
Theoretical Study on the Copper-Catalyzed ortho-Selective C-H Functionalization of Naphthols with α-Phenyl-α-Diazoesters


Abstract
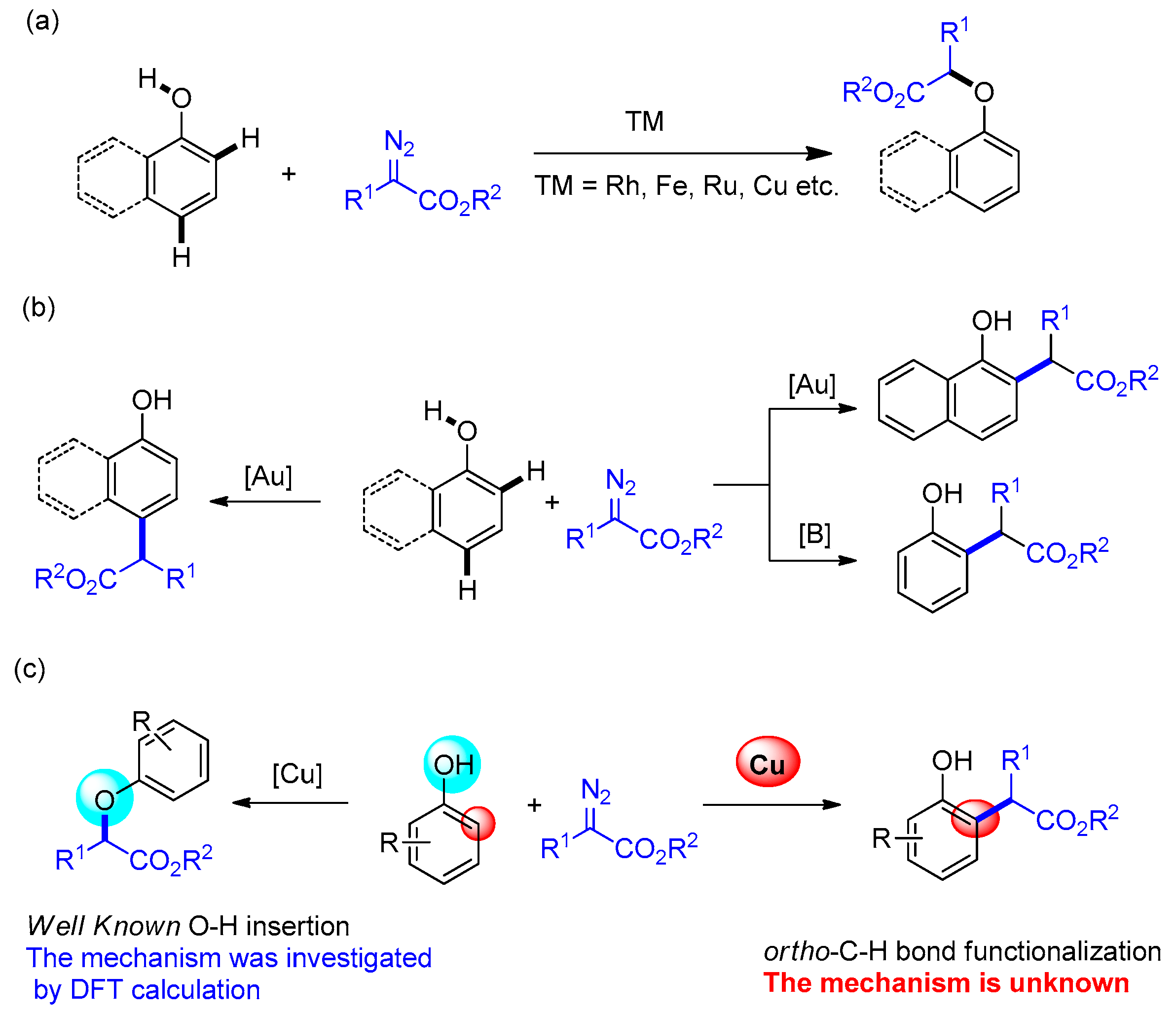
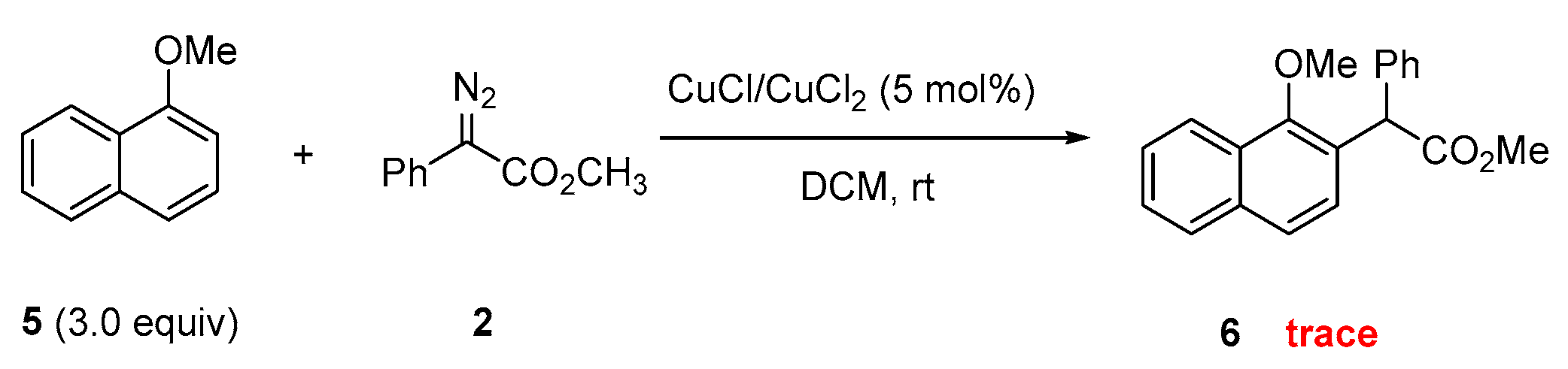
:1. Introduction
2. Results and Discussion
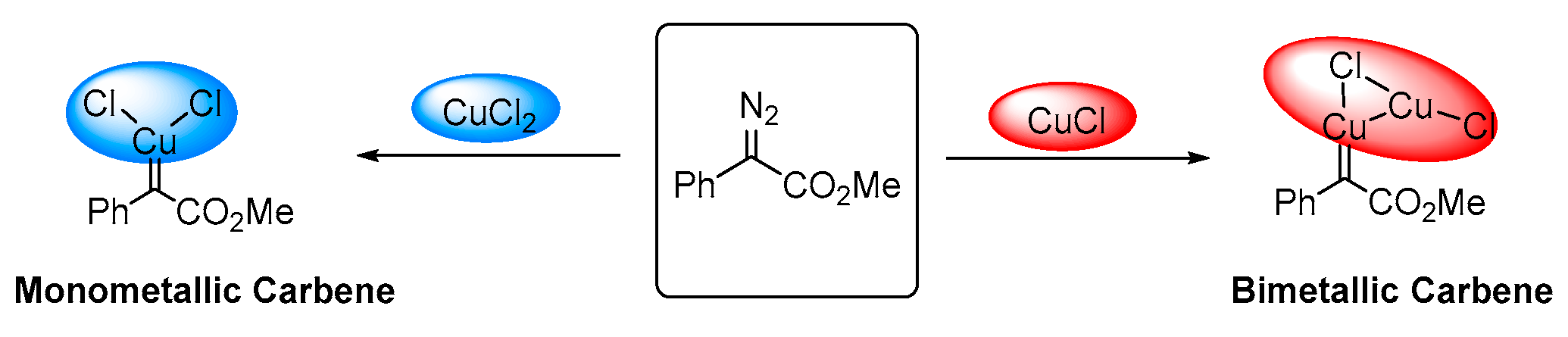
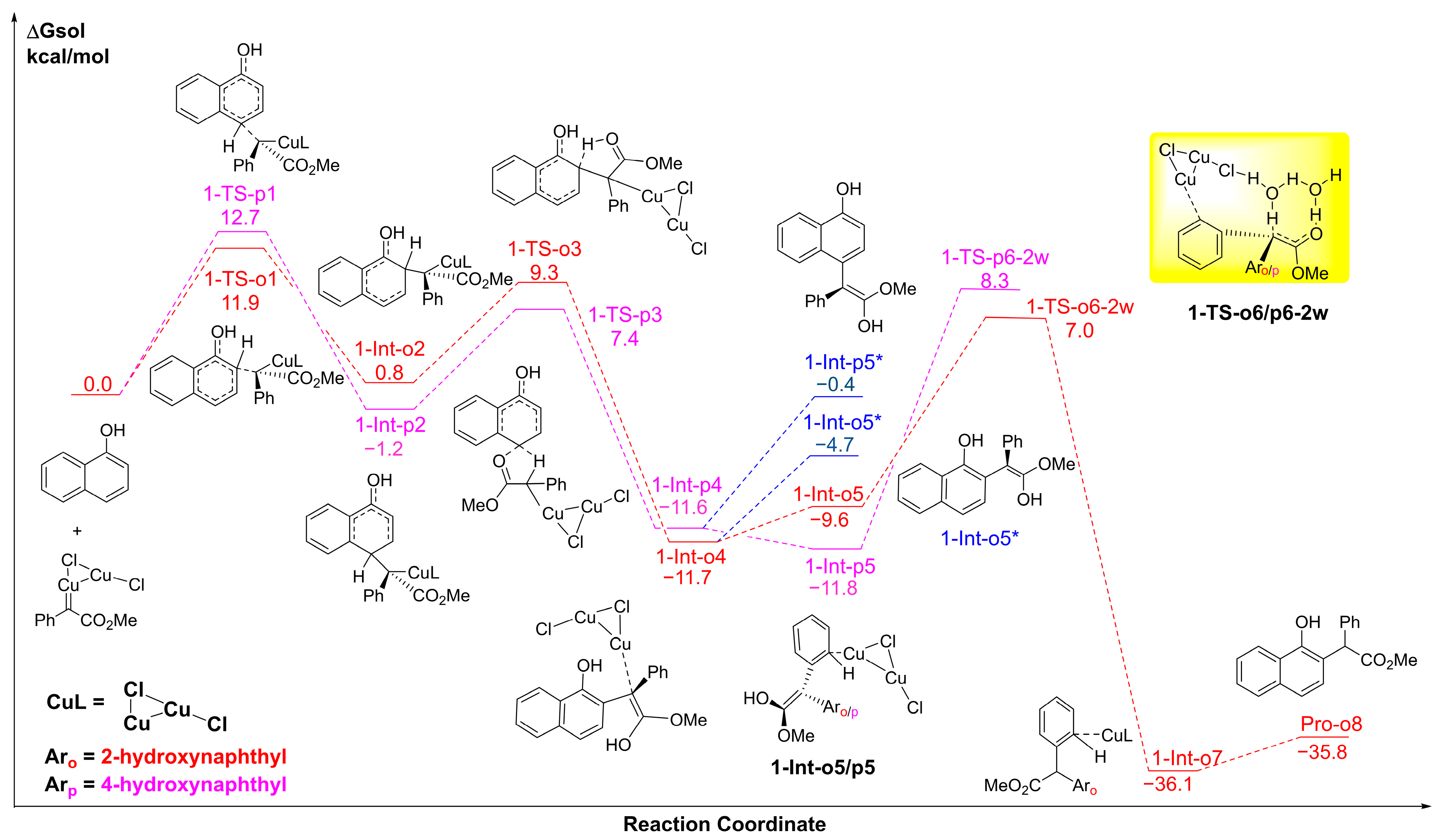
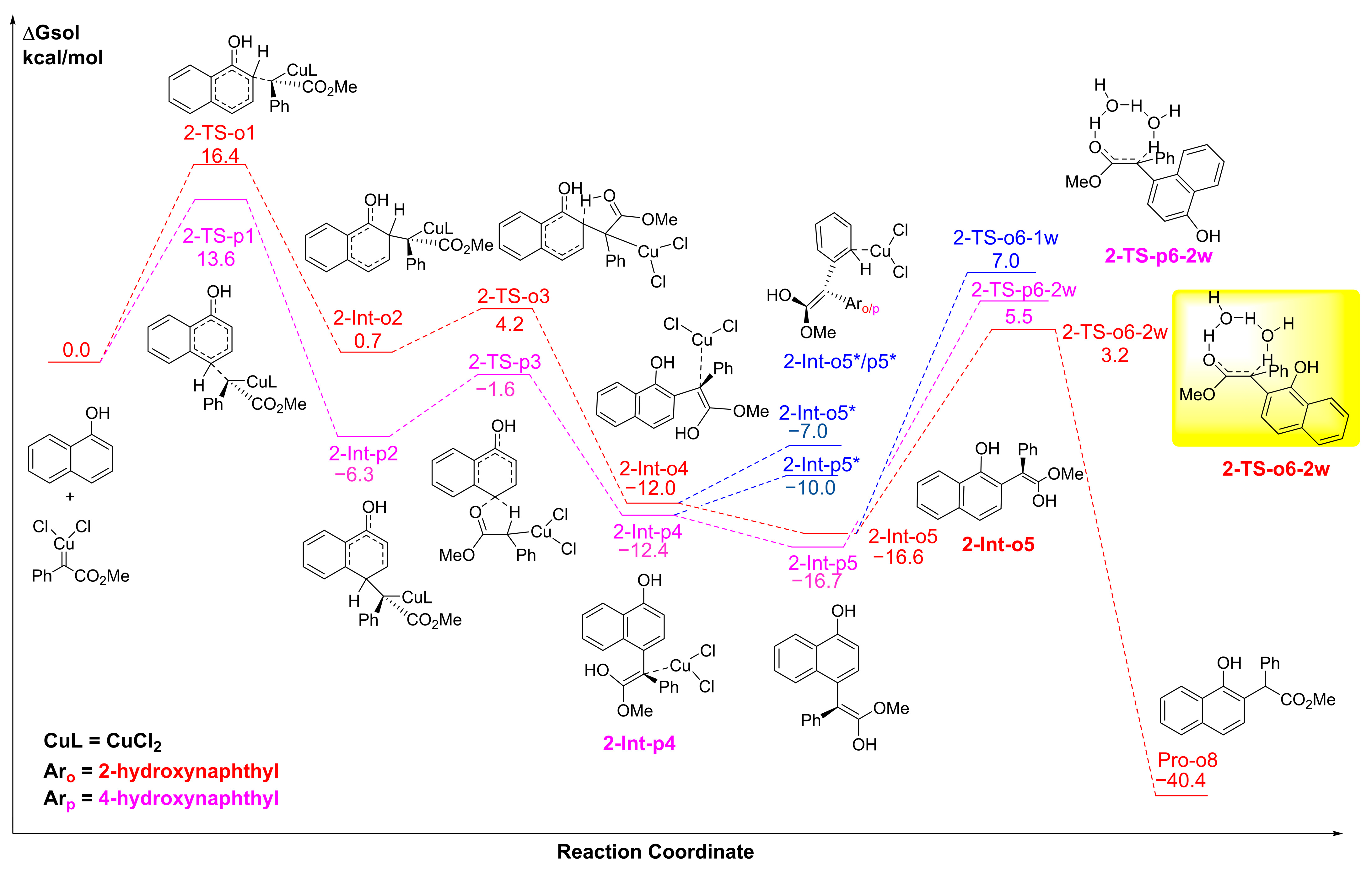
2.1. The C-H Insertion of Naphthols Catalyzed by the (CuCl)2 Dimer
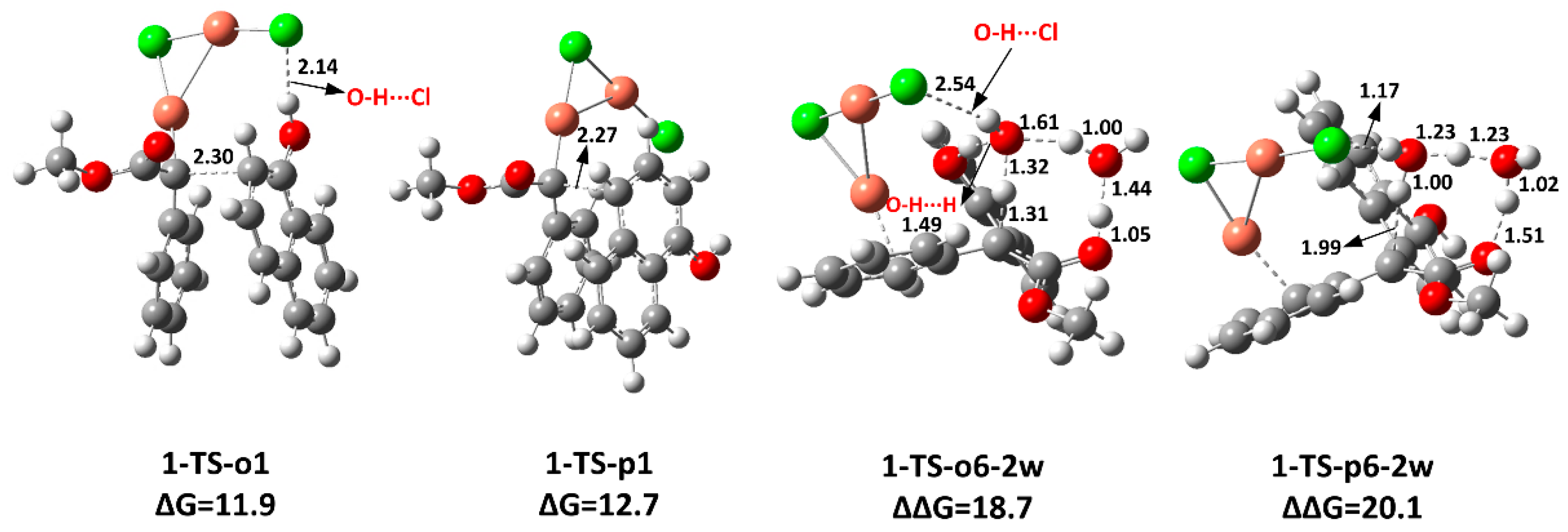
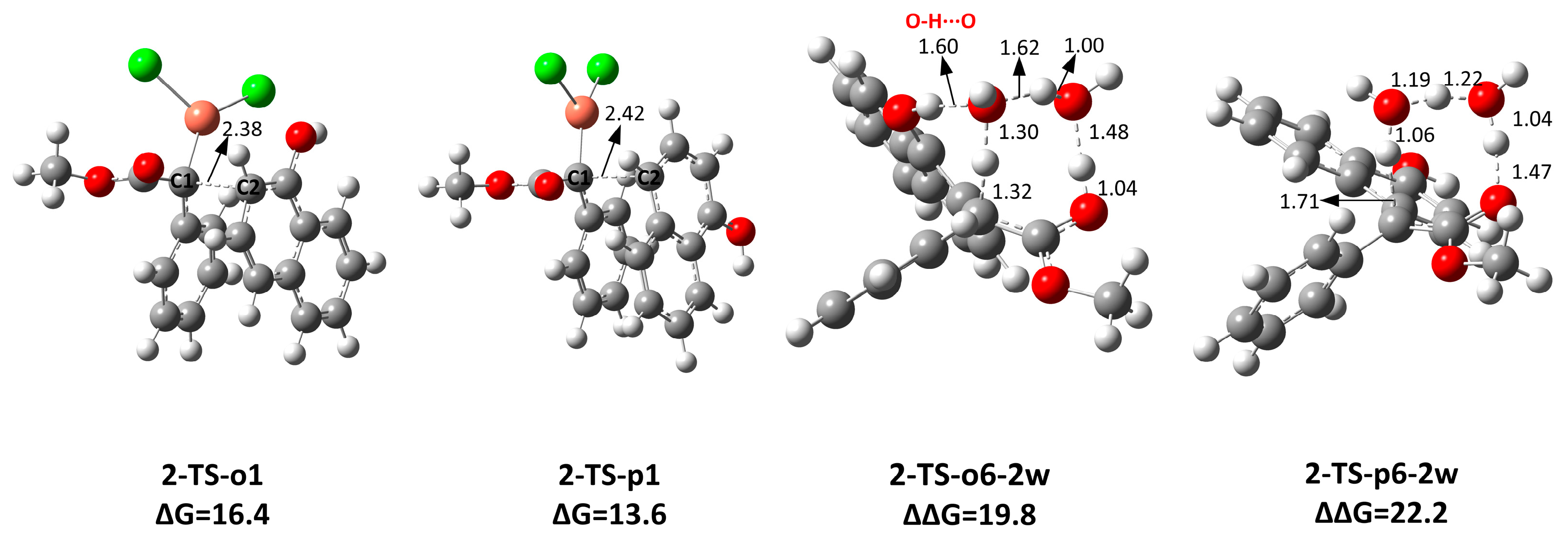
2.2. The Key H-Bond Interactions Formed with (CuCl)2
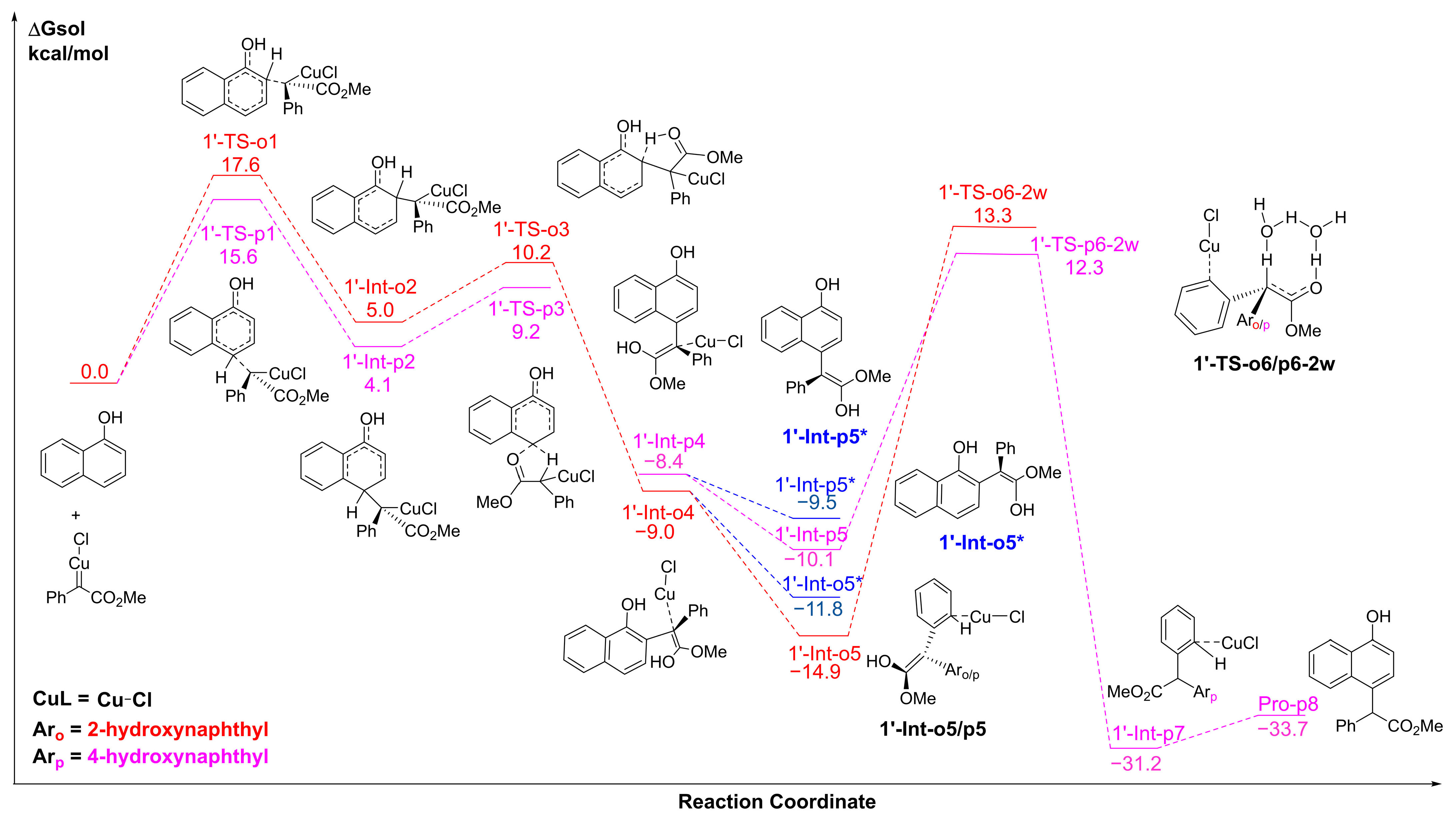
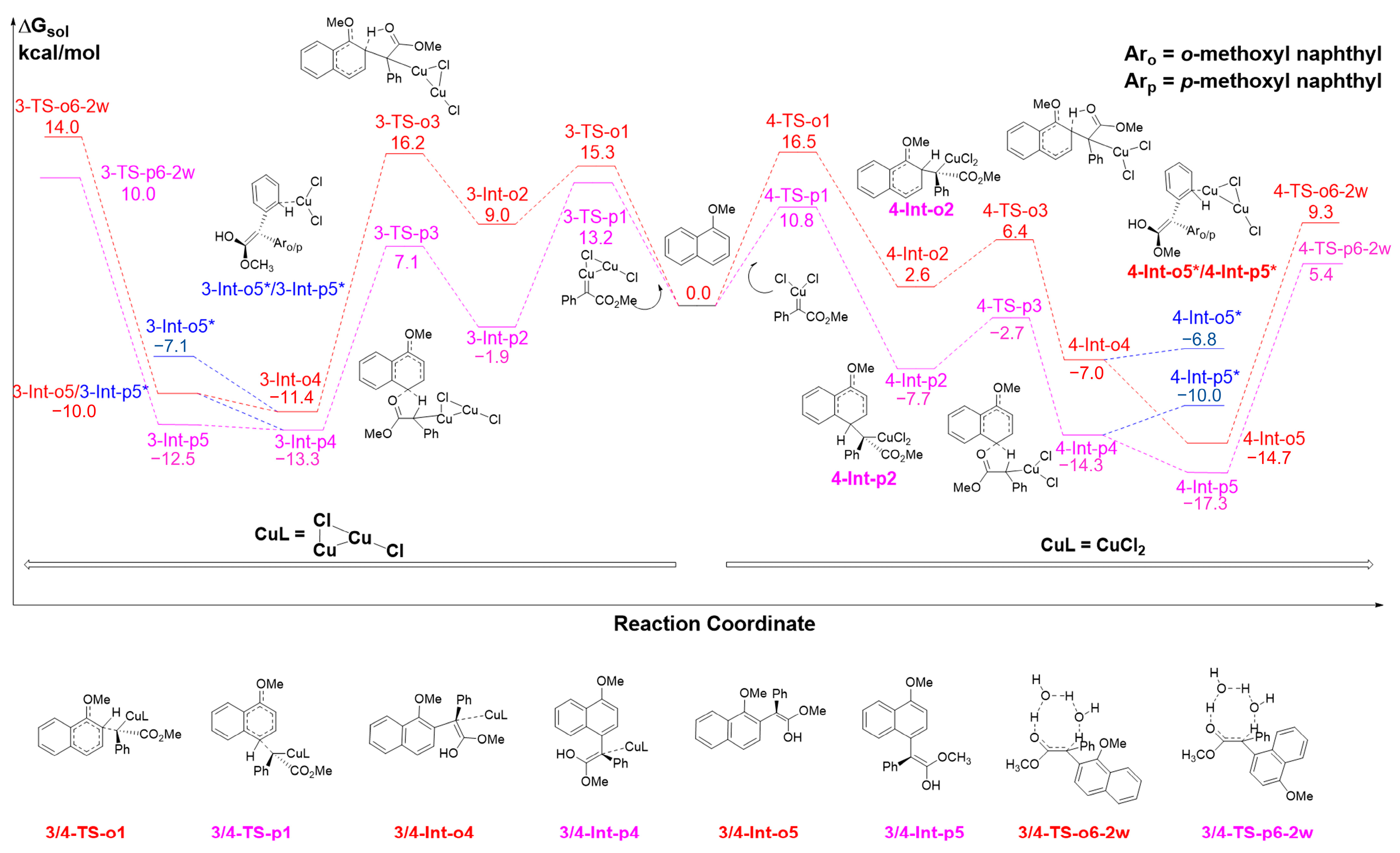
2.3. The C-H Insertion of Naphthols Catalyzed by the CuCl Monomer
2.4. The C-H Insertion of Naphthols Catalyzed by the CuCl2 Monomer
2.5. Experimental Reactivity of 1-Methoxynaphthalene Catalyzed by Cu Catalysts
3. Materials and Methods
3.1. General Imformation
3.2. Synthetic Procedure for the Reaction of 1-Methoxynaphthalene and Diazoester
3.3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Doyle, M.P. Catalytic Methods for Metal Carbene Transformations. Chem. Rev. 1986, 86, 919–939. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Denton, J.R. Application of donor/acceptor-carbenoids to the synthesis of natural products. Chem. Soc. Rev. 2009, 38, 3061–3071. [Google Scholar] [CrossRef] [PubMed]
- Maas, G. New syntheses of diazo compounds. Angew. Chem. Int. Ed. 2009, 48, 8186–8195. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.; Miel, H.; Ring, A.; Slattery, C.N.; Maguire, A.R.; McKervey, M.A. Modern Organic Synthesis with α-Diazocarbonyl Compounds. Chem. Rev. 2015, 115, 9981–10080. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, J. Gold-catalyzed transformations of α-diazocarbonyl compounds: Selectivity and diversity. Chem. Soc. Rev. 2016, 45, 506–516. [Google Scholar] [CrossRef]
