

Investigation on Gold–Ligand Interaction for Complexes from Gold Leaching: A DFT Study




Abstract
:1. Introduction
2. Results and Discussion
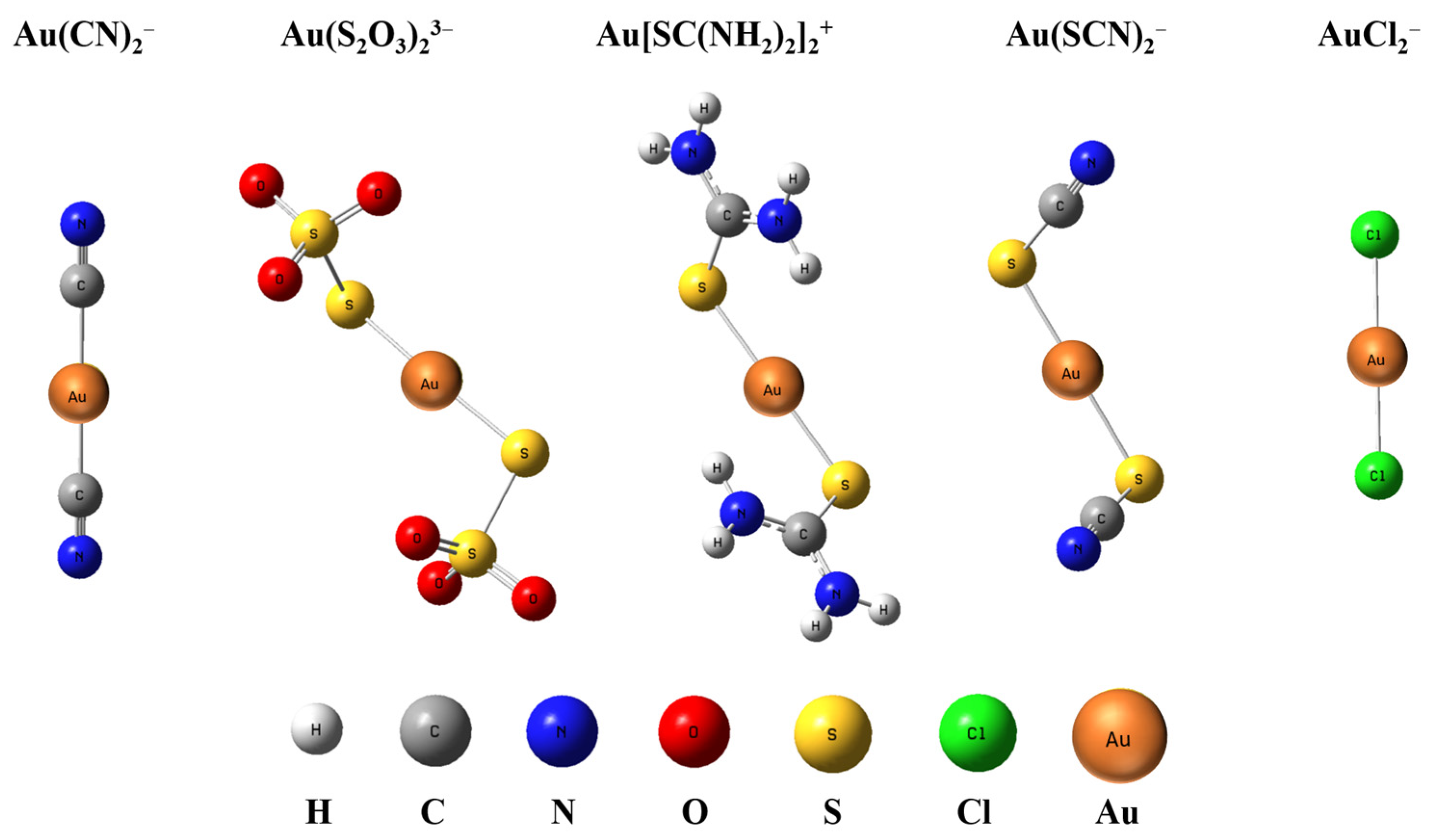
2.1. Optimized Geometries of Gold Complexes
