
Identification of New L-Fucosyl and L-Galactosyl Amides as Glycomimetic Ligands of TNF Lectin Domain of BC2L-C from Burkholderia cenocepacia

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
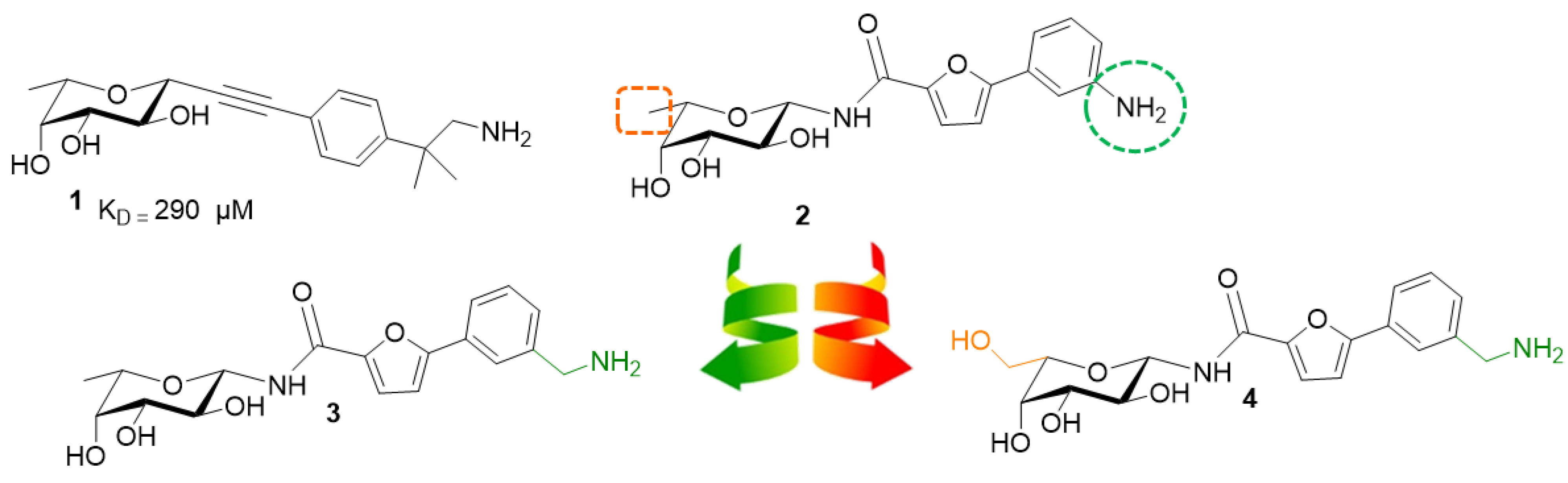
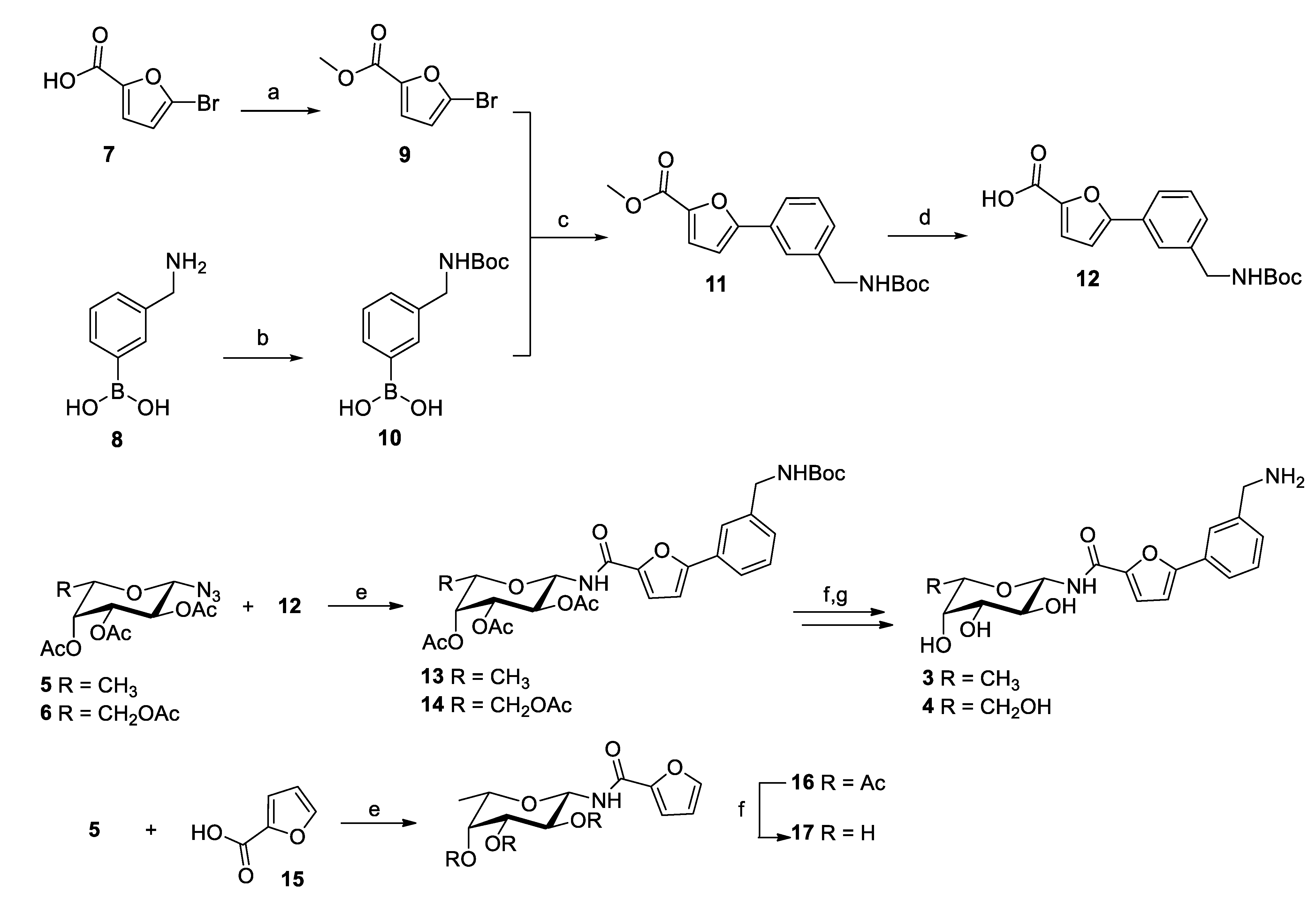
2.1. Synthesis
2.2. Biophysical Evaluation
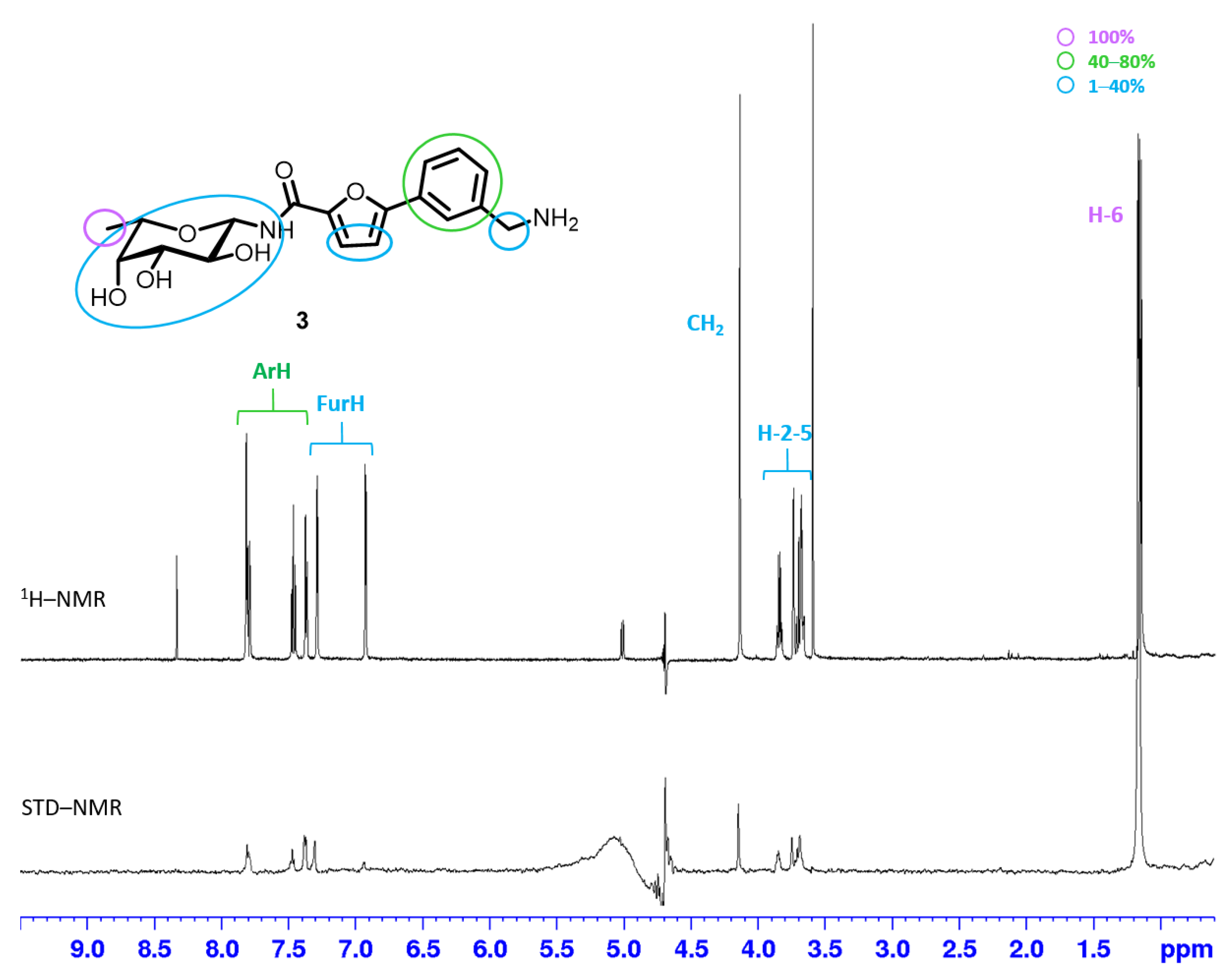
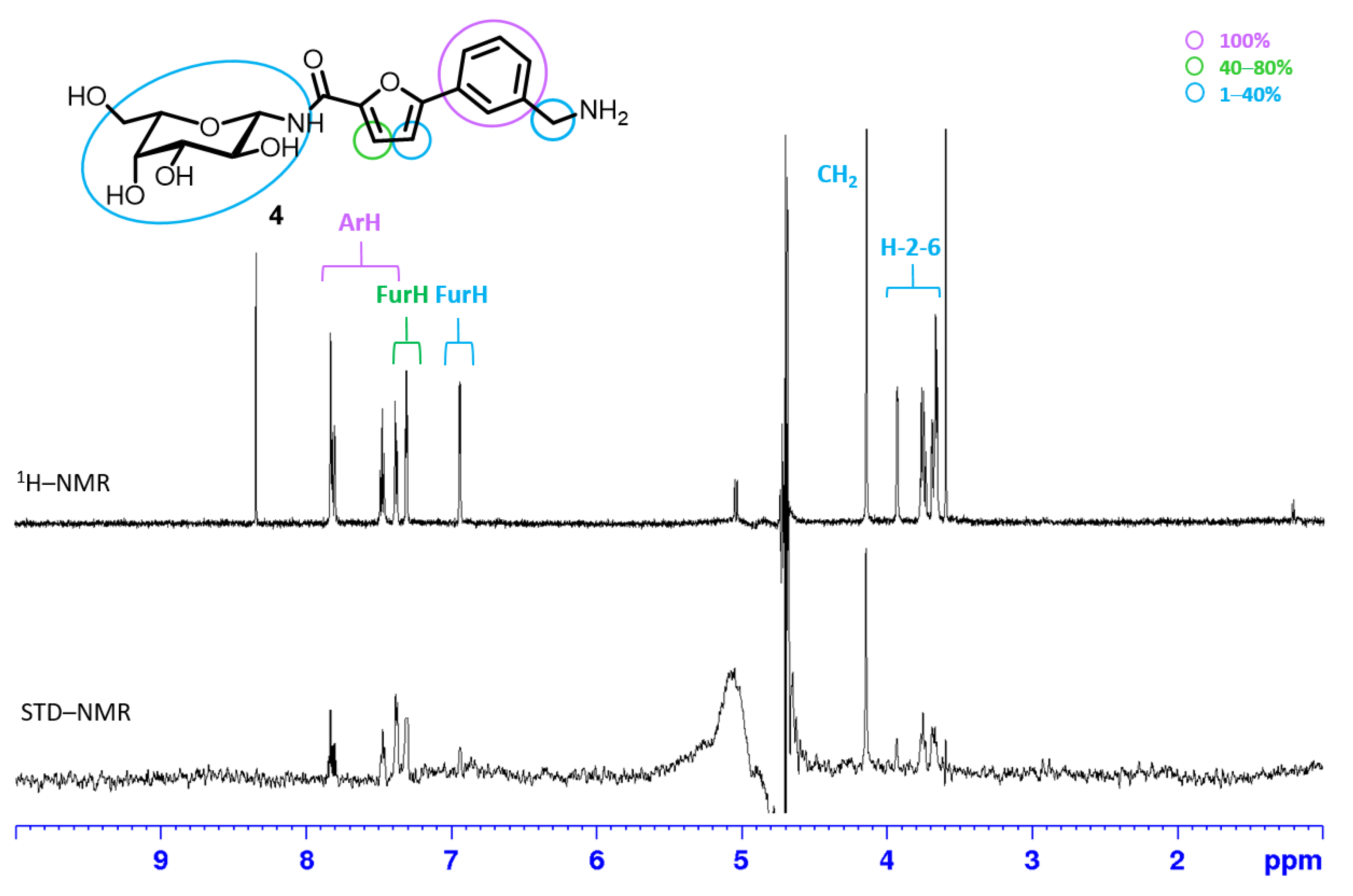
2.2.1. STD NMR
2.2.2. Isothermal Titration Calorimetry
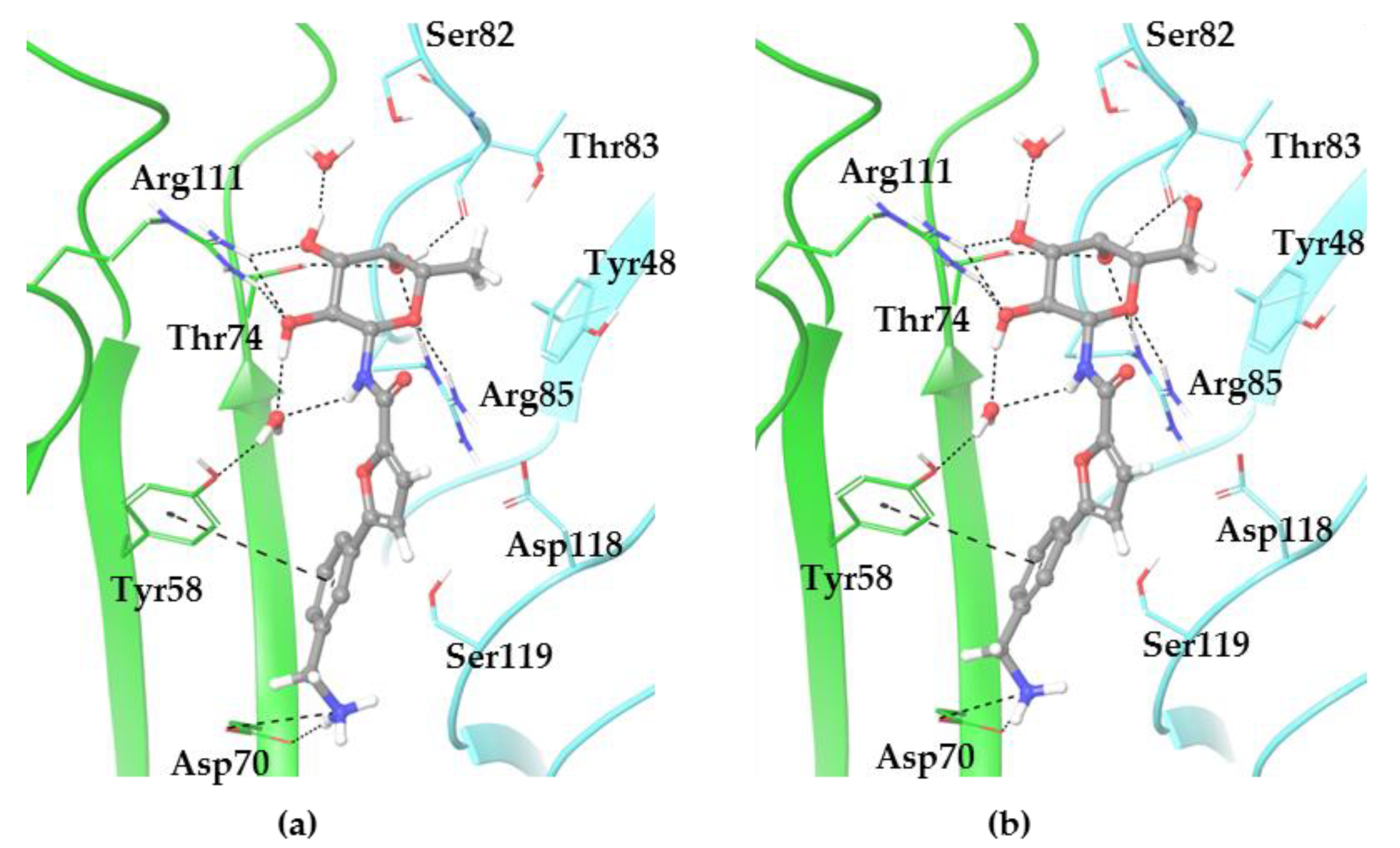
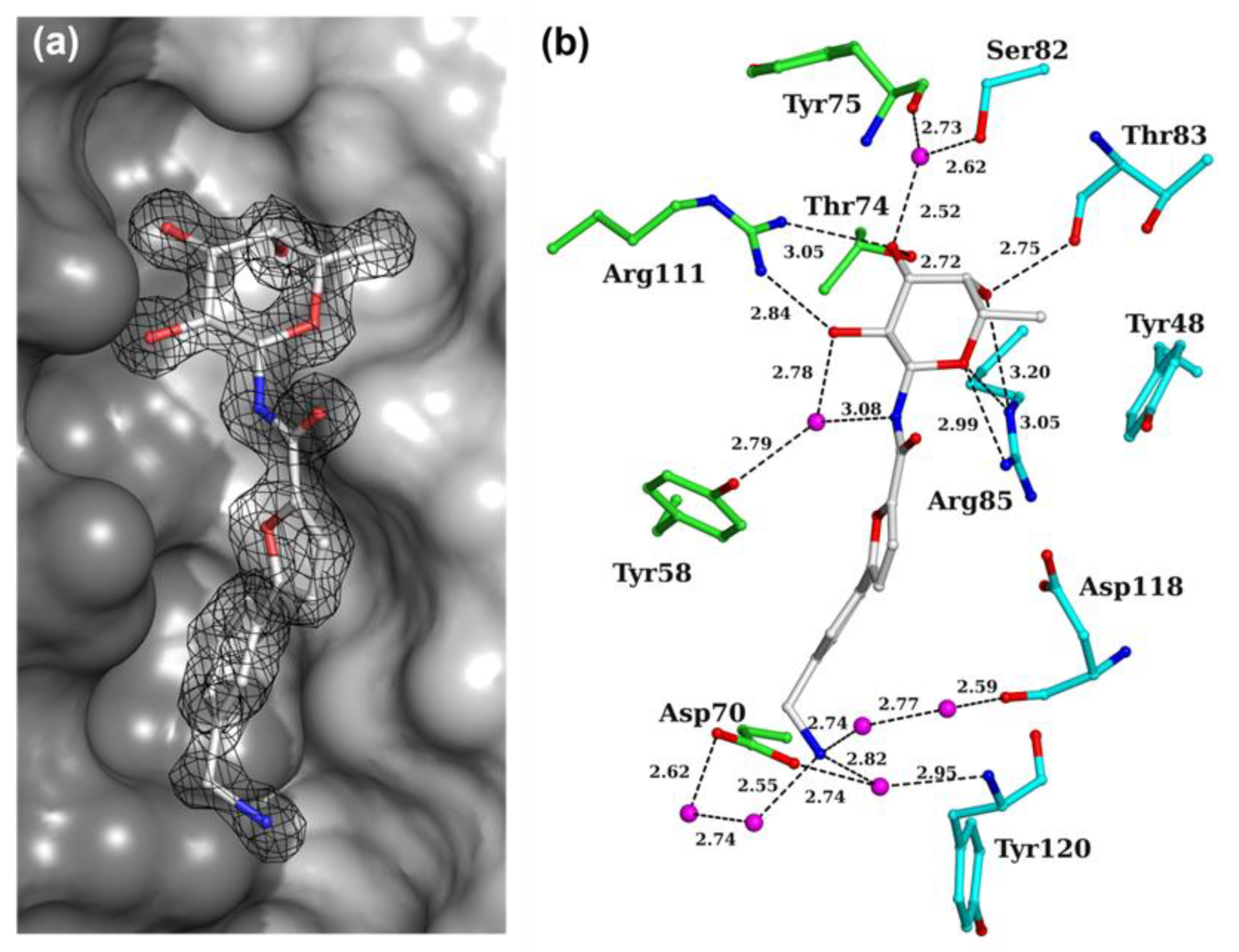
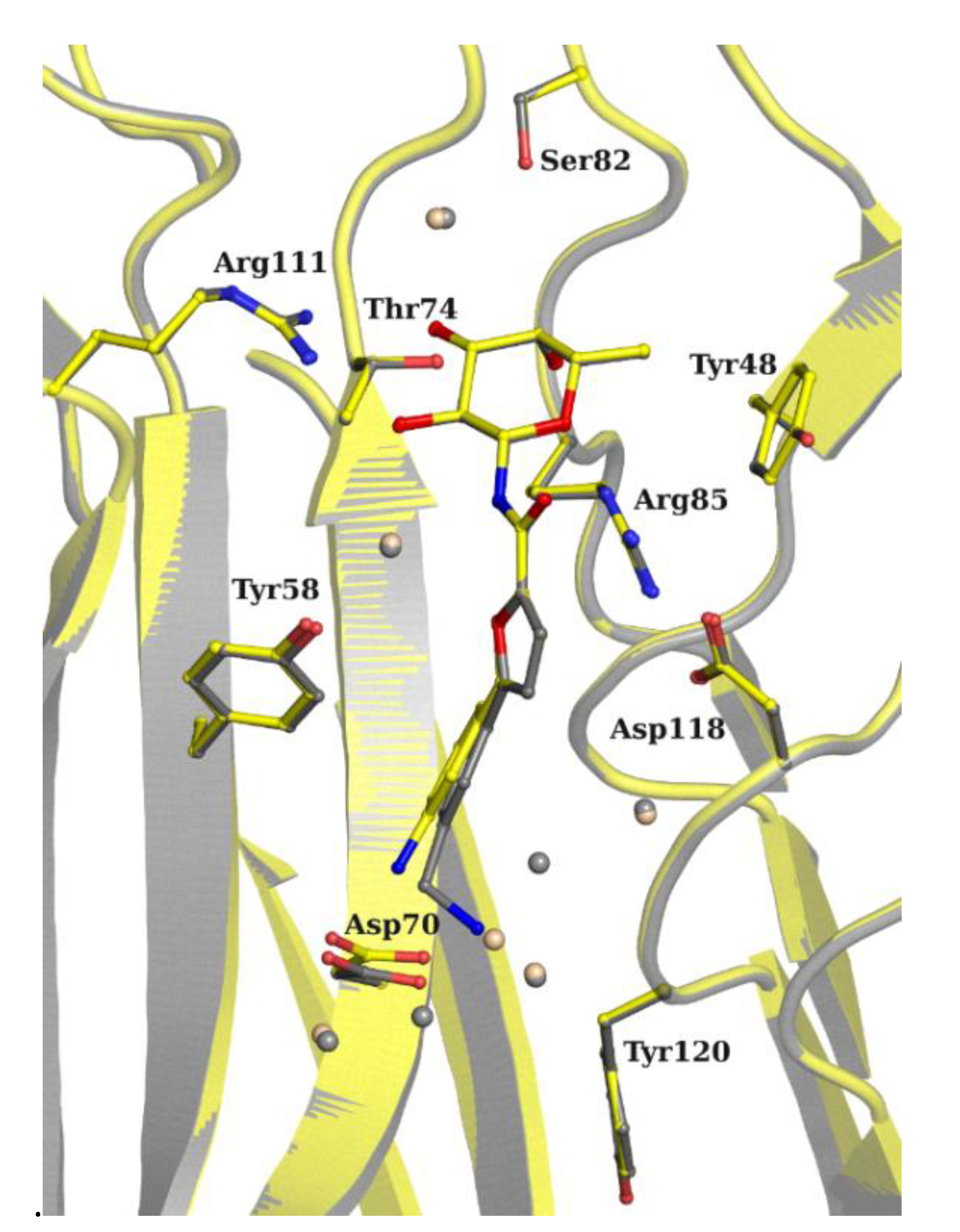
2.3. X-ray Crystallography
3. Materials and Methods
3.1. Synthesis
3.1.1. Synthesis of Methyl 5-Bromofuran-2-Carboxylate (9)
3.1.2. Synthesis of (3-(tert-Butoxycarbonyl)aminomethyl)phenylboronic Acid (10)
3.1.3. Synthesis of Methyl 5-(3-((tert-Butoxycarbonyl)aminomethyl)phenyl)furan-2-carboxylate (11)
3.1.4. Synthesis of 5-(3-((Tert-butoxycarbonyl)aminomethyl)phenyl)furan-2-carboxylic acid (12)
