
Remote Steric Control of the Tetrahedral Coordination Geometry around Heteroleptic Copper(I) Bis(Diimine) Complexes
Abstract
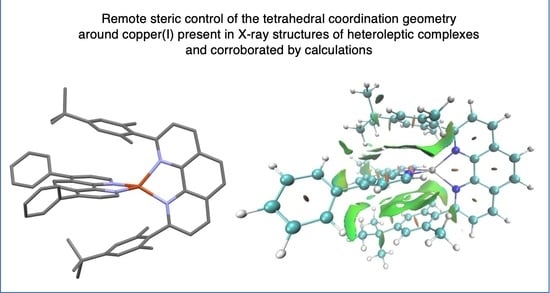
:
1. Introduction
2. Results
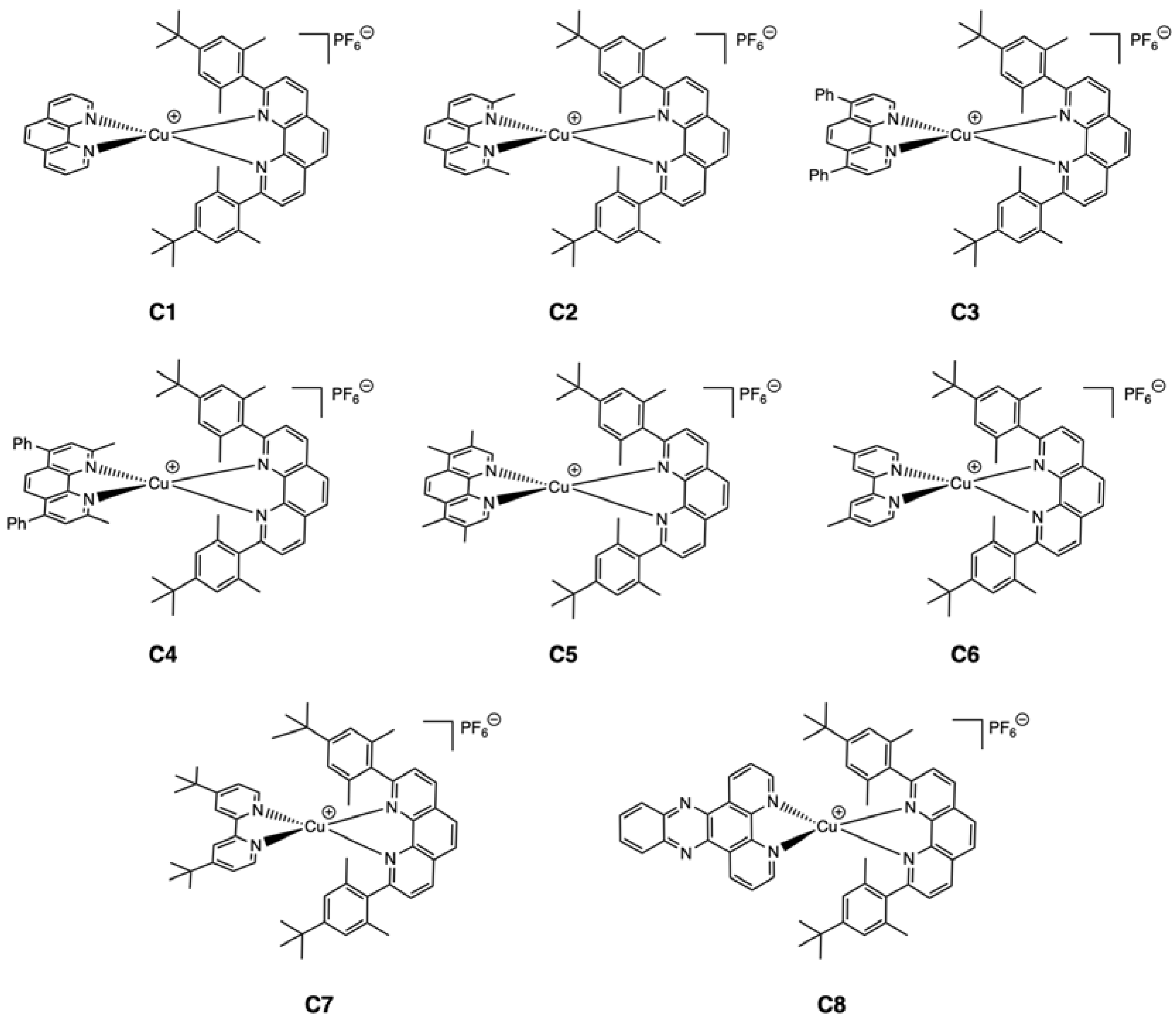
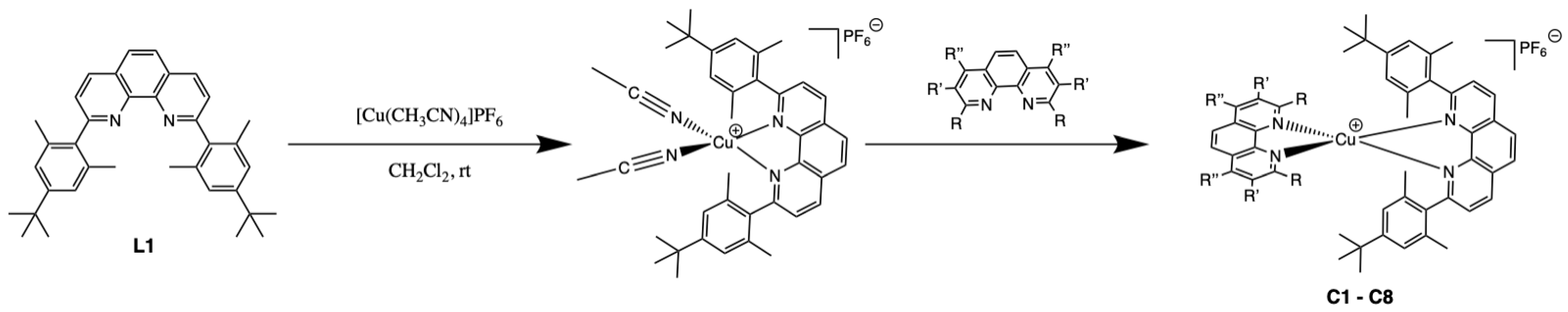
2.1. Synthesis
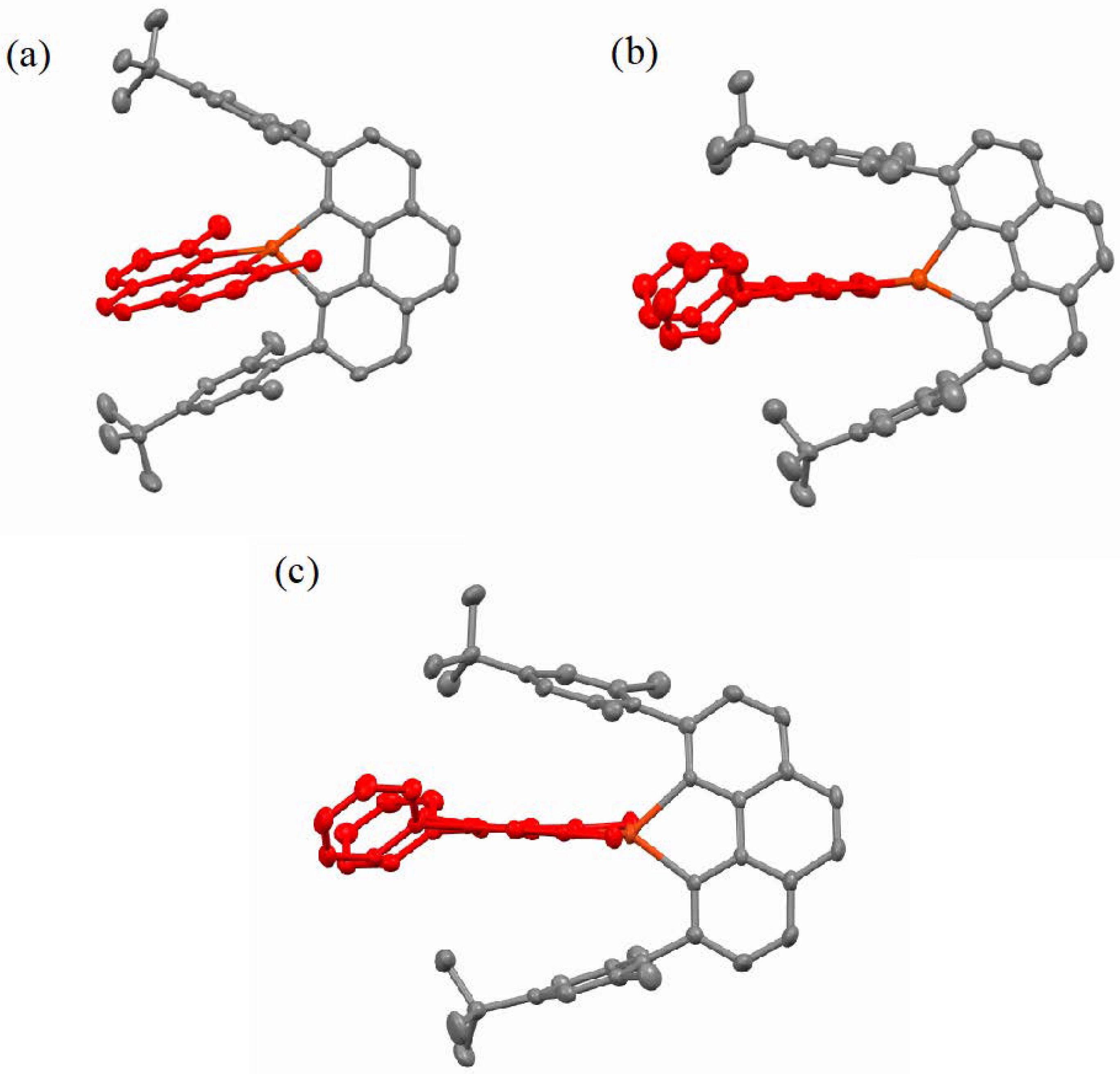
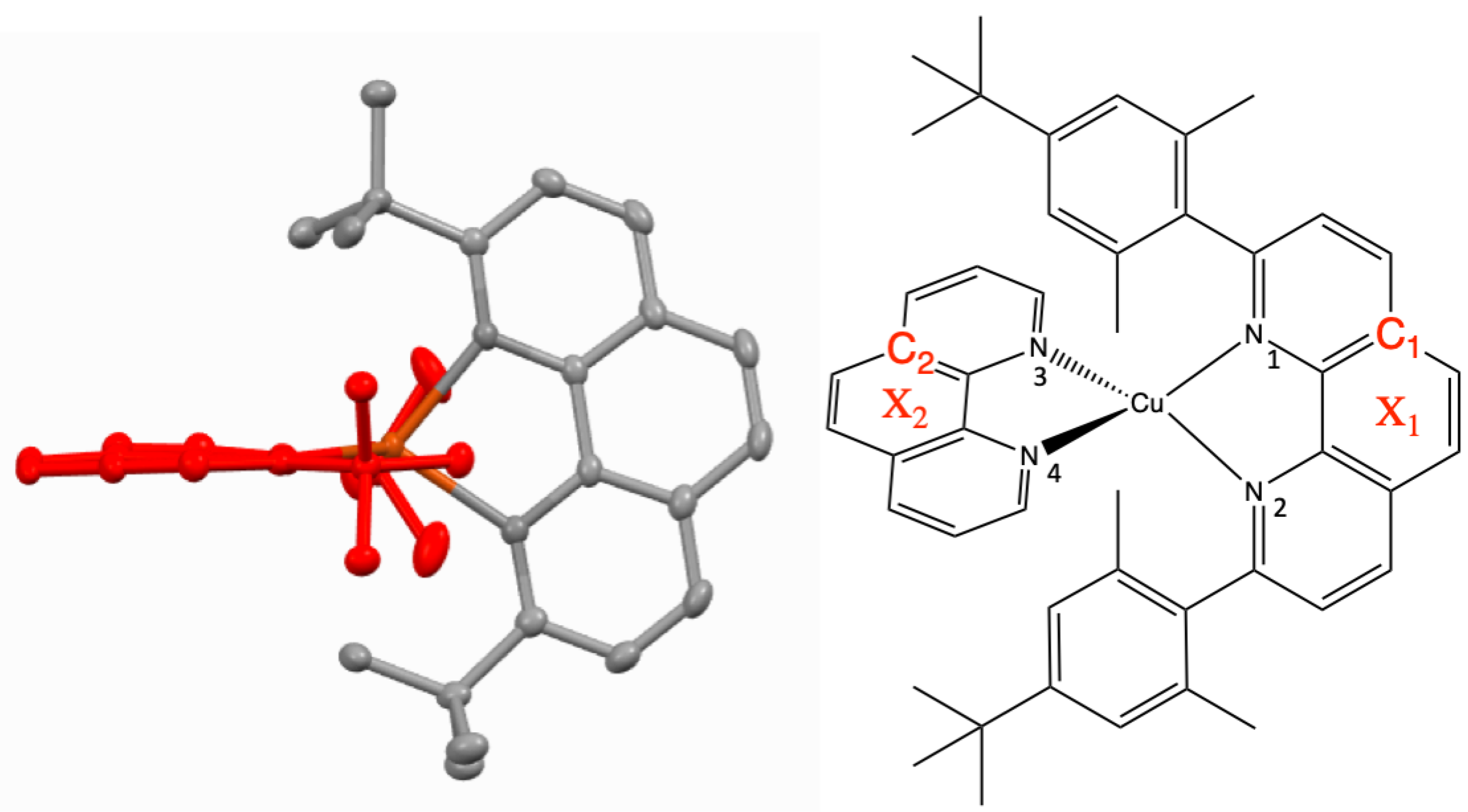
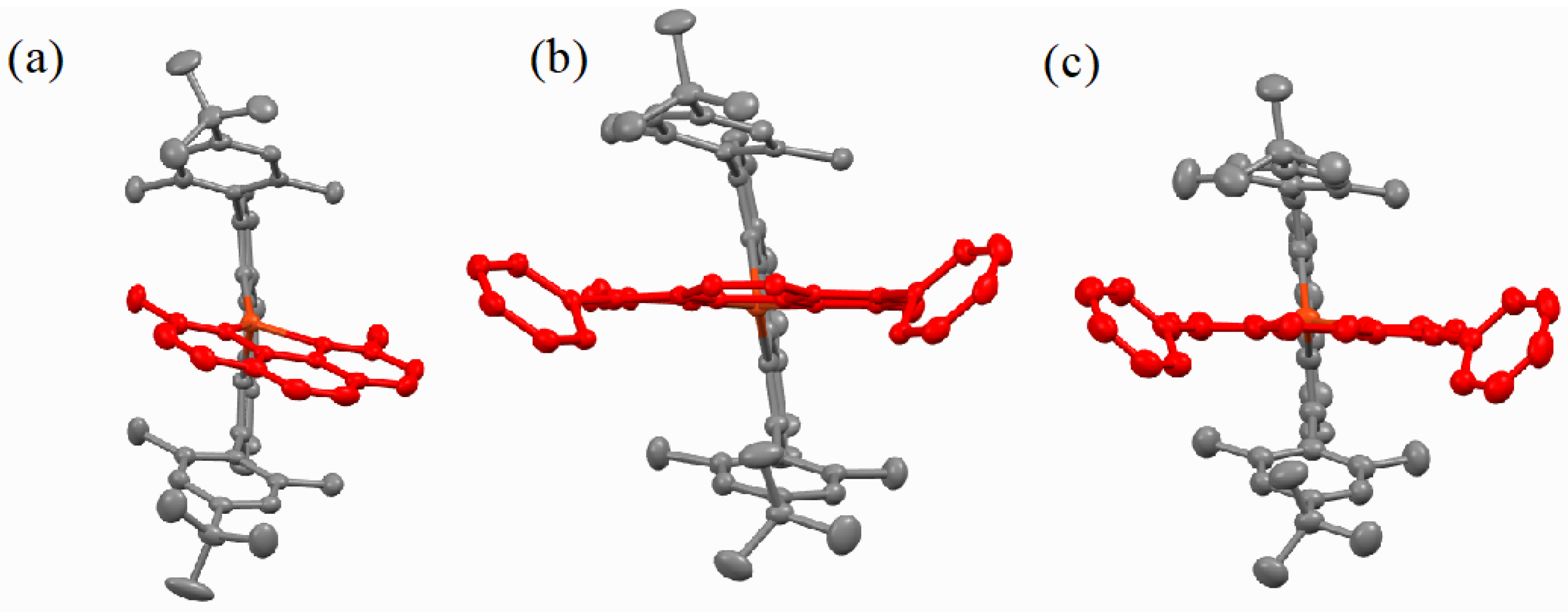
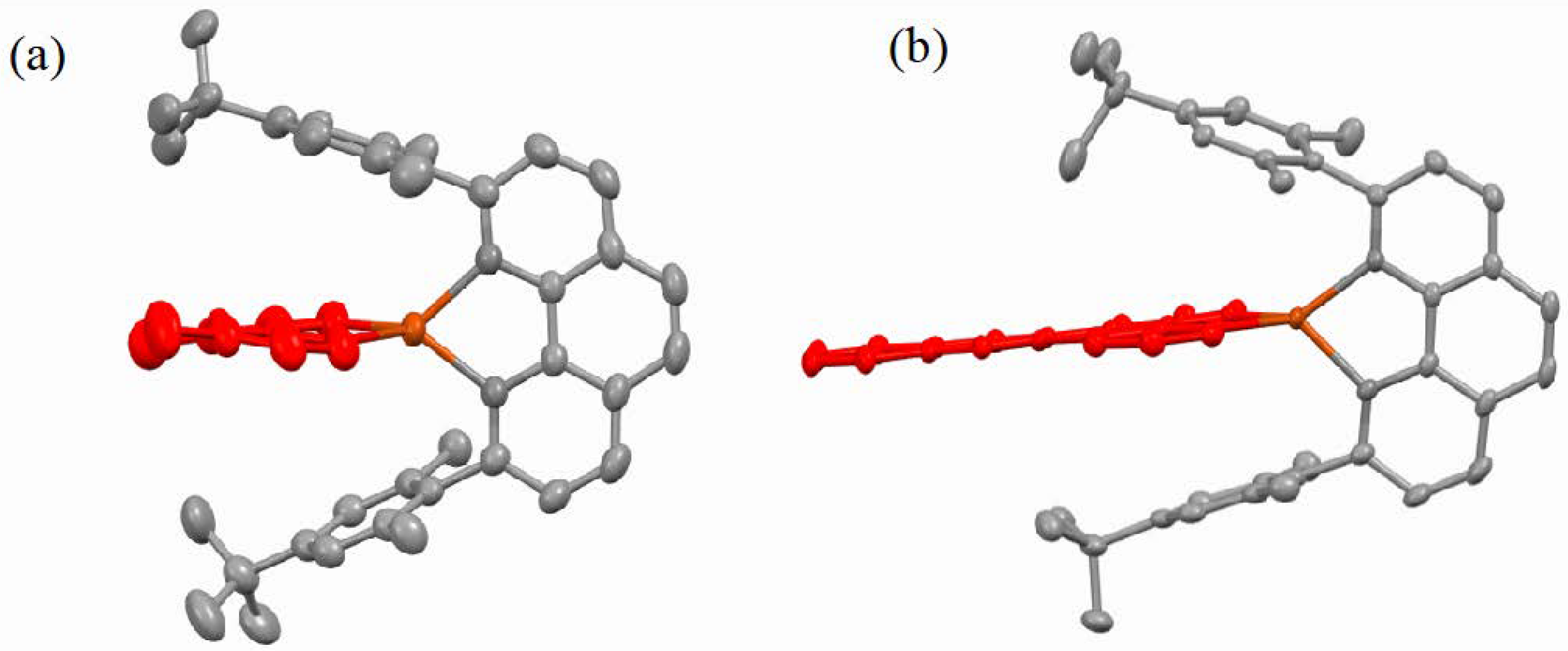
2.2. Structural Properties
3. Discussion
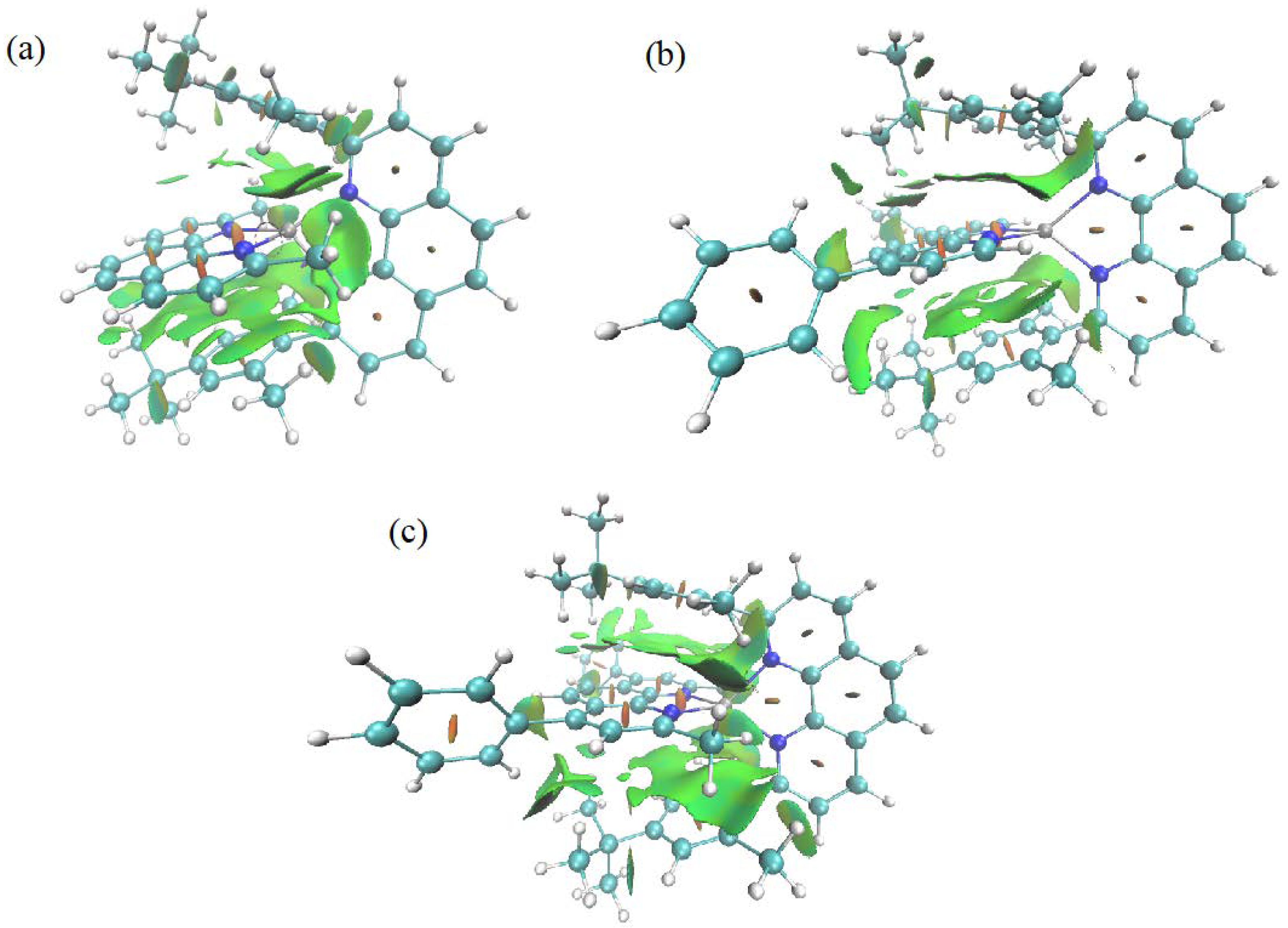
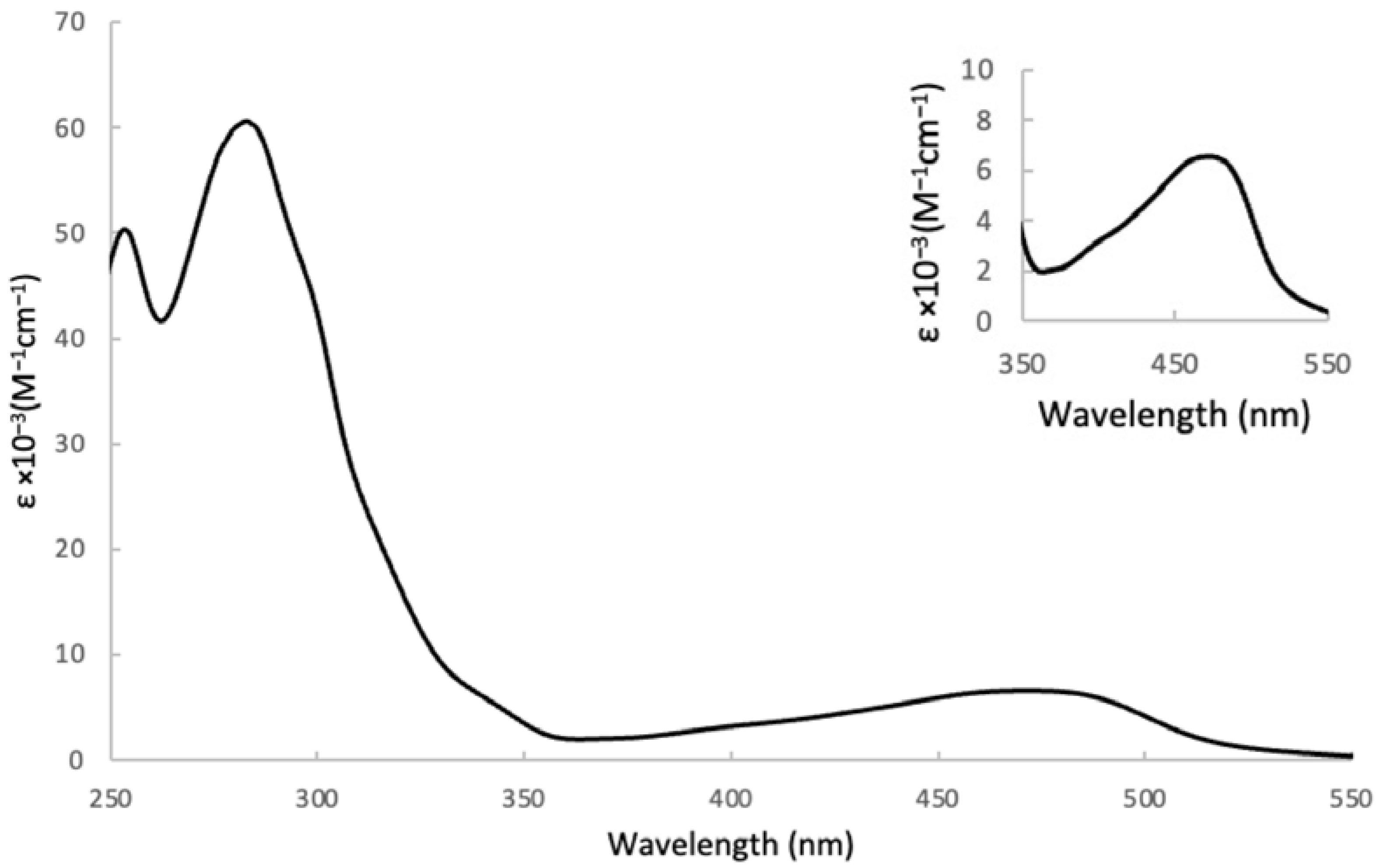
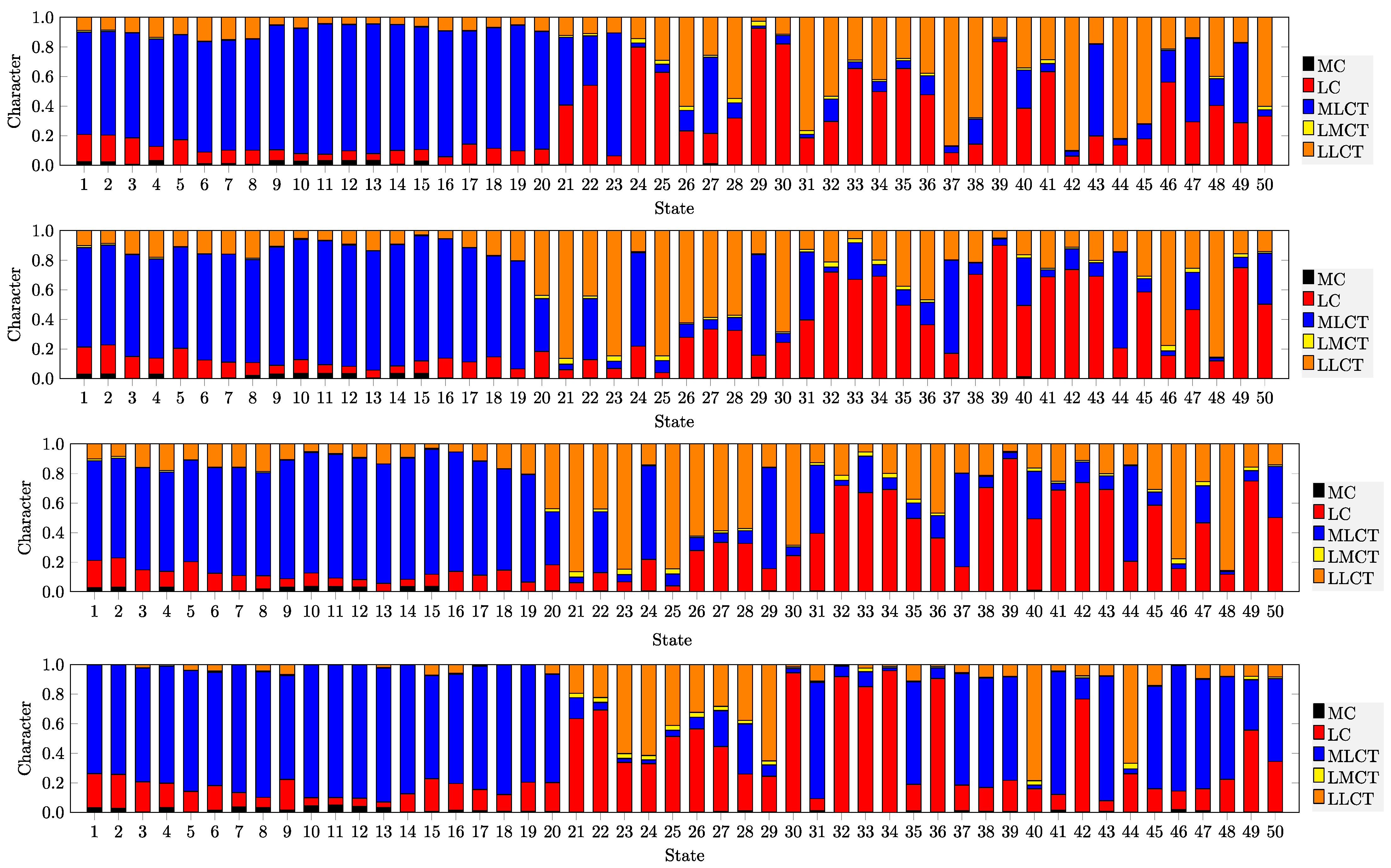
3.1. Electronic Properties
3.2. Photoluminescent Properties
4. Materials and Methods
DFT Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fujishama, K.; Honda, A. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, B.; Grätzel, M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. [Google Scholar] [CrossRef]
- Alonso-Vante, N.; Nierengarten, J.-F.; Sauvage, J.-P. Spectral Sensitization of Large-band-gap Semiconductors (Thin Films and Ceramics) by a Carboxylated Bis(1,10-Phenanthroline)copper(l) Complex. J. Chem. Soc. Dalton Trans. 1994, 11, 1649–1654. [Google Scholar] [CrossRef]
- Kern, J.-M.; Sauvage, J.-P. Photoassisted C-C coupling via electron transfer to benzylic halides by a bis(diimine) copper(I) complex. J. Chem. Soc. Chem. Commun. 1987, 8, 546–548. [Google Scholar] [CrossRef]