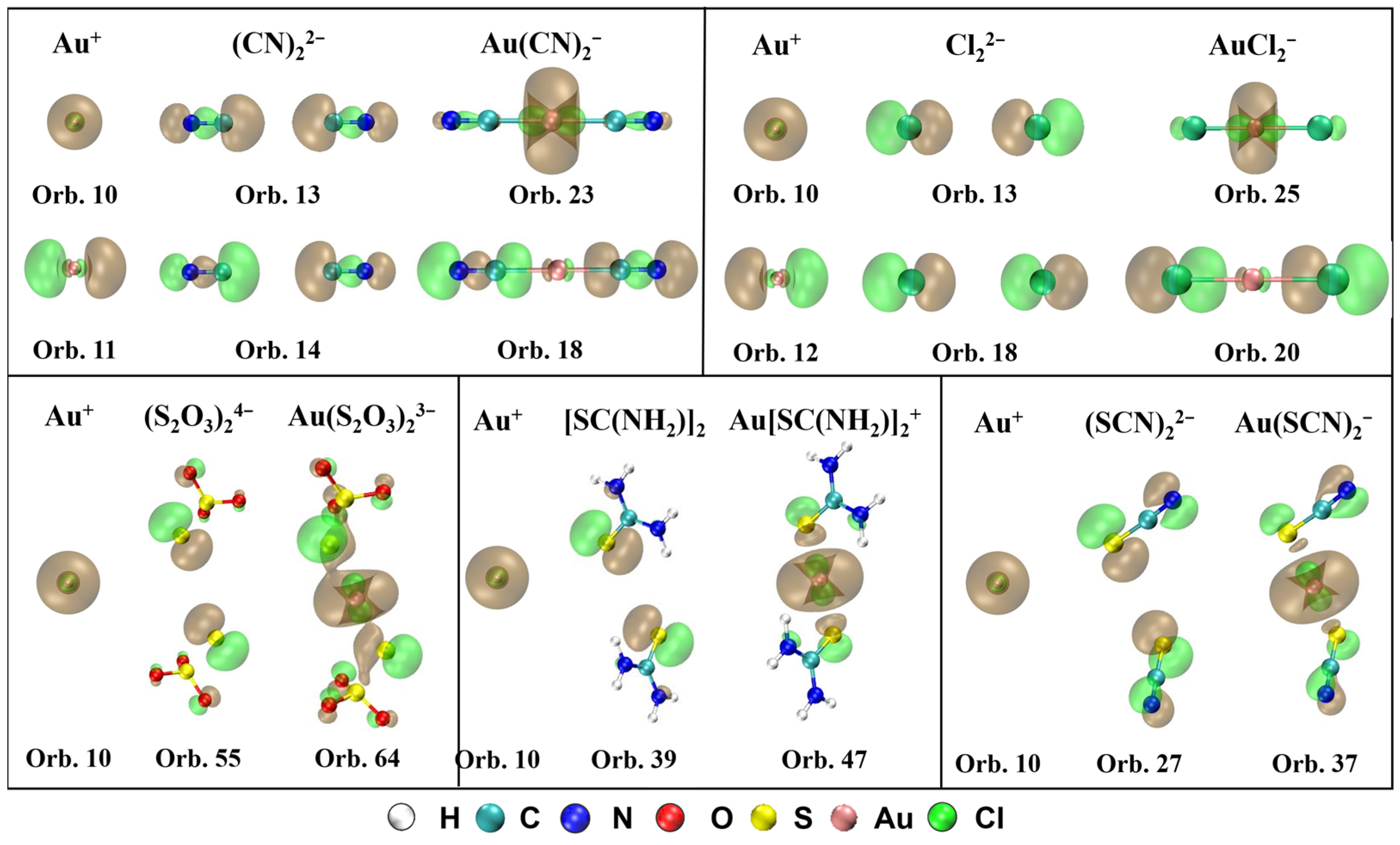
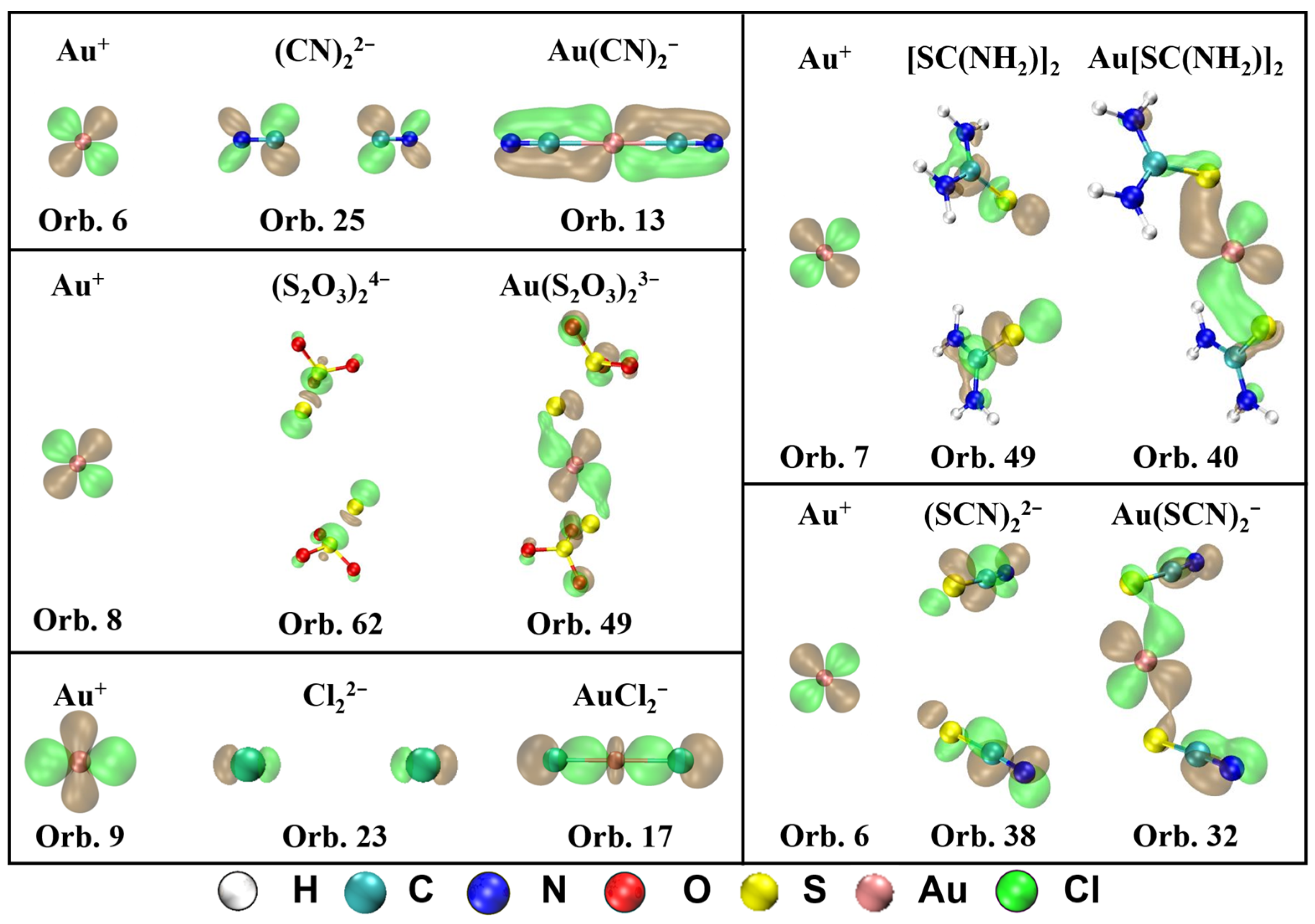
2.2. Charge Decomposition Analysis (CDA)
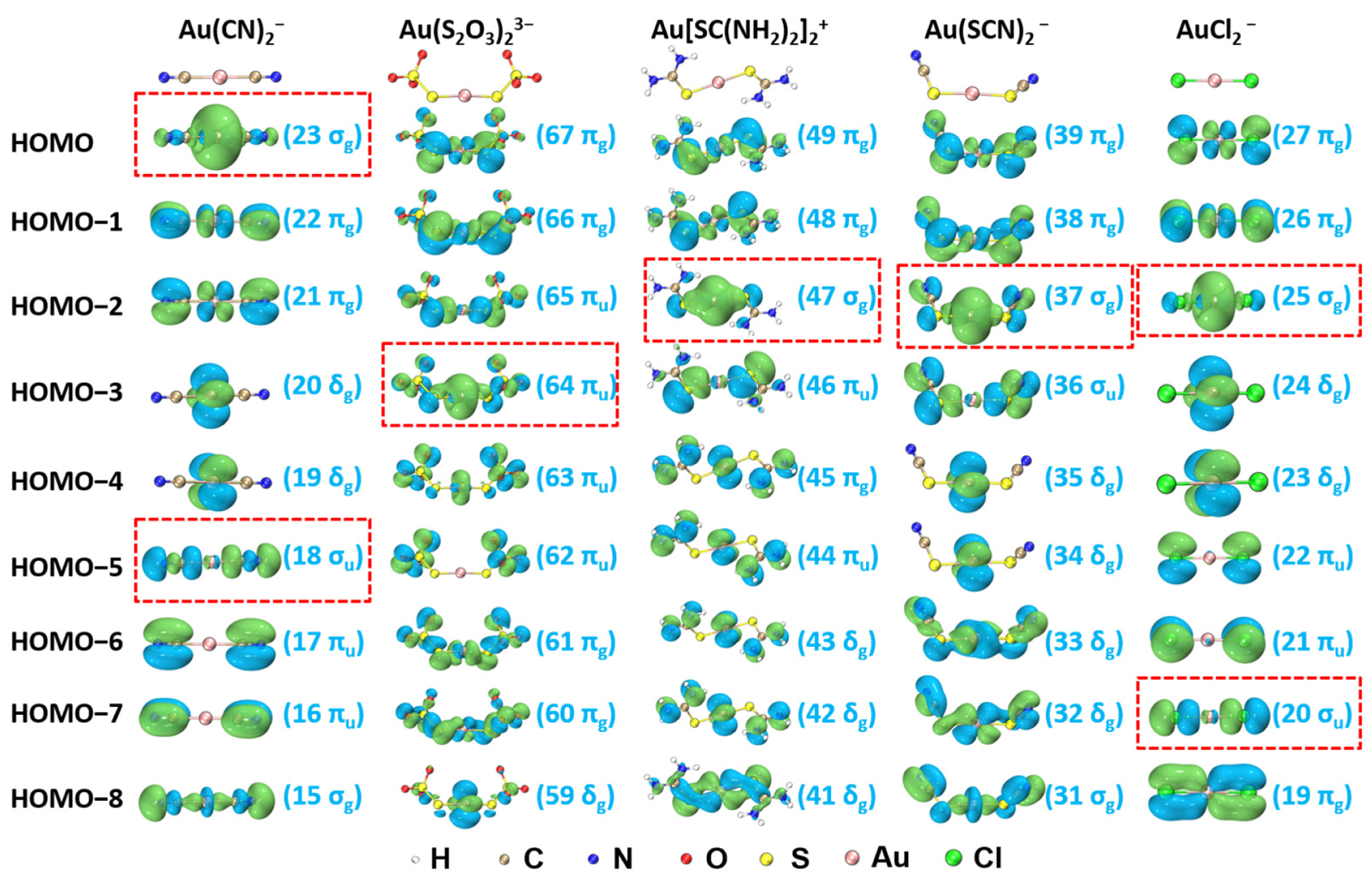
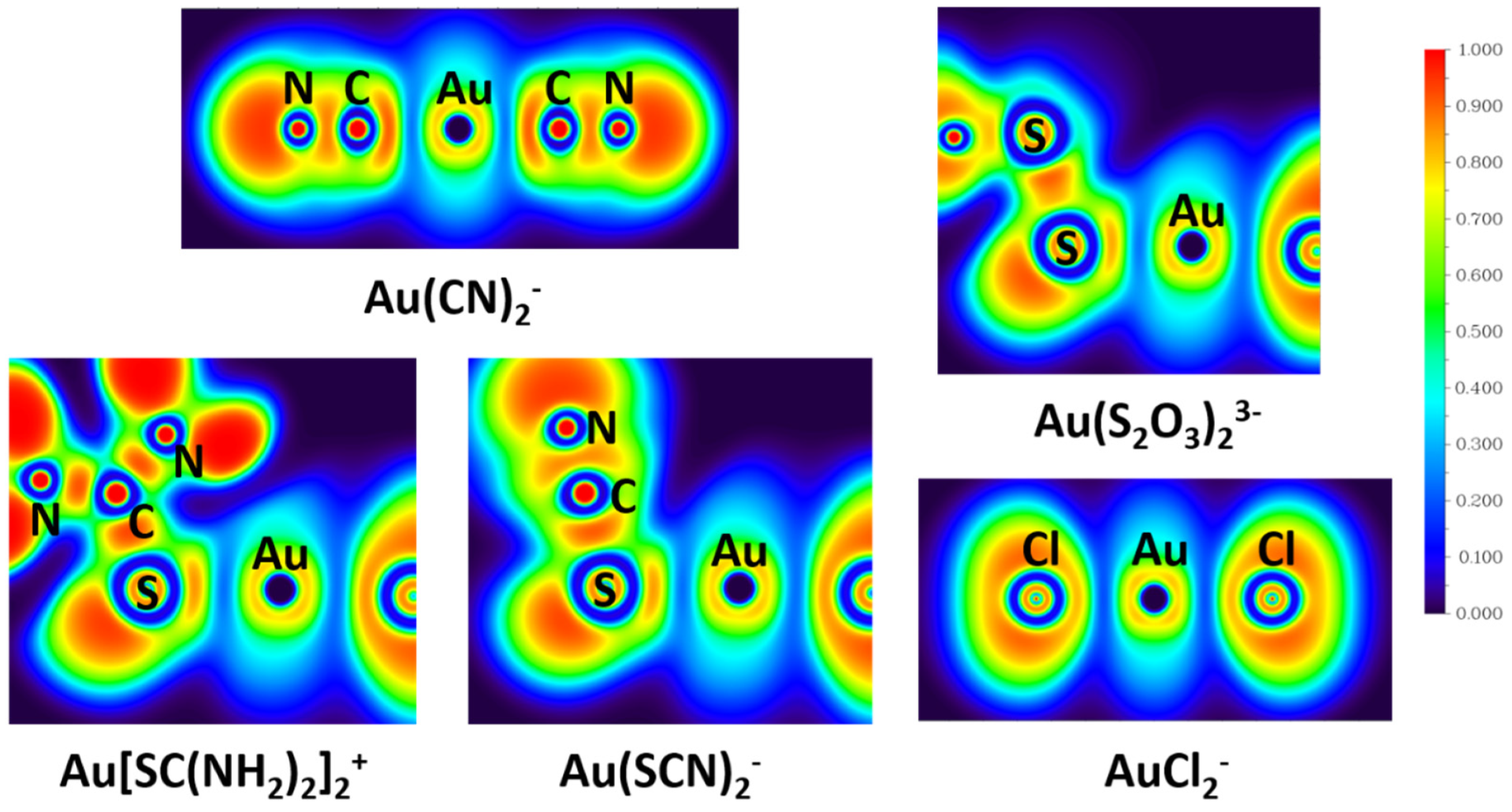
2.3. Chemical Bonding Analyses
2.4. Energy Decomposition Analysis (EDA)
3. Computational Methods
4. Conclusions
- (1)
- Based on the CDA results, the orbital interaction and electron transferring between Au+ and ligands for Au(CN)2−, Au(S2O3)23−, Au[SC(NH2)2]2+, Au(SCN)2−, and AuCl2− are interpreted. There is not only σ-donation from ligand to Au+, but also electron backdonation from Au+ to ligands, which strengthens the coordinate bond between them. The percentage of π-backdonation in Au(CN)2− is the largest contributing to its high stability.
