
Direct Regioselective C-H Cyanation of Purines


Abstract
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Information
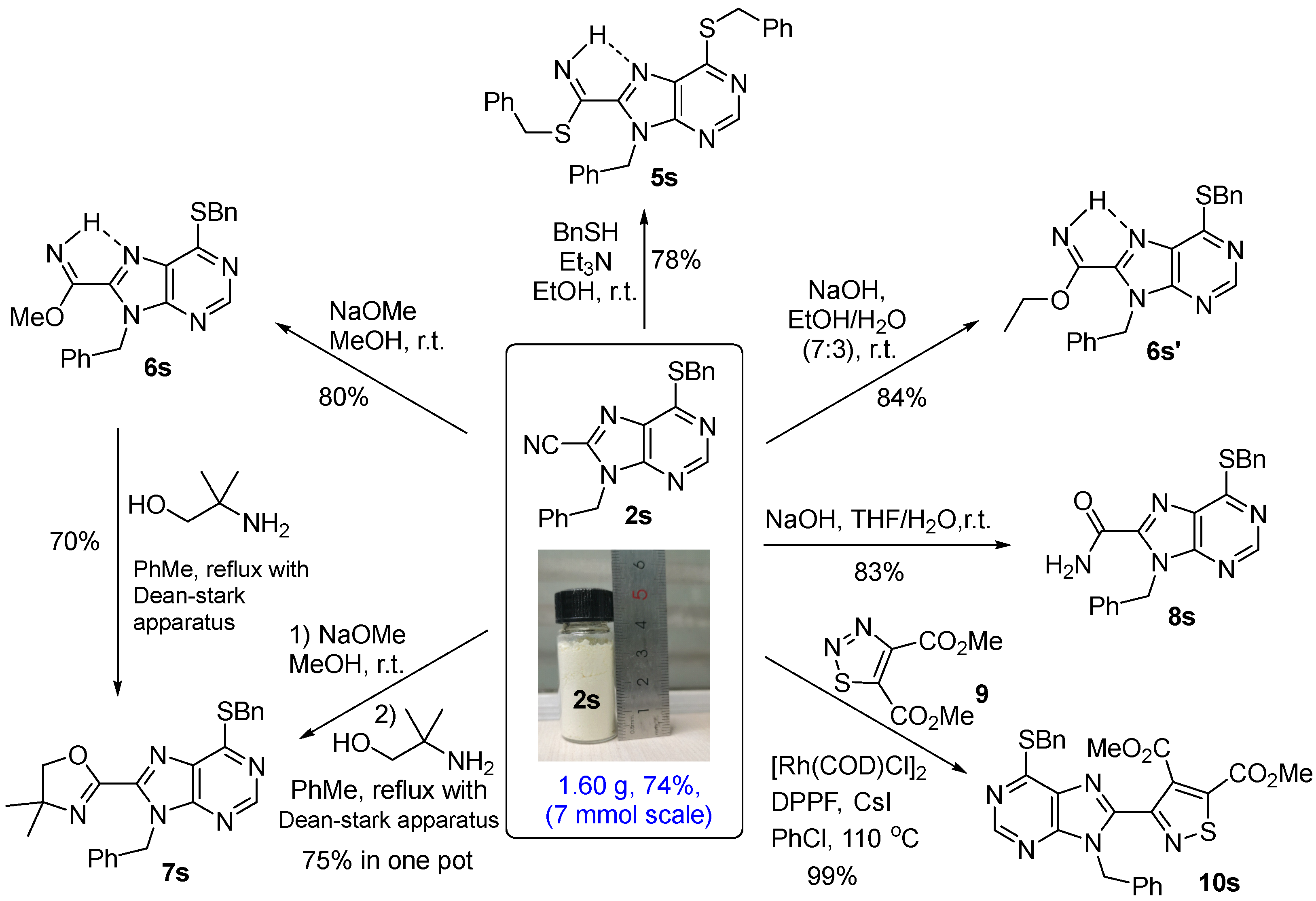
3.2. General Procedure for the 8-Cyanation of N-Benzylpurine, 6-Chloropurines and 6-Alkoxy, Alkylthio, and Aryl Purines 2
3.2.1. 9-Benzyl-9H-purine-8-carbonitrile (2a)
3.2.2. 6-Chloro-9-propyl-9H-purine-8-carbonitrile (2b)
3.2.3. 6-Chloro-9-isopropyl-9H-purine-8-carbonitrile (2c)
3.2.4. 9-Allyl-6-chloro-9h-purine-8-carbonitrile (2d)
3.2.5. 6-Chloro-9-(propargyl)-9H-purine-8-carbonitrile (2e)
3.2.6. 9-Benzyl-6-chloro-9H-purine-8-carbonitrile (2f)
3.2.7. 6-Chloro-9-(4-methylbenzyl)-9h-purine-8-carbonitrile (2g)
3.2.8. 6-Chloro-9-(4-nitrobenzyl)-9H-purine-8-carbonitrile (2h)
3.2.9. 6-Chloro-9-(4-methoxybenzyl)-9H-purine-8-carbonitrile (2i)
3.2.10. Ethyl-2-(6-chloro-8-cyano-9H-purin-9-yl)acetate (2j)
3.2.11. 6-Chloro-9-(2-(4-chlorophenyl)-2-oxoethyl)-9H-purine-8-carbonitrile (2k)
3.2.12. 6-Chloro-9-phenyl-9H-purine-8-carbonitrile (2l)
3.2.13. 6-Chloro-9-(p-tolyl)-9H-purine-8-carbonitrile (2m)
3.2.14. 6-Chloro-9-(4-methoxyphenyl)-9H-purine-8-carbonitrile (2n)
3.2.15. 6-Chloro-9-(4-(methylthio) phenyl)-9H-purine-8-carbonitrile (2o)
3.2.16. 9-(4-bromophenyl)-6-chloro-9H-purine-8-carbonitrile (2p)
3.2.17. 6-Chloro-9-(3-chlorophenyl)-9H-purine-8-carbonitrile (2q)
3.2.18. 9-Benzyl-2,6-dichloro-9h-purine-8-carbonitrile (2r) and 9-Benzyl-2,6-dichloro-7-((trifluoromethyl)sulfonyl)-8,9-dihydro-7H-purine-8-carbonitrile (3r)
3.2.19. 9-Benzyl-6-(benzylthio)-9H-purine-8-carbonitrile (2s)
3.2.20. 9-Benzyl-6-methoxy-9H-purine-8-carbonitrile (2t)
3.2.21. 9-Benzyl-6-(p-tolyl)-9H-purine-8-carbonitrile (2u) and 9-Benzyl-6-(p-tolyl)-9H-purine-2,8-dicarbonitrile (4u)
3.2.22. 9-Benzyl-6-(diethylamino)-9H-purine-2-carbonitrile (2v’)
3.3. Synthesis of 9-Benzyl-6-(diethylamino)-9H-purine-8-carbonitrile (2v) and 9-Benzyl-6-(diethylamino)-N,N-diethyl-9H-purine-8-carboximidamide (5f)
3.4. Synthesis of 9-Benzyl-6-((4-methoxyphenyl)ethynyl)-9H-purine-8-carbonitrile (6f)
3.5. Synthesis of 2u from 2f
3.6. Synthesis of 2t from 2f
3.7. Synthesis of 2s and Benzyl 9-Benzyl-6-(benzylthio)-2-cyano-9H-purine-8-carbimidothioate (5s) from 2f
3.8. Synthesis of 5s from 2s
3.9. Synthesis of Methyl 9-Benzyl-6-(benzylthio)-9H-purine-8-carbimidate (6s) from 2s
3.10. Synthesis of 2-(9-Benzyl-6-(benzylthio)-9H-purin-8-yl)-4,4-dimethyl-4,5-dihydrooxazole (7s) from 6s, and One-Pot Procedure from 2s
3.11. Synthesis of Ethyl 9-Benzyl-6-(benzylthio)-9H-purine-8-carbimidate (6s’) from 2s
3.12. Synthesis of 9-Benzyl-6-(benzylthio)-9H-purine-8-carboxamide (8s) from 2s