3.1.5. General Procedure for the Synthesis of Compounds 13, 14 and 16 through Staudinger Ligation
(5-(3-((Tert-butoxycarbonyl)aminomethyl)phenyl)furan-2-carboxamido)-2,3,4-tri-O-acetyl-β-L-fucopyranose (13)
(5-(3-((Tert-butoxycarbonyl)aminomethyl)phenyl)furan-2-carboxamido)-2,3,4,6-tetra-O-acetyl-β-L-galactopyranose (14)
(Furan-2-carboxamido)-2,3,4-tri-O-acetyl-β-L-fucopyranose (16)
3.1.6. General Procedure for Synthesis of Compounds 3 and 4 and 17
General Procedure for Zemplén Deacetylation
General Procedure for Boc Removal
Synthesis of (5-(3′-Aminomethyl)phenyl)furan-2-carboxamido)-β-L-fucopyranose (3)
Synthesis of (5-(3′-Aminomethyl)phenyl)furan-2-carboxamido)-β-L-galactopyranose (4)
Synthesis of Furan-2-carboxamido-β-L-fucopyranose (17)
3.2. STD NMR
3.3. Isothermal Microcalorimetry
3.4. X-ray Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Poole, J.; Day, C.J.; Von Itzstein, M.; Paton, J.C.; Jennings, M.P. Glycointeractions in Bacterial Pathogenesis. Nat. Rev. Microbiol. 2018, 16, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Krachler, A.M.; Orth, K. Targeting the Bacteria-Host Interface Strategies in Anti-Adhesion Therapy. Virulence 2013, 4, 284–294. [Google Scholar] [CrossRef]
- Cozens, D.; Read, R.C. Anti-Adhesion Methods as Novel Therapeutics for Bacterial Infections. Expert Rev. Anti. Infect. Ther. 2012, 10, 1457–1468. [Google Scholar] [CrossRef]
- Damalanka, V.C.; Reddy Maddirala, A.; Janetka, J.W. Novel Approaches to Glycomimetic Design: Development of Small Molecular Weight Lectin Antagonists. Expert Opin. Drug Discov. 2021, 16, 513–536. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Lindhorst, T.K. The Bacterial Lectin FimH, a Target for Drug Discovery—Carbohydrate Inhibitors of Type 1 Fimbriae-Mediated Bacterial Adhesion. Eur. J. Org. Chem. 2011, 2011, 3583–3609. [Google Scholar] [CrossRef]