- (2)
- From the perspective of NRT and ELF, compared with Cl−, ligands CN−, S2O32−, SC(NH2)2, and SCN− have very large covalent contribution to the coordinate bond with Au+, which explains the special stability of Au-CN and Au-S bonds.
- (3)
- The decomposition of the bonding energy between Au and the ligand provides further insight into the nature of the Au–ligand bonding. The EDA results show the increased value of orbital interaction from Au(CN)2−, Au(S2O3)23−, Au[SC(NH2)2]2+, Au(SCN)2−, to AuCl2−, corresponding to the decreased degree of covalency in Au–ligand bonding, which interprets the stability of the five complexes: Au(CN)2− > Au(S2O3)23− > Au[SC(NH2)2]2+ > Au(SCN)2− > AuCl2−.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, J.; Miller, J.D. A Review of Gold Leaching in Acid Thiourea Solutions. Miner. Process. Extr. Metall. Rev. 2006, 27, 177–214. [Google Scholar] [CrossRef]
- Li, L.; Pan, C.; Shan, J.; She, W.; You, X.; Ji, C.; Gao, Q. Pit-Induced Electrochemical Layer Dissolution and Wave Propagation on an Au(111) Surface in an Acidic Thiourea Solution. J. Phys. Chem. C 2020, 124, 19112–19118. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, B.; Cui, M.; Li, Q.; Liu, X.; Jiang, T.; Lyu, X. Thiosulfate leaching of gold catalyzed by hexaamminecobalt(III): Electrochemical behavior and mechanisms. Electrochim. Acta 2021, 399, 139393. [Google Scholar] [CrossRef]
- Li, W.-J.; Zhou, H.; Bai, A.-P.; Song, Y.-S.; Cai, L.-L.; Zheng, S.-L.; Zhang, Q.-D.; Cao, S. Electrochemical adsorption and passivation on gold surface in alkaline thiourea solutions. Rare Met. 2020, 39, 951–958. [Google Scholar] [CrossRef]
- Birich, A.; Stopic, S.; Friedrich, B. Kinetic Investigation and Dissolution Behavior of Cyanide Alternative Gold Leaching Reagents. Sci. Rep. 2019, 9, 7191. [Google Scholar] [CrossRef]
- Zhang, X.M.; Senanayake, G. A Review of Ammoniacal Thiosulfate Leaching of Gold: An Update Useful for Further Research in Non-cyanide Gold Lixiviants. Miner. Process. Extr. Metall. Rev. 2016, 37, 385–411. [Google Scholar] [CrossRef]
- Whitehead, J.; Zhang, J.; McCluskey, A.; Lawrance, G. Comparative leaching of a sulfidic gold ore in ionic liquid and aqueous acid with thiourea and halides using Fe(III) or HSO5−oxidant. Hydrometallurgy 2009, 98, 276–280. [Google Scholar] [CrossRef]
- Azizitorghabeh, A.; Wang, J.; Ramsay, J.A.; Ghahreman, A. A review of thiocyanate gold leaching—Chemistry, thermodynamics, kinetics and processing. Miner. Eng. 2021, 160, 106689. [Google Scholar] [CrossRef]
- Ilyas, S.; Srivastava, R.R.; Kim, H. Gold recovery from secondary waste of PCBs by electro-Cl2 leaching in brine solution and solvo-chemical separation with tri-butyl phosphate. J. Clean. Prod. 2021, 295, 126389. [Google Scholar] [CrossRef]
- Senanayake, G. Gold leaching by thiosulphate solutions: A critical review on copper(II)–thiosulphate–oxygen interactions. Miner. Eng. 2005, 18, 995–1009. [Google Scholar] [CrossRef]
- Grosse, A.C.; Dicinoski, G.W.; Shaw, M.J.; Haddad, P. Leaching and recovery of gold using ammoniacal thiosulfate leach liquors (a review). Hydrometallurgy 2003, 69, 1–21. [Google Scholar] [CrossRef]
- Sitando, O.; Senanayake, G.; Dai, X.; Nikoloski, A.; Breuer, P. A review of factors affecting gold leaching in non-ammoniacal thiosulfate solutions including degradation and in-situ generation of thiosulfate. Hydrometallurgy 2018, 178, 151–175. [Google Scholar] [CrossRef]