- Zhu, D.; Chen, L.; Fan, H.; Yao, Q.; Zhu, S. Recent progress on donor and donor-donor carbenes. Chem. Soc. Rev. 2020, 49, 908–950. [Google Scholar] [CrossRef]
- Yadagiri, D.; Anbarasan, P. Catalytic Functionalization of Metallocarbenes Derived from α-Diazocarbonyl Compounds and Their Precursors. Chem. Rec. 2021, 21, 3872–3883. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Qiu, H.; Liu, L.; Zhang, J. Gold-Catalyzed Construction of Two Adjacent Quaternary Stereocenters via Sequential C-H Functionalization and Aldol Annulation. Chem. Commun. 2016, 52, 2257–2260. [Google Scholar] [CrossRef]
- Ma, B.; Wu, Z.; Huang, B.; Liu, L.; Zhang, J. Gold-catalysed facile access to indene scaffold via sequential C-H functionalization and 5-endo-dig carbocyclization. Chem. Commun. 2016, 52, 9351–9354. [Google Scholar] [CrossRef]
- Ma, B.; Wu, J.; Liu, L.; Zhang, J. Gold-catalyzed para-selective C-H bond alkylation of benzene derivatives with donor/acceptor-substituted diazo compounds. Chem. Commun. 2017, 53, 10164–10167. [Google Scholar] [CrossRef]
- Li, Y.; Tang, Z.; Zhang, J.; Liu, L. Gold-Catalyzed Intermolecular [4+1] Spiroannulation via Site Selective Aromatic C(sp2)-H Functionalization and Dearomatization of Phenol Derivatives. Chem. Commun. 2020, 56, 8202–8205. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Li, G.; Zhang, J.; Liu, L. Iron-catalysed chemo- and ortho-selective C-H bond functionalization of phenols with α-aryl-α-diazoacetates. Org. Chem. Front. 2021, 8, 3770–3775. [Google Scholar] [CrossRef]