- Paria, S.; Reiser, O. Copper in Photocatalysis. ChemCatChem 2014, 6, 2477–2483. [Google Scholar] [CrossRef]
- Housecroft, C.E.; Constable, E. Solar energy conversion using first row d-block metal coordination compound sensitizers and redox mediators. Chem. Sci. 2022, 13, 1225–1262. [Google Scholar] [CrossRef]
- Wegeberg, C.; Wenger, O. Luminescent first-row transition metal complexes. JACS Au 2021, 1, 1860–1876. [Google Scholar] [CrossRef]
- McMillin, D.R.; Buckner, M.T.; Tae Ahn, B. A Light-induced redox reaction of Bis(2,9-dimethyl-l,10-phenanthroIine)copper(I). Inorg. Chem. 1977, 16, 943–945. [Google Scholar] [CrossRef]
- McMillin, D.R.; Kirchhoff, J.R.; Goodwin, K.V. Exciplex quenching of photo-excited copper complexes. Coord. Chem. Rev. 1985, 64, 83–92. [Google Scholar] [CrossRef]
- Dietrich-Buchecker, C.O.; Marnot, P.A.; Sauvage, J.-P. Direct synthesis of disubstituted aromatic polyimine chelates. Tetrahedron Lett. 1982, 23, 5291–5294. [Google Scholar] [CrossRef]
- Dietrich-Buchecker, C.O.; Marnot, P.A.; Sauvage, J.P.; Kirchhoff, J.R.; McMillin, D.R. Bis(2,9-diphenyl-1,10-phenanthroline)copper(l): A copper complex with a long-lived charge-transfer excited state. J. Chem. Soc. Chem. Commun. 1983, 9, 513–515. [Google Scholar] [CrossRef]
- Ichinaga, A.K.; Kirchhoff, J.R.; McMillin, D.R.; Dietrich-Buchecker, C.O.; Marnot, P.A.; Sauvage, J.-P. Charge-transfer Absorption and emission of Cu(NN)2+ systems. Inorg. Chem. 1987, 26, 4290–4292. [Google Scholar] [CrossRef]