- Xu, B.; Kong, W.; Li, Q.; Yang, Y.; Jiang, T.; Liu, X. A Review of Thiosulfate Leaching of Gold: Focus on Thiosulfate Consumption and Gold Recovery from Pregnant Solution. Metals 2017, 7, 222. [Google Scholar] [CrossRef]
- Aylmore, M.G.; Muir, D.M. Thiosulfate leaching of gold—A review. Miner. Eng. 2001, 14, 135–174. [Google Scholar] [CrossRef]
- Yu, B.; Liu, Y.; Peng, X.; Hua, S.; Zhou, G.; Yan, K.; Liu, Y. Synthesis, characterization, and antitumor properties of Au(i)-thiourea complexes. Metallomics 2020, 12, 104–113. [Google Scholar] [CrossRef]
- Yang, X.; Moats, M.S.; Miller, J.D.; Wang, X.; Shi, X.; Xu, H. Thiourea–thiocyanate leaching system for gold. Hydrometallurgy 2011, 106, 58–63. [Google Scholar] [CrossRef]
- Kholmogorov, A.; Kononova, O.; Pashkov, G.; Kononov, Y. Thiocyanate solutions in gold technology. Hydrometallurgy 2002, 64, 43–48. [Google Scholar] [CrossRef]
- Azizitorghabeh, A.; Mahandra, H.; Ramsay, J.; Ghahreman, A. Gold leaching from an oxide ore using thiocyanate as a lixiviant: Process optimization and kinetics. ACS Omega 2021, 6, 17183–17193. [Google Scholar] [CrossRef]
- Nikoloski, A.; Stockton, B. Application of alternative lixiviants for secondary heap leaching of gold. In Proceedings of the 7th Mill Operators Conference, Kalgoorlie, Australia, 12–14 October 2000. [Google Scholar]
- Dönmez, B.; Sevim, F.; Çolak, S. A Study on Recovery of Gold from Decopperized Anode Slime. Chem. Eng. Technol. 2001, 24, 91–95. [Google Scholar] [CrossRef]
- Wang, S.; Li, L.; Wang, H.; Wu, G.-D. Extraction of platinum and gold from copper anode slimes by a process of chlorinating roasting followed by chlorinating leaching. J. Min. Met. Sect. B Met. 2020, 56, 193–202. [Google Scholar] [CrossRef]
- Rodríguez, M.; Ayala, L.; Robles, P.; Sepúlveda, R.; Torres, D.; Carrillo-Pedroza, F.R.; Jeldres, R.I.; Toro, N. Leaching Chalcopyrite with an Imidazolium-Based Ionic Liquid and Bromide. Metals 2020, 10, 183. [Google Scholar] [CrossRef]
- Li, G.-Z.; Kou, J.; Xing, Y.; Hu, Y.; Han, W.; Liu, Z.-Y.; Sun, C.-B. Gold-leaching performance and mechanism of sodium dicyanamide. Int. J. Miner. Met. Mater. 2021, 28, 1759–1768. [Google Scholar] [CrossRef]
- Liu, Z.; Kou, J.; Xing, Y.; Sun, C. Recovery of Gold from Ore with Potassium Ferrocyanide Solution under UV Light. Minerals 2021, 11, 387. [Google Scholar] [CrossRef]
- Pyykkö, P.; Atsumi, M. Molecular single-bond covalent radii for elements 1-118. Chemistry 2009, 15, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.G.; Wang, Y.L.; Xu, C.Q.; Qiu, Y.H.; Wang, L.S.; Li, J. On the gold-ligand covalency in linear [AuX2](−) complexes. Dalton Trans. 2015, 44, 5535–5546. [Google Scholar] [CrossRef]
- Xu, C.Q.; Xiong, X.G.; Li, W.L.; Li, J. Periodicity and Covalency of [MX2]− (M=Cu, Ag, Au, Rg;X=H, Cl, CN) Complexes. Eur. J. Inorg. Chem. 2016, 2016, 1395–1404. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Atomic dipole moment corrected Hirshfeld population method. J. Theor. Comput. Chem. 2012, 11, 163–183. [Google Scholar] [CrossRef]
- Marenich, A.V.; Jerome, S.V.; Cramer, C.J.; Truhlar, D.G. Charge Model 5: An Extension of Hirshfeld Population Analysis for the Accurate Description of Molecular Interactions in Gaseous and Condensed Phases. J. Chem. Theory Comput. 2012, 8, 527–541. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Chatt, J.; Duncanson, L.A. 586. Olefin co-ordination compounds. Part III. Infrared spectra and structure: Attempted preparation of acetylene complexes. J. Chem. Soc. 1953, 2939–2947. [Google Scholar] [CrossRef]