- Liu, X.; Tang, Z.; Li, Z.; Li, M.; Xu, L.; Liu, L. Modular and stereoselective synthesis of tetrasubstituted vinyl sulfides leading to a library of AIEgens. Nat. Commun. 2021, 12, 7298. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, M.; Dong, K.; Peng, S.; Liu, L. Highly Stereoselective Synthesis of Tetrasubstituted Vinyl Selenides via Rhodium-Catalyzed [1,4]-Acyl Migration of Selenoesters and Diazo Compounds. Org. Lett. 2022, 24, 2175–2180. [Google Scholar] [CrossRef]
- Liu, X.; Tang, Z.; Si, Z.-Y.; Zhang, Z.; Zhao, L.; Liu, L. Enantioselective para-C(sp2)-H Functionalization of Alkyl Benzene Derivatives via Cooperative Catalysis of Gold/Chiral Brønsted Acid. Angew. Chem. Int. Ed. 2022, 61, e202208874. [Google Scholar]
- Dong, K.; Liu, X.-S.; Wei, X.; Zhao, Y.; Liu, L. Borane-catalysed S–H insertion reaction of thiophenols and thiols with α-aryl-α-diazoesters. Green Synth. Catal. 2021, 2, 385–388. [Google Scholar] [CrossRef]
- Maas, G. Ruthenium-catalysed carbenoid cyclopropanation reactions with diazo compounds. Chem. Soc. Rev. 2004, 33, 183–190. [Google Scholar] [CrossRef]
- Morandi, B.; Carreira, E.M. Iron-catalyzed cyclopropanation in 6M KOH with in situ generation of diazomethane. Science 2012, 335, 1471–1474. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Beckwith, R.E. Catalytic enantioselective C-H activation by means of metal-carbenoid-induced C-H insertion. Chem. Rev. 2003, 103, 2861–2904. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Manning, J.R. Catalytic C-H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451, 417–424. [Google Scholar] [CrossRef]
- Diaz-Requejo, M.M.; Perez, P.J. Coinage metal catalyzed C-H bond functionalization of hydrocarbons. Chem. Rev. 2008, 108, 3379–3394. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P.; Duffy, R.; Ratnikov, M.; Zhou, L. Catalytic carbene insertion into C-H bonds. Chem. Rev. 2010, 110, 704–724. [Google Scholar] [CrossRef] [PubMed]
- Wencel-Delord, J.; Droge, T.; Liu, F.; Glorius, F. Towards mild metal-catalyzed C-H bond activation. Chem. Soc. Rev. 2011, 40, 4740–4761. [Google Scholar] [CrossRef]