3.13. Synthesis of Dimethyl 3-(9-Benzyl-6-(benzylthio)-9H-purin-8-yl)isothiazole-4,5-dicarboxylate (10s) from 2s
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Mallari, J.P.; Guiguemde, W.A.; Guy, R.K. Antimalarial activity of thiosemicarbazones and purine derived nitriles. Bioorg. Med. Chem. Lett. 2009, 19, 3546–3549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallari, J.P.; Shelat, A.A.; Obrien, T.; Caffrey, C.R.; Kosinsk, A.; Connelly, M.; Harbut, M.; Creenbaum, D.; McKerrow, J.H.; Guy, R.K. Development of potent purine-perived nitrile inhibitors of the Trypanosomal Protease TbcatB. J. Med. Chem. 2008, 51, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, A.; Suzuki, Y.; Ohta, K.; Higashino, T. Preparation of heteroarenecarbonitriles by reaction of haloheteroarenes with potassium cyanide with sodium p-toluenesulfinate as catalyst. Heterocycles 1994, 39, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Herdewijn, P.; Van Aerschot, A.; Pfleiderer, W. Synthesis of 2-amino-6-(acetamidomethyl)-9-(β-D-ribofuranosyl)purine. Synthesis 1989, 961–962. [Google Scholar] [CrossRef]
- Gundersen, L.-L. Synthesis of purinecarbonitriles by Pd(0)-catalyzed coupling of halopurines with zinc cyanide. Acta Chem. Scand. 1996, 50, 58–63. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Girardet, J.-L.; Hong, Z.; Lai, V.C.H.; An, H.; Koh, Y.-h.; Shaw, S.Z.; Zhong, W. Synthesis of 9-(2-β-C-methyl-β-D-ribofuranosyl)-6-substituted purine derivatives as inhibitors of HCV RNA replication. Bioorg. Med. Chem. Lett. 2005, 15, 709–713. [Google Scholar] [CrossRef]
- Ai, C.; Zhang, W.; Zhou, L.; Cai, X.; Zheng, Z. Molecular modeling of three-dimensional structure of hTRPV4 protein and experimental verification of its antagonist binding sites. J. Mol. Struct. 2021, 1227, 129421. [Google Scholar] [CrossRef]
- Tanji, K.; Higashino, T. Purines. IX. Reaction of 9-phenyl-9H-purine-2-carbonitriles with Grignard reagents. Heterocycles 1990, 30, 435–440. [Google Scholar] [CrossRef]
- Altmann, E.; Cowan-Jacob, S.W.; Missbach, M. Novel purine nitrile derived inhibitors of the Cysteine Protease Cathepsin K. J. Med. Chem. 2004, 47, 5833–5836. [Google Scholar] [CrossRef]
- Zulfiqar, F.; Kojima, H.; Nakanishi, M.; Ando, T.; Kitade, Y. Synthesis of carbocyclic 2-substituted adenine nucleoside and related Analogs. Nucleosides Nucleotides Nucleic Acids 2008, 27, 1153–1157. [Google Scholar] [CrossRef]
- Braendvang, M.; Gundersen, L.-L. Synthesis, biological activity, and SAR of antimycobacterial 2- and 8-substituted 6-(2-furyl)-9-(p-methoxybenzyl)purines. Bioorg. Med. Chem. 2007, 15, 7144–7165. [Google Scholar] [CrossRef] [PubMed]