- Dapprich, S.; Frenking, G. Investigation of donor-acceptor interactions: A charge decomposition analysis using fragment molecular orbitals. J. Phys. Chem. 1995, 99, 9352–9362. [Google Scholar] [CrossRef]
- Hernández, M.G.; Beste, A.; Frenking, G.; Illas, F. Charge decomposition analysis of the chemisorption bond. Chem. Phys. Lett. 2000, 320, 222–228. [Google Scholar] [CrossRef]
- Ehlers, A.W.; Dapprich, S.; Vyboishchikov, S.F.; Frenking, G. Structure and Bonding of the Transition-Metal Carbonyl Complexes M (CO) 5L (M= Cr, Mo, W) and M (CO) 3L (M= Ni, Pd, Pt; L= CO, SiO, CS, N2, NO+, CN−, NC−, HCCH, CCH2, CH2, CF2, H2). Organometallics 1996, 15, 105–117. [Google Scholar] [CrossRef]
- Szilagyi, R.K.; Frenking, G.J.O. Structure and Bonding of the Isoelectronic Hexacarbonyls [Hf(CO)6]2−, [Ta(CO)6]−, W(CO)6, [Re(CO)6]+, [Os(CO)6]2+, and [Ir(CO)6]3+: A Theoretical Study. Organometallics 1997, 16, 4807–4815. [Google Scholar] [CrossRef]
- Crabtree, R.H. The Organometallic Chemistry of the Transition Metals; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Pyykko, P.J.C.R. Relativistic effects in structural chemistry. Chem. Rev. 1988, 88, 563–594. [Google Scholar] [CrossRef]
- Ning, C.G.; Xiong, X.G.; Wang, Y.L.; Li, J.; Wang, L.S. Probing the electronic structure and chemical bonding of the “staple” motifs of thiolate gold nanoparticles: Au(SCH3)2− and Au2(SCH3)3. Phys. Chem. Chem. Phys. 2012, 14, 9323–9329. [Google Scholar] [CrossRef]
- Glendening, E.D.; Weinhold, F. Natural resonance theory: I. General formalism. J. Comput. Chem. 1998, 19, 593–609. [Google Scholar] [CrossRef]
- Poater, J.; Duran, M.; Solà, M.; Silvi, B. Theoretical Evaluation of Electron Delocalization in Aromatic Molecules by Means of Atoms in Molecules (AIM) and Electron Localization Function (ELF) Topological Approaches. Chem. Rev. 2005, 105, 3911–3947. [Google Scholar] [CrossRef]
- Frisch, M.E.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.P.G.A.; Petersson, G.A.; Nakatsuji, H.J.R.A.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Wilson, A.K.; Woon, D.E.; Peterson, K.A.; Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. IX. The atoms gallium through krypton. J. Chem. Phys. 1999, 110, 7667–7676. [Google Scholar] [CrossRef]
- Figgen, D.; Rauhut, G.; Dolg, M.; Stoll, H. Energy-consistent pseudopotentials for group 11 and 12 atoms: Adjustment to multi-configuration Dirac–Hartree–Fock data. Chem. Phys. 2005, 311, 227–244. [Google Scholar] [CrossRef]
- Peterson, K.A.; Puzzarini, C. Systematically convergent basis sets for transition metals. II. Pseudopotential-based correlation consistent basis sets for the group 11 (Cu, Ag, Au) and 12 (Zn, Cd, Hg) elements. Theor. Chem. Acc. 2005, 114, 283–296. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Cammi, R.; Tomasi, J. Remarks on the use of the apparent surface charges (ASC) methods in solvation problems: Iterative versus matrix-inversion procedures and the renormalization of the apparent charges. J. Comput. Chem. 1995, 16, 1449–1458. [Google Scholar] [CrossRef]
- Weinhold, F.; Glendening, E.D. NBO 5.0 Program Manual: Natural Bond Orbital Analysis Programs; Theoretical Chemistry Institute and Department of Chemistry, University of Wisconsin: Madison, WI, USA, 2001; p. 53706. [Google Scholar]
- Xiao, M.; Lu, T. Generalized charge decomposition analysis (GCDA) method. J. Adv. Phys. Chem. 2015, 4, 111–124. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Au(CN)2− | Au(S2O3)23− | Au[SC(NH2)2]2+ | Au(SCN)2− | AuCl2− |