- Zheng, C.; You, S. Recent development of direct asymmetric functionalization of inert C–H bonds. RSC Adv. 2014, 4, 6173–6214. [Google Scholar] [CrossRef]
- Arockiam, P.B.; Bruneau, C.; Dixneuf, P.H. Ruthenium(II)-catalyzed C-H bond activation and functionalization. Chem. Rev. 2012, 112, 5879–5918. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Liu, L.; Zhang, J. Gold-Catalyzed Site-Selective C−H Bond Functionalization with Diazo Compounds. Asian J. Org. Chem. 2018, 7, 2015–2025. [Google Scholar] [CrossRef]
- Ma, B.; Chu, Z.; Huang, B.; Liu, Z.; Liu, L.; Zhang, J. Highly para-selective C-H Alkylation of Benzene Derivatives with 2,2,2-Trifluoroethyl α-Aryl-α-diazoesters. Angew. Chem. Int. Ed. 2017, 56, 2749–2753. [Google Scholar] [CrossRef]
- Ji, X.; Zhang, Z.; Wang, Y.; Han, Y.; Peng, H.; Li, F.; Liu, L. Catalyst-free synthesis of α,α-disubstituted carboxylic acid derivatives under ambient conditions via a Wolff rearrangement reaction. Org. Chem. Front. 2021, 8, 6916–6922. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Su, Y.; Li, R.; Li, C.; Liu, L.; Zhang, J. Phosphine-Catalyzed [3+2] Cycloaddition reaction of α-Diazoacetates and β-Trifluoromethyl Enones: A Facile Access to Multisubstituted 4-CF3 Pyrazolines. Org. Lett. 2018, 20, 6444–6448. [Google Scholar] [CrossRef]
- Gillingham, D.; Fei, N. Catalytic X-H insertion reactions based on carbenoids. Chem. Soc. Rev. 2013, 42, 4918–4931. [Google Scholar] [CrossRef]
- Shi, Y.; Gulevich, A.V.; Gevorgyan, V. Rhodium-catalyzed NH insertion of pyridyl carbenes derived from pyridotriazoles: A general and efficient approach to 2-picolylamines and imidazo[1,5-a]pyridines. Angew. Chem. Int. Ed. 2014, 53, 14191–14195. [Google Scholar] [CrossRef] [Green Version]