- Butora, G.; Schmitt, C.; Levorse, D.A.; Streckfuss, E.; Doss, G.A.; MacCoss, M. The elusive 8-fluoroadenosine: A simple non-enzymatic synthesis and characterization. Tetrahedron 2007, 63, 3782–3789. [Google Scholar] [CrossRef]
- El Safadi, Y.; Marquet, R.; Aubertin, A.-M.; Vivet-Boudou, V. Synthesis and primary evaluation of novel HIV-1 Inhibitors. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1161–1165. [Google Scholar] [CrossRef] [PubMed]
- Vivet-Boudou, V.; Isel, C.; Sleiman, M.; Smyth, R.; Ben Gaied, N.; Barhoum, P.; Laumond, G.; Bec, G.; Gotte, M.; Mak, J.; et al. 8-Modified-2’-deoxyadenosine analogues induce delayed polymerization arrest during HIV-1 reverse transcription. PLoS ONE 2011, 6, e27456. [Google Scholar] [CrossRef] [Green Version]
- Butler, R.S.; Myers, A.K.; Bellarmine, P.; Abboud, K.A.; Castellano, R.K. Highly fluorescent donor-acceptor purines. J. Mater. Chem. 2007, 17, 1863–1865. [Google Scholar] [CrossRef]
- Butler, R.S.; Cohn, P.; Tenzel, P.; Abboud, K.A.; Castellano, R.K. Synthesis, photophysical behavior, and electronic structure of push-pull purines. J. Am. Chem. Soc. 2009, 131, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, E.; Shimada, N.; Matsuoka, Y. Purines. 1. Reaction of 9-phenyl-9H-purine and 7-phenyl-7H-purine with Grignard reagents. Yakugaku Zasshi 1979, 99, 114–119. [Google Scholar] [CrossRef] [Green Version]
- D’Errico, S.; Piccialli, V.; Oliviero, G.; Borbone, N.; Amato, J.; D’Atri, V.; Piccialli, G. Probing the reactivity of nebularine N1-oxide. A novel approach to C-6 C-substituted purine nucleosides. Tetrahedron 2011, 67, 6138–6144. [Google Scholar] [CrossRef]
- D’Errico, S.; Oliviero, G.; Amato, J.; Borbone, N.; Cerullo, V.; Hemminki, A.; Piccialli, V.; Zaccaria, S.; Mayol, L.; Piccialli, G. Synthesis and biological evaluation of unprecedented ring-expanded nucleosides (RENs) containing the imidazo[4,5-d][1,2,6]oxadiazepine ring system. Chem. Commun. 2012, 48, 9310–9312. [Google Scholar] [CrossRef]
- D’Errico, S.; Oliviero, G.; Borbone, N.; Piccialli, V.; D’Atri, V.; Mayol, L.; Piccialli, G. Synthesis of 2,6-dialkyl(aryl)purine nucleosides by exploiting the reactivity of Nebularine N1-oxide towards Grignard reagents. Eur. J. Org. Chem. 2013, 2013, 6948–6954. [Google Scholar] [CrossRef]