- Sommer, R.; Rox, K.; Wagner, S.; Hauck, D.; Henrikus, S.S.; Newsad, S.; Arnold, T.; Ryckmans, T.; Brönstrup, M.; Imberty, A.; et al. Anti-Biofilm Agents against Pseudomonas aeruginosa: A Structure-Activity Relationship Study of C-Glycosidic LecB Inhibitors. J. Med. Chem. 2019, 62, 9201–9216. [Google Scholar] [CrossRef] [PubMed]
- Lal, K.; Bermeo, R.; Cramer, J.; Vasile, F.; Ernst, B.; Imberty, A.; Bernardi, A.; Varrot, A.; Belvisi, L. Prediction and Validation of a Druggable Site on Virulence Factor of Drug Resistant Burkholderia cenocepacia. Chem. -A Eur. J. 2021, 27, 10341–10348. [Google Scholar] [CrossRef] [PubMed]
- Bermeo, R.; Lal, K.; Ruggeri, D.; Lanaro, D.; Mazzotta, S.; Vasile, F.; Imberty, A.; Belvisi, L.; Varrot, A.; Bernardi, A. Targeting a Multidrug-Resistant Pathogen: First Generation Antagonists of Burkholderia cenocepacia’s BC2L-C Lectin. ACS Chem. Biol. 2022, 17, 2899–2910. [Google Scholar] [CrossRef] [PubMed]