- Miller, M.T.; Gantzel, P.K.; Karpishin, T.B. Effects of sterics and electronic delocalization on the photophysical, structural, and electrochemical properties of 2,9-disubstituted-1,10-phenanthroline copper(I) complexes. Inorg. Chem. 1999, 38, 3414–3422. [Google Scholar] [CrossRef]
- Miller, M.T.; Gantzel, P.K.; Karpishin, T.B. A photoluminescent copper(I) complex with an exceptionnally high CuII/CuI redox potential: [Cu(bfp)2]+ (bfp = 2,9-bis-(trifluoromethyl)-1,10-phenanthroline). Angew. Chem. Int. Ed. 1998, 37, 1556–1558. [Google Scholar] [CrossRef]
- Schmittel, M.; Ganz, A. Stable mixed phenanthroline copper(I) complexes. Key building blocks for supramolecular coordination chemistry. Chem. Commun. 1997, 11, 999–1000. [Google Scholar] [CrossRef]
- Cuttell, D.G.; Kuang, S.-M.; Fanwick, P.E.; McMillin, M.; Walton, R.A. Simple Cu(I) complexes with unprecedented excited-state lifetimes. J. Am. Chem. Soc. 2002, 124, 6–7. [Google Scholar] [CrossRef]
- Kalsani, V.; Schmittel, M.; Listorti, A.; Accorsi, G.; Armaroli, N. Novel phenanthroline ligands and their kinetically locked copper(I) complexes with unexpected photophysical properties. Inorg. Chem. 2006, 45, 2061–2067. [Google Scholar] [CrossRef]
- Felder, D.; Nierengarten, J.-F.; Barigelletti, F.; Ventura, B.; Armaroli, N. Highly luminescent Cu(I)-phenanthroline complexes in rigid matrix and temperature dependence of the photophysical properties. J. Am. Chem. Soc. 2001, 123, 6291–6299. [Google Scholar] [CrossRef]