|---|---|---|---|---|---|
| logβi | 38.3 | 28.7 | 21.3 | 17.1 | 9.1 |
| E0/V | −0.57 | 0.17 | 0.35 | 0.66 | 1.11 |
| Complex | Symm. | R(Au − X)(X = C, Cl, S) | ∠(X – Au − X)(X = C, Cl, S) | ||
|---|---|---|---|---|---|
| a | b | a | b | ||
| Au(CN)2− | D∞h | 1.987 | 1.987 | 180.00 | 180.00 |
| Au(S2O3)23− | C2 | 2.299 | 2.297 | 176.45 | 176.86 |
| Au[SC(NH2)2]2+ | C2 | 2.304 | 2.299 | 176.18 | 176.15 |
| Au(SCN)2− | C2 | 2.310 | 2.305 | 176.94 | 177.16 |
| AuCl2− | D∞h | 2.291 | 2.287 | 180.00 | 180.00 |
| Complex | Atom | Charge | |||
|---|---|---|---|---|---|
| Hirshfeld [28] | ADCH [29] | CM5 [30] | CHELPG [31] | ||
| Au(CN)2− | Au | 0.089 | 0.100 | 0.207 | 0.197 |
| C | −0.173 | −0.077 | −0.109 | −0.020 | |
| Au(S2O3)23− | Au | −0.092 | 0.031 | 0.042 | 0.290 |
| S | −0.409 | −0.502 | −0.469 | −0.727 | |
| Au[SC(NH2)2]2+ | Au | 0.135 | 0.153 | 0.271 | 0.427 |
| S | −0.178 | −0.224 | −0.247 | −0.438 | |
| Au(SCN)2− | Au | 0.066 | 0.267 | 0.195 | 0.262 |
| S | −0.178 | −0.238 | −0.252 | −0.393 | |
| AuCl2− | Au | −0.036 | −0.004 | 0.056 | 0.270 |
| Cl | −0.482 | −0.498 | −0.528 | −0.636 | |
| Complex | Donation | Backdonation | Backdonation/Donation (%) |
|---|---|---|---|
| Au(CN)2− | 0.8035 | 0.1464 | 18.22 |
| Au(S2O3)23− | 0.5294 | 0.0829 | 15.66 |
| Au[SC(NH2)2]2+ | 0.4458 | 0.0722 | 16.20 |
| Au(SCN)2− | 0.6673 | 0.0938 | 14.06 |
| AuCl2− | 0.6226 | 0.0453 | 7.28 |
| Complex | NBO Analysis | NRT BO | ||
|---|---|---|---|---|
| Covalent | Ionic | Covalent/Total | ||
| Au(CN)2− | 25.37% (sd0.27)Au + 74.63% (sp0.87)C | 0.215 | 0.312 | 0.408 |
| Au(S2O3)23− | 24.01% (sp0.01d0.18)Au + 75.99% (sp5.82d0.04)S | 0.215 | 0.257 | 0.456 |
| Au[SC(NH2)2]2+ | 24.70% (sp0.01d0.18)Au + 75.30% (sp6.54d0.04)S | 0.216 | 0.265 | 0.449 |
| Au(SCN)2− | 24.22% (sp0.01d0.18)Au + 75.78% (sp9.76d0.05)S | 0.194 | 0.253 | 0.434 |
| AuCl2− | 19.41% (sp0.01d0.21)Au + 80.59% (sp6.61d0.02)Cl | 0.176 | 0.824 | 0.176 |
| Complex | Steric Interaction | Orbital Interaction | Total | Orb./Total |
|---|---|---|---|---|
| Au(CN)2− | 1.32 | −4.09 | −2.77 | 1.477 |
| Au(S2O3)23− | 1.49 | −3.64 | −2.14 | 1.696 |
| Au[SC(NH2)2]2+ | 1.46 | −3.43 | −1.97 | 1.740 |
| Au(SCN)2− | 1.34 | −3.17 | −1.83 | 1.737 |
| AuCl2− | 0.83 | −2.70 | −1.86 | 1.446 |
| Complex | Steric Interaction | Orbital Interaction | Total | Orb./Total |
|---|---|---|---|---|
| Au(CN)2− | 3.41 | −9.37 | −5.97 | 1.571 |
| Au(S2O3)23− | 2.68 | −7.84 | −5.16 | 1.520 |
| Au[SC(NH2)2]2+ | 3.17 | −7.43 | −4.26 | 1.744 |
| Au(SCN)2− | 2.84 | −6.90 | −4.05 | 1.701 |
| AuCl2− | 2.35 | −6.22 | −3.87 | 1.606 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, N.; Kou, J.; Sun, C. Investigation on Gold–Ligand Interaction for Complexes from Gold Leaching: A DFT Study. Molecules 2023, 28, 1508. https://doi.org/10.3390/molecules28031508
Zhang N, Kou J, Sun C. Investigation on Gold–Ligand Interaction for Complexes from Gold Leaching: A DFT Study. Molecules. 2023; 28(3):1508. https://doi.org/10.3390/molecules28031508
Chicago/Turabian StyleZhang, Na, Jue Kou, and Chunbao Sun. 2023. "Investigation on Gold–Ligand Interaction for Complexes from Gold Leaching: A DFT Study" Molecules 28, no. 3: 1508. https://doi.org/10.3390/molecules28031508
APA StyleZhang, N., Kou, J., & Sun, C. (2023). Investigation on Gold–Ligand Interaction for Complexes from Gold Leaching: A DFT Study. Molecules, 28(3), 1508. https://doi.org/10.3390/molecules28031508