- Šulák, O.; Cioci, G.; Delia, M.; Lahmann, M.; Varrot, A.; Imberty, A.; Wimmerová, M. A TNF-like Trimeric Lectin Domain from Burkholderia cenocepacia with Specificity for Fucosylated Human Histo-Blood Group Antigens. Structure 2010, 18, 59–72. [Google Scholar] [CrossRef]
- Šulák, O.; Cioci, G.; Lameignère, E.; Balloy, V.; Round, A.; Gutsche, I.; Malinovská, L.; Chignard, M.; Kosma, P.; Aubert, D.F.; et al. Burkholderia cenocepacia Bc2l-c Is a Super Lectin with Dual Specificity and Proinflammatory Activity. PLoS Pathog. 2011, 7, e1002238. [Google Scholar] [CrossRef] [Green Version]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef]
- Bermeo, R.; Bernardi, A.; Varrot, A. BC2L-C N-Terminal Lectin Domain Complexed with Histo Blood Group Oligosaccharides Provides New Structural Information. Molecules 2020, 25, 248. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Duléry, V.; Renaudet, O.; Philouze, C.; Dumy, P. α and β L-Fucopyranosyl Oxyamines: Key Intermediates for the Preparation of Fucose-Containing Glycoconjugates by Oxime Ligation. Carbohydr. Res. 2007, 342, 894–900. [Google Scholar] [CrossRef]
- Palomo, C.; Aizpurua, J.M.; Balentová, E.; Azcune, I.; Santos, J.I.; Jiménez-Barbero, J.; Cañada, J.; Miranda, J.I. “Click” Saccharide/β-Lactam Hybrids for Lectin Inhibition. Org. Lett. 2008, 10, 2227–2230. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Tiwari, P.; Maulik, P.R.; Misra, A.K. A Generalized Procedure for the One-Pot Preparation of Glycosyl Azides and Thioglycosides Directly from Unprotected Reducing Sugars under Phase-Transfer Reaction Conditions. Eur. J. Org. Chem. 2005, 2006, 74–79. [Google Scholar] [CrossRef]
- Mayer, M.; Meyer, B. Characterization of Ligand Binding by Saturation Transfer Difference NMR Spectroscopy. Angew. Chemie -Int. Ed. 1999, 38, 1784–1788. [Google Scholar] [CrossRef]