- Gandhi, B.A.; Green, O.; Burstyn, J.N. Facile oxidation-based synthesis of sterically encumbered four-coordinate bis(2,9-di-tert-butyl-1,10-phenanthroline)copper(I) and related three-coordinate copper(I) complexes. Inorg. Chem. 2007, 46, 3816–3825. [Google Scholar] [CrossRef]
- Green, O.; Gandhi, B.A.; Burstyn, J.N. Photophysical characteristics and reactivity of Bis(2,9-di-tert-butyl-1,10-phenanthroline) copper(I). Inorg. Chem. 2009, 48, 5704–5714. [Google Scholar] [CrossRef]
- Nicholls, T.P.; Caporale, C.; Massi, M.; Gardiner, M.G.; Bissember, A.E. Synthesis and characterization of homoleptic 2,9-diaryl-1,10-phenanthroline copper(I) complexes: Influencing selectivity in photoredox-catalysed atom-transfer radical addition reactions. Dalton Trans. 2019, 48, 7290–7301. [Google Scholar] [CrossRef]
- McCusker, C.E.; Castellano, F.N. Design of a long-lifetime, earth-abundant, aqueous compatible Cu(I) photosensitizer using cooperative steric effects. Inorg. Chem. 2013, 52, 8114–8120. [Google Scholar] [CrossRef] [PubMed]
- Garakyaraghi, S.; Crapps, P.D.; McCusker, C.E.; Castellano, F.N. Cuprous phenanthroline MLCT chromophore featuring synthetically tailored photophysics. Inorg. Chem. 2016, 55, 10628–10636. [Google Scholar] [CrossRef]