- Xia, R.; Xie, M.-S.; Niu, H.-Y.; Qu, G.-R.; Guo, H.-M. Radical route for the alkylation of purine nucleosides at C6 via Minisci reaction. Org. Lett. 2014, 16, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-C.; Xia, R.; Xie, M.-S.; Qu, G.-R.; Guo, H.-M. Synthesis of cycloalkyl substituted purine nucleosides via a metal-free radical route. Org. Biomol. Chem. 2016, 14, 4189–4193. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.-M.; Shang, R.; Fu, M.-C.; Fu, Y. Photoredox-catalysed decarboxylative alkylation of N-heteroarenes with N-(acyloxy)phthalimides. Chem. Eur. J. 2017, 23, 2537–2541. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Yu, M.; Shi, T.; Hu, J.; Li, S.; Xu, J.X.; Chen, N.; Du, H.G. Silver-catalyzed direct C6-H arylation of purines and purine nucleosides with arylboronic acids. Eur. J. Org. Chem. 2017, 2017, 3415–3420. [Google Scholar] [CrossRef]
- Yu, M.; Zhou, Z.; Zou, C.; Wang, Z.; Wang, W.; Sun, K. Traceless proton aided regioselective C(sp2)–C(sp2) construction to synthesize C6-acylated purines and purine nucleosides without metal catalysts. Org. Chem. Front. 2022, 9, 4460–4465. [Google Scholar] [CrossRef]
- Yu, M.; Zhou, Z.; Chen, Y.; Wang, Z.; Wang, W.; Sun, K. Regioselective C6–H hydroxyalkylation of purines and purine nucleosides via α-C–H functionalization of alcohols at room temperature. Org. Lett. 2022, 24, 4886–4891. [Google Scholar] [CrossRef]
- Corey, E.J.; Tian, Y. Selective 4-arylation of pyridines by a nonmetalloorganic process. Org. Lett. 2005, 7, 5535–5537. [Google Scholar] [CrossRef]
- Hilton, M.C.; Dolewski, R.D.; McNally, A. Selective functionalization of pyridines via heterocyclic phosphonium salts. J. Am. Chem. Soc. 2016, 138, 13806–13809. [Google Scholar] [CrossRef]
- Elbert, B.L.; Farley, A.J.M.; Gorman, T.W.; Johnson, T.C.; Genicot, C.; Lallemand, B.; Pasau, P.; Flasz, J.; Castro, J.L.; MacCoss, M.; et al. C–H cyanation of 6-ring N-containing heteroaromatics. Chem. Eur. J. 2017, 23, 14733–14737. [Google Scholar] [CrossRef] [Green Version]
- Aly, A.A.; Brase, S.; Gomaa, A.A.-M. Amidines: Their synthesis, reactivity, and applications in heterocycle synthesis. Arkivoc 2018, vi, 85–138. [Google Scholar] [CrossRef]
- Faizi, D.J.; Nava, N.A.; Al-Amin, M.; Blum, S.A. Oxyboration: Synthesis of borylated benzofurans. Org. Synth. 2016, 93, 228–244. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Li, M.-P.; Li, L.-J.; Huang, Q.; Hu, M.-Y.; Zhu, S.-F. Phenanthroline-imine ligands for iron-catalyzed alkene hydrosilylation. Chem. Sci. 2022, 13, 2721–2728. [Google Scholar] [CrossRef] [PubMed]
- Keegstra, M.A.; Peters, T.H.A.; Brandsma, L. Copper(I) halide catalyzed synthesis of alkyl aryl and alkyl heteroaryl ethers. Tetrahedron 1992, 48, 3633–3652. [Google Scholar] [CrossRef]