- Rousseau, G.; Breit, B. Removable directing groups in organic synthesis and catalysis. Angew. Chem. Int. Ed. 2011, 50, 2450–2494. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Kwon, K.; Yi, C.S. Dehydrative C-H Alkylation and Alkenylation of Phenols with Alcohols: Expedient Synthesis for Substituted Phenols and Benzofurans. J. Am. Chem. Soc. 2012, 134, 7325–7328. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.G.; de Azambuja, F.; Glorius, F. Direct functionalization with complete and switchable positional control: Free phenol as a role model. Angew. Chem. Int. Ed. 2014, 53, 7710–7712. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Shao, N.; Zhang, J.; Jia, R.; Wang, D. Cu(II)-Catalyzed ortho-Selective Aminomethylation of Phenols. J. Am. Chem. Soc. 2017, 139, 12390–12393. [Google Scholar] [CrossRef]
- Huang, Z.; Lumb, J. Phenol-Directed C–H Functionalization. ACS Catal. 2018, 9, 521–555. [Google Scholar] [CrossRef]
- Yu, C.; Patureau, F.W. Cu-Catalyzed Cross-Dehydrogenative ortho-Aminomethylation of Phenols. Angew. Chem. Int. Ed. 2018, 57, 11807–11811. [Google Scholar] [CrossRef]
- Murai, M.; Yamamoto, M.; Takai, K. Rhenium-Catalyzed Regioselective ortho-Alkenylation and [3+2+1] Cycloaddition of Phenols with Internal Alkynes. Org. Lett. 2019, 21, 3441–3445. [Google Scholar] [CrossRef]
- Miller, D.J.; Moody, C.J. Synthetic applications of the O-H insertion reactions of carbenes and carbenoids derived from diazocarbonyl and related diazo compounds. Tetrahedron 1995, 51, 10811–10843. [Google Scholar] [CrossRef]
- Zhu, S.; Zhou, Q. Transition-metal-catalyzed enantioselective heteroatom-hydrogen bond insertion reactions. Acc. Chem. Res. 2012, 45, 1365–1377. [Google Scholar] [CrossRef]
- Guo, X.; Hu, W. Novel multicomponent reactions via trapping of protic onium ylides with electrophiles. Acc. Chem. Res. 2013, 46, 2427–2440. [Google Scholar] [CrossRef]