- Khnayzer, R.S.; McCusker, C.E.; Olaiya, B.S.; Castellano, F.N. Robust cuprous phenanthroline sensitizer for solar hydrogen photocatalysis. J. Am. Chem. Soc. 2013, 135, 14068–14070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holler, M.; Delavaux-Nicot, B.; Nierengarten, J.-F. Topological and steric constraints to stabilize heteroleptic copper(I) complexes combining phenanthroline ligands and phosphines. Chem. Eur. J. 2019, 25, 4543–4550. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, L.; Phelan, B.T.; Sprague-Klein, E.A.; Roisnel, T.; Blart, E.; Gourlaouen, C.; Chen, L.X.; Pellegrin, Y. Bulky and stable copper(I)-Phenanthroline complex: Impact of steric strain and symmetry on the excited-state properties. Inorg. Chem. 2022, 61, 7296–7307. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, L.; Blart, E.; Rebilly, J.-N.; Coupeau, M.; Allain, M.; Roisnel, T.; Quarré de Verneuil, A.; Gourlaouen, C.; Daniel, C.; Pellegrin, Y. Non-symmetrical sterically challenged phenanthroline ligands and their homoleptic copper(I) complexes with improved excited-state properties. Chem. Eur. J. 2020, 26, 11887–11899. [Google Scholar] [CrossRef] [PubMed]
- Sandroni, M.; Kayanuma, M.; Rebarz, M.; Akdas-Kilig, H.; Pellegrin, Y.; Blart, E.; Le Bozec, H.; Daniel, C.; Odobel, F. Heteroleptic diimine copper(I) complexes with large extinction coefficients: Synthesis, quantum chemistry calculations and physico-chemical properties. Dalton Trans. 2013, 42, 14628–14638. [Google Scholar] [CrossRef] [Green Version]