- Hu, L.B.; Zhu, H.; Du, D.-M.; Xu, J.X. Efficient Synthesis of taurine and structurally diverse substituted taurines from aziridines. J. Org. Chem. 2007, 72, 4543–4546. [Google Scholar] [CrossRef]
- Lu, D.-F.; Zhu, C.-L.; Jia, Z.-X.; Xu, H. Iron(II)-catalyzed intermolecular amino-oxygenation of olefins through the N–O bond cleavage of functionalized hydroxylamines. J. Am. Chem. Soc. 2014, 136, 13186–13189. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.Y.; Chen, C.Z.; Wang, J.; Xu, J.X.; Yang, Z.H. Dual roles of bisphosphines and epoxides: Rh-catalyzed highly chemoselective and diastereoselective (3 + 2) transannulations of 1,2,3-thiadiazoles with cyanoepoxides. Org. Chem. Front. 2021, 8, 6687–6698. [Google Scholar] [CrossRef]
- Wu, Q.Y.; Chen, N.; Xu, J.X. Chemoselectivity in the transannulaction of 1,2,3-thiadiazoles and alk-2-enenitriles: Specific synthesis of 3-(Alk-1-enyl)isothiazoles. Chemistryselect 2022, 7, e202103943. [Google Scholar] [CrossRef]
- Hu, J.B.; Wang, C.X.; Yu, M.W.; Zhang, S.J.; Chen, N.; Du, H.G. Palladium-catalyzed N3-directed C–H halogenation of N9-arylpurines and azapurines. Eur. J. Org. Chem. 2022, 2022, e202101266. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
|---|---|---|
| Entry | Variation from the Standard Conditions | 2a (%) a |
| 1 | - | 54 |
| 2 | TFA, TfOH, or TMSOTf instead of Tf2O | 0 |
| 3 | BF3.OEt2 or HBF4 instead of Tf2O | 0 |
| 4 | Ac2O, Ts2O, or Boc2o instead of Tf2O | 0 |
| 5 | THF or MeCN as the solvent | 0 |
| 6 | DCM as the solvent | 30 |
| 7 | PhCl as the solvent | 47 |
| 8 | PhMe as the solvent | 49 |
| 9 | CHCl3 as the solvent | 50 |
| 10 | Py instead of DBU | 40 |
| 11 | Et3N instead of DBU | 40 |
| 12 | DABCO instead of DBU | 26 |
| 13 | N-Methylmorpholine (NMM) instead of DBU | 47 |
| 14 | Quinuclidine instead of DBU | 26 |
| 15 | DBN instead of DBU | 41 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Hu, J.; Fu, Y.; Shi, X.; Du, H.; Xu, J.; Chen, N. Direct Regioselective C-H Cyanation of Purines. Molecules 2023, 28, 914. https://doi.org/10.3390/molecules28030914
Li L, Hu J, Fu Y, Shi X, Du H, Xu J, Chen N. Direct Regioselective C-H Cyanation of Purines. Molecules. 2023; 28(3):914. https://doi.org/10.3390/molecules28030914
Chicago/Turabian StyleLi, Luyong, Jie Hu, Yuqing Fu, Xiaolin Shi, Hongguang Du, Jiaxi Xu, and Ning Chen. 2023. "Direct Regioselective C-H Cyanation of Purines" Molecules 28, no. 3: 914. https://doi.org/10.3390/molecules28030914
APA StyleLi, L., Hu, J., Fu, Y., Shi, X., Du, H., Xu, J., & Chen, N. (2023). Direct Regioselective C-H Cyanation of Purines. Molecules, 28(3), 914. https://doi.org/10.3390/molecules28030914