- Xie, X.; Zhu, S.; Guo, J.; Cai, Y.; Zhou, Q. Enantioselective palladium-catalyzed insertion of α-aryl-α-diazoacetates into the O-H bonds of phenols. Angew. Chem. Int. Ed. 2014, 53, 2978–2981. [Google Scholar] [CrossRef]
- Gao, X.; Wu, B.; Huang, W.; Chen, M.; Zhou, Y. Enantioselective palladium-catalyzed C-H functionalization of indoles using an axially chiral 2,2′-bipyridine metal. Angew. Chem. Int. Ed. 2015, 54, 11956–11960. [Google Scholar] [CrossRef]
- Zhang, Y.; Yao, Y.; He, L.; Liu, Y.; Shi, Y. Rhodium(II)/Chiral Phosphoric Acid-Cocatalyzed Enantioselective O-H Bond Insertion of α-Diazo Esters. Adv. Synth. Catal. 2017, 359, 2754–2761. [Google Scholar] [CrossRef]
- Yu, Z.; Ma, B.; Chen, M.; Wu, H.; Liu, L.; Zhang, J. Highly site-selective direct C-H bond functionalization of phenols with α-aryl-α-diazoacetates and diazooxindoles via gold catalysis. J. Am. Chem. Soc. 2014, 136, 6904–6907. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Li, Y.; Shi, J.; Ma, B.; Liu, L.; Zhang, J. B(C6F5)3 Catalyzed Chemoselective and ortho-Selective Substitution of Phenols with α-Aryl α-Diazoesters. Angew. Chem. Int. Ed. 2016, 55, 14807–14811. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Li, Y.; Zhang, P.; Liu, L.; Zhang, J. Ligand and counteranion enabled regiodivergent C-H bond functionalization of naphthols with α-aryl-α-diazoesters. Chem. Sci. 2019, 10, 6553–6559. [Google Scholar] [CrossRef]
- Harada, S.; Sakai, C.; Tanikawa, K.; Nemoto, T. Gold-catalyzed chemoselective formal (3þ2)-Annulation reaction between β-naphthols and methyl aryldiazoacetate. Tetrahedron 2019, 75, 3650. [Google Scholar] [CrossRef]