- Schmittel, M.; Lüning, U.; Meder, M.; Ganz, A.; Michel, C.; Herderich, M. Synthesis of sterically encumbered 2,9-diaryl substituted phenanthrolines. Key building blocks for the preparation of mixed (bis-heteroleptic) phenanthroline copper(I) complexes. Heterocycl. Commun. 1997, 3, 493–498. [Google Scholar] [CrossRef]
- Schmittel, M.; Ganz, A.; Fenske, D.; Herderich, M. Heteroleptic silver(I) and zinc(II) bis(phenanthroline complexes. J. Chem. Soc. Dalton Trans. 2000, 3, 353–359. [Google Scholar] [CrossRef]
- Huang, J.; Buyukcakir, O.; Mara, M.W.; Coskun, A.; Dimitrijevic, N.M.; Barin, G.; Kokhan, O.; Stickrath, A.B.; Ruppert, R.; Tiede, D.M.; et al. Highly Efficient Ultrafast Electron Injection from the Singlet MLCT Excited State of Copper(I) Diimine Complexes to TiO2 Nanoparticles. Angew. Chem. Int. Ed. 2012, 51, 12711–12715. [Google Scholar] [CrossRef] [PubMed]
- Mara, M.W.; Bowman, D.N.; Buyukcakir, O.; Shelby, M.L.; Haldrup, K.; Huang, J.; Harpham, M.R.; Stickrath, A.B.; Zhang, X.; Stoddart, J.F.; et al. Electron injection from copper diimine sensitizers into TiO2: Structural effects and their implications for solar energy conversion devices. J. Am. Chem. Soc. 2015, 137, 9670–9684. [Google Scholar] [CrossRef] [PubMed]
- Eberhart, M.S.; Phelan, B.T.; Niklas, J.; Sprague-Klein, E.A.; Kaphan, D.M.; Gosztola, D.J.; Chen, L.X.; Tiede, D.M.; Poluektov, O.G.; Mulfort, K.L. Surface immobilized copper(I) diimine photosensitizers as molecular probes for elucidating the effects of confinement at interfaces for solar energy conversion. Chem. Commun. 2020, 56, 12130–12133. [Google Scholar] [CrossRef] [PubMed]