- Chiarelli, L.R.; Mori, M.; Barlocco, D.; Beretta, G.; Gelain, A.; Pini, E.; Porcino, M.; Mori, G.; Stelitano, G.; Costantino, L.; et al. Discovery and Development of Novel Salicylate Synthase (MbtI) Furanic Inhibitors as Antitubercular Agents. Eur. J. Med. Chem. 2018, 155, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Pilgram, O.; Keils, A.; Benary, G.E.; Müller, J.; Merkl, S.; Ngaha, S.; Huber, S.; Chevillard, F.; Harbig, A.; Magdolen, V.; et al. Improving the Selectivity of 3-Amidinophenylalanine-Derived Matriptase Inhibitors. Eur. J. Med. Chem. 2022, 238, 114437. [Google Scholar] [CrossRef]
- Mała, P.; Siebs, E.; Meiers, J.; Rox, K.; Varrot, A.; Imberty, A.; Titz, A. Discovery of N-β- l -Fucosyl Amides as High-Affinity Ligands for the Pseudomonas aeruginosa Lectin LecB. J. Med. Chem. 2022, 65, 14180–14200. [Google Scholar] [CrossRef]
- Legrand, P. XDSME: XDS Made Easier. GitHub Repos. 2017. Available online: https://github.com/legrandp/xdsme (accessed on 30 January 2023).