- Zhang, C.; Tang, C.; Jiao, N. Recent advances in copper-catalyzed dehydrogenative functionalization via a single electron transfer (SET) process. Chem. Soc. Rev. 2012, 41, 3464–3484. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, Y.; Wang, J. Recent developments in copper-catalyzed reactions of diazo compounds. Chem. Commun. 2012, 48, 10162–10173. [Google Scholar] [CrossRef]
- Guo, X.; Gu, D.; Wu, Z.; Zhang, W. Copper-catalyzed C-H functionalization reactions: Efficient synthesis of heterocycles. Chem. Rev. 2015, 115, 1622–1651. [Google Scholar] [CrossRef]
- Zhu, X.; Chiba, S. Copper-catalyzed oxidative carbon-heteroatom bond formation: A recent update. Chem. Soc. Rev. 2016, 45, 4504–4523. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, W.; Wang, Y.; Wang, X.; Cao, F.; Shi, T.; Wang, Z. Recent Advances in Copper Promoted Inert C(sp3)–H Functionalization. ACS Catal. 2021, 11, 967–984. [Google Scholar] [CrossRef]
- Yates, P. The Copper-Catalyzed Decomposition of Diazoketones. J. Am. Chem. Soc. 1952, 74, 5376–5381. [Google Scholar] [CrossRef]
- Maier, T.C.; Fu, G.C. Catalytic enantioselective O-H insertion reactions. J. Am. Chem. Soc. 2006, 128, 4594–4595. [Google Scholar] [CrossRef]
- Chen, C.; Zhu, S.; Liu, B.; Wang, L.; Zhou, Q. Highly enantioselective insertion of carbenoids into O-H bonds of phenols: An efficient approach to chiral α-aryloxycarboxylic esters. J. Am. Chem. Soc. 2007, 129, 12616–12617. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhu, S.; Xie, X.; Zhou, Q. Enantioselective copper-catalyzed intramolecular phenolic O-H bond insertion: Synthesis of chiral 2-carboxy dihydrobenzofurans, dihydrobenzopyrans, and tetrahydrobenzooxepines. Angew. Chem. Int. Ed. 2013, 52, 2555–2558. [Google Scholar] [CrossRef]
- Morilla, M.E.; Molina, M.J.; Díaz-Requejo, M.M.; Belderraín, T.R.; Nicasio, M.C.; Trofimenko, S.; Pérez, P.J. Copper-catalyzed carbene insertion into O-H bonds: High selective conversion of alcohols into ethers. Organometallics 2003, 22, 2914–2918. [Google Scholar] [CrossRef]