- Appleton, J.L.; Silber, V.; Karmazin, L.; Bailly, C.; Chambron, J.-C.; Weiss, J.; Ruppert, R. A new phenanthroline ligand and the spontaneous resolution of its homoleptic copper(I) complex. Eur. J. Org. Chem. 2020, 2020, 7320–7326. [Google Scholar] [CrossRef]
- Czerwieniec, R.; Yersin, H. Diversity of Copper(I) Complexes Showing Thermally Activated Delayed Fluorescence: Basis Photophysical Analysis. Inorg. Chem. 2015, 54, 4322–4327. [Google Scholar] [CrossRef]
- Friedman, A.; Chambron, J.-C.; Sauvage, J.-P.; Turro, N.J.; Barton, J. A molecular light switch for DNA: Ru(bpy)2(dppz)2+. J. Am. Chem. Soc. 1990, 112, 4960–4962. [Google Scholar] [CrossRef]
- Zeglis, B.M.; Pierre, V.C.; Barton, J.K. Metallo-intercalators and metallo-insertors. Chem. Commun. 2007, 44, 4565–4579. [Google Scholar] [CrossRef] [Green Version]
- Kohler, L.; Hadt, R.G.; Hayes, D.; Chen, L.X.; Mulfort, K.L. Synthesis, structure, and excited state kinetics of heteroleptic Cu(I) complexes with a new sterically demanding phenanthroline ligand. Dalton Trans. 2017, 46, 10388–13100. [Google Scholar] [CrossRef] [Green Version]
- Garakyaraghi, S.; McCusker, C.E.; Khan, S.; Koutnik, P.; Bui, A.T.; Castellano, F.N. Enhancing the visible-light absorption and excited-state properties of Cu(I) MLCT excited states. Inorg. Chem. 2018, 57, 2296–2307. [Google Scholar] [CrossRef]
- M86-EXX229V1 APEX3 User Manual; BRUKER AXS Inc.: Madison, WI, USA, 2016.
