 | 100 | 500 | 1000 | 2500 |
| HT-2/T-2 | 1 | 5 | 10 | 50 | 100 | 250 |
| OTA | 1 | 5 | 10 | 50 | 100 | 250 |
| Pesticides | 0.2 | 1 | 2 | 10 | 20 | 50 |
| TEA | 100 | 500 | 1000 | 5000 | 10,000 | 25,000 |
| ZON | 1 | 5 | 10 | 50 | 100 | 250 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tölgyesi, Á.; Cseh, A.; Simon, A.; Sharma, V.K. Development of a Novel LC-MS/MS Multi-Method for the Determination of Regulated and Emerging Food Contaminants Including Tenuazonic Acid, a Chromatographically Challenging Alternaria Toxin. Molecules 2023, 28, 1468. https://doi.org/10.3390/molecules28031468
Tölgyesi Á, Cseh A, Simon A, Sharma VK. Development of a Novel LC-MS/MS Multi-Method for the Determination of Regulated and Emerging Food Contaminants Including Tenuazonic Acid, a Chromatographically Challenging Alternaria Toxin. Molecules. 2023; 28(3):1468. https://doi.org/10.3390/molecules28031468
Chicago/Turabian StyleTölgyesi, Ádám, Attila Cseh, Andrea Simon, and Virender K. Sharma. 2023. "Development of a Novel LC-MS/MS Multi-Method for the Determination of Regulated and Emerging Food Contaminants Including Tenuazonic Acid, a Chromatographically Challenging Alternaria Toxin" Molecules 28, no. 3: 1468. https://doi.org/10.3390/molecules28031468
APA StyleTölgyesi, Á., Cseh, A., Simon, A., & Sharma, V. K. (2023). Development of a Novel LC-MS/MS Multi-Method for the Determination of Regulated and Emerging Food Contaminants Including Tenuazonic Acid, a Chromatographically Challenging Alternaria Toxin. Molecules, 28(3), 1468. https://doi.org/10.3390/molecules28031468