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef]
- McCoy, A.J. Solving Structures of Protein Complexes by Molecular Replacement with Phaser. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 63, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Ligand | n | KD (μM) | |
|---|---|---|---|---|
| 1 | H-type 1 tetrasaccharide |  | 0.81 | 56 ± 4.5 |
| 2 | 3 |  | 1 | 159 ± 7 |
| 3 | 4 |  | 1 | 390.5 ± 15.5 |
| 4 | 17 |  | 1 | 2845 ± 45 |
| 5 | α-methyl fucoside |  | 2700 ± 700 c | |
| Data Collection | Refinement | ||
|---|---|---|---|
| Beamline | SOLEIL Proxima-1 | Resolution (Å) | 37.25–1.55 |
| Wavelength | 0.97856 | No. reflections | 13,457 |
| Space group | P63 | No. free reflections | 734 |
| Unit cell dimensions | 43.01 43.01 94.22 90.00 90.00 120.00 | Rwork/Rfree | 0.142/0.169 |
| Resolution (Å) | 37.25–1.55 (1.58–1.55) | R.m.s Bond lengths (Å) | 0.0153 |
| Rmerge ** | 0.064 (0.447) | Rmsd Bond angles (°) | 1.821 |
| Rmeas ** | 0.076 (0.540) | Rmsd Chiral (Å3) | 0.103 |
| Rpim ** | 0.040 (0.297) | Rmsd Chiral (Å3) | 0.103 |
| Mean I/σI | 17.8 (3.5) | Clashscore | 2.49 |
| Completeness (%) | 99.1 (99.7) | No. atoms/Bfac (Å2) | |
| Redundancy | 5.6 (4.6) | Protein | 993/11.53 |
| CC1/2 | 0.998 (0.888) | Ligand | 26/8.97 |
| Nb reflections | 79,558 (3313) | Water | 128/21.57 |
| Nb unique reflections | 14,192 (716) | Ramachandran (%) | |
| Allowed | 100 | ||
| Favored | 97.1 | ||
| Outliers | 0 | ||
| PDB Code | 8BRO | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazzotta, S.; Antonini, G.; Vasile, F.; Gillon, E.; Lundstrøm, J.; Varrot, A.; Belvisi, L.; Bernardi, A. Identification of New L-Fucosyl and L-Galactosyl Amides as Glycomimetic Ligands of TNF Lectin Domain of BC2L-C from Burkholderia cenocepacia. Molecules 2023, 28, 1494. https://doi.org/10.3390/molecules28031494
Mazzotta S, Antonini G, Vasile F, Gillon E, Lundstrøm J, Varrot A, Belvisi L, Bernardi A. Identification of New L-Fucosyl and L-Galactosyl Amides as Glycomimetic Ligands of TNF Lectin Domain of BC2L-C from Burkholderia cenocepacia. Molecules. 2023; 28(3):1494. https://doi.org/10.3390/molecules28031494
Chicago/Turabian StyleMazzotta, Sarah, Giulia Antonini, Francesca Vasile, Emilie Gillon, Jon Lundstrøm, Annabelle Varrot, Laura Belvisi, and Anna Bernardi. 2023. "Identification of New L-Fucosyl and L-Galactosyl Amides as Glycomimetic Ligands of TNF Lectin Domain of BC2L-C from Burkholderia cenocepacia" Molecules 28, no. 3: 1494. https://doi.org/10.3390/molecules28031494
APA StyleMazzotta, S., Antonini, G., Vasile, F., Gillon, E., Lundstrøm, J., Varrot, A., Belvisi, L., & Bernardi, A. (2023). Identification of New L-Fucosyl and L-Galactosyl Amides as Glycomimetic Ligands of TNF Lectin Domain of BC2L-C from Burkholderia cenocepacia. Molecules, 28(3), 1494. https://doi.org/10.3390/molecules28031494