- Sheldrick, G.M. SHELXT - Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional thermochemistry. 3. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Anichtchenko, D.; Errandonea, D. Comparative study of the compressibility of M3V2O8 (M = Cd, Zn, Mg, Ni) orthovanadates. Crystals 2022, 12, 1544. [Google Scholar] [CrossRef]
- ADF, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. Available online: https://www.scm.com/doc/ADF/index.html (accessed on 1 June 2019).
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Laurent, A.D.; Jacquemin, D. TD-DFT benchmarks: A review. Int. J. Quantum Chem. 2013, 113, 2019–2039. [Google Scholar] [CrossRef]
- Plasser, F. TheoDORE: A toolbox for a detailed and automated analysis of electronic excited state computations. J. Chem. Phys. 2020, 152, 8418. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Garcia, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W.T. NCIPLOT: A program for plotting noncovalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental | C2 | C3 | C4 | [Cu(ditBuphen)2]+ |
|---|---|---|---|---|
| Cu-N1 (Å) | 2.015 | 2.016 | 2.076 | 2.096 |
| Cu-N2 (Å) | 2.083 | 2.063 | 2.049 | 2.129 |
| Cu-N3 (Å) | 1.996 | 2.038 | 2.035 | 2.103 |
| Cu-N4 (Å) | 2.085 | 2.023 | 2.050 | 2.120 |
| X1-Cu-X2° | 148 | 169 | 173 | 175 |
| C1-X1-X2-C2° | 51 | 80 | 81 | 80 |
| Computational | C2 | C3 | C4 |
|---|---|---|---|
| Cu-N1 (Å) | 2.040 | 2.056 | 2.090 |
| Cu-N2 (Å) | 2.111 | 2.078 | 2.062 |
| Cu-N3 (Å) | 2.033 | 2.056 | 2.069 |
| Cu-N4 (Å) | 2.097 | 2.052 | 2.045 |
| X1-Cu-X2° | 154 | 174 | 169 |
| C1-X1-X2-C2° | 79 | 83 | 77 |
| Experimental Parameters | C6 | C8 |
|---|---|---|
| Cu-N1 (Å) | 2.051 | 2.047 |
| Cu-N2 (Å) | 2.044 | 2.013 |
| Cu-N3 (Å) | 2.031 | 2.035 |
| Cu-N4 (Å) | 2.043 | 2.022 |
| X1-Cu-X2° | 175 | 172 |
| C1-X1-X2-C2° | 94 | 98 |
| Computational Parameters | C6 | C8 |
|---|---|---|
| Cu-N1 (Å) | 2.065 | 2.055 |
| Cu-N2 (Å) | 2.071 | 2.073 |
| Cu-N3 (Å) | 2.037 | 2.061 |
| Cu-N4 (Å) | 2.064 | 2.057 |
| X1-Cu-X2° | 170.3 | 175.3 |
| |C1-X1-X2-C2|° | 88.3 | 78.6 |
| Anodic Peak Potential (Volts) | Cathodic Peak Potential (Volts) | Potential Difference (pd) | |
|---|---|---|---|
| C1 | 0.38 | 0.26 | 0.12 |
| C2 | 0.47 | 0.36 | 0.11 |
| C3 | 0.51 | 0.28 | 0.23 |
| C4 | 0.60 | 0.36 | 0.24 |
| C5 | 0.23 | 0.13 | 0.10 |
| C6 | 0.32 | 0.065 | 0.26 |
| C7 | 0.42 | 0.076 | 0.34 |
| C8 | 0.54 | 0.32 | 0.22 |
| Complex | C1 | C2 | C3 | C4 | C5 | C6 | C7 | C8 |
|---|---|---|---|---|---|---|---|---|
| λmax (nm) | 276 461 | 277 468 | 285 496 | 286 477 | 279 470 | 275 475 | 276 478 | 275 476 |
| ε × 10−3 (M−1cm−1) | 47,900 6700 | 56,100 5600 | 69,200 8100 | 59,300 6500 | 57,200 6300 | 43,600 6300 | 43,500 6500 | 81,200 6800 |
| C2 | C4 | |
|---|---|---|
| Lifetime (ns) | 58 | 44 |
| Emission (nm) | 680 | 697 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Appleton, J.L.; Gourlaouen, C.; Ruppert, R. Remote Steric Control of the Tetrahedral Coordination Geometry around Heteroleptic Copper(I) Bis(Diimine) Complexes. Molecules 2023, 28, 983. https://doi.org/10.3390/molecules28030983
Appleton JL, Gourlaouen C, Ruppert R. Remote Steric Control of the Tetrahedral Coordination Geometry around Heteroleptic Copper(I) Bis(Diimine) Complexes. Molecules. 2023; 28(3):983. https://doi.org/10.3390/molecules28030983
Chicago/Turabian StyleAppleton, Jordan L., Christophe Gourlaouen, and Romain Ruppert. 2023. "Remote Steric Control of the Tetrahedral Coordination Geometry around Heteroleptic Copper(I) Bis(Diimine) Complexes" Molecules 28, no. 3: 983. https://doi.org/10.3390/molecules28030983
APA StyleAppleton, J. L., Gourlaouen, C., & Ruppert, R. (2023). Remote Steric Control of the Tetrahedral Coordination Geometry around Heteroleptic Copper(I) Bis(Diimine) Complexes. Molecules, 28(3), 983. https://doi.org/10.3390/molecules28030983