- Liang, Y.; Zhou, H.; Yu, Z. Why is copper(I) complex more competent than dirhodium(II) complex in catalytic asymmetric O-H insertion reactions? A computational study of the metal carbenoid O-H insertion into water. J. Am. Chem. Soc. 2009, 131, 17783–17785. [Google Scholar] [CrossRef]
- Cai, Y.; Lu, Y.; Yu, C.; Lyu, H.; Miao, Z. Combined C-H functionalization/O-H insertion reaction to form tertiary β-alkoxy substituted β-aminophosphonates catalyzed by [Cu(MeCN)4]PF6. Org. Biomol. Chem. 2013, 11, 5491–5499. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, Z.; Zhang, J.Z.; Xia, F. DFT Calculations on the Mechanism of Transition-Metal-Catalyzed Reaction of Diazo Compounds with Phenols: O-H Insertion versus C-H Insertion. J. Phys. Chem. A 2016, 120, 6485–6492. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yu, Z.; Zhang, J.Z.; Liu, L.; Xia, F.; Zhang, J. Origins of unique gold-catalysed chemo- and site-selective C-H functionalization of phenols with diazo compounds. Chem. Sci. 2016, 7, 1988–1995. [Google Scholar] [CrossRef]
- Ma, B.; Tang, Z.; Zhang, J.; Liu, L. Copper-catalysed ortho-selective C-H bond functionalization of phenols and naphthols with α-aryl-α-diazoesters. Chem. Commun. 2020, 56, 9485–9488. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, J.Z.; Xia, F. How CuCl and CuCl2 Insert into C-N Bonds of Diazo Compounds: An Electronic Structure and Mechanistic Study. J. Phys. Chem. A 2020, 124, 2029–2035. [Google Scholar] [CrossRef]
- Fructos, M.R.; Belderrain, T.R.; Fremont, P.; Scott, N.M.; Nolan, S.P.; Diaz-Requejo, M.M.; Perez, P.J. A gold catalyst for carbene-transfer reactions from ethyl diazoacetate. Angew. Chem. Int. Ed. 2005, 44, 5284–5288. [Google Scholar] [CrossRef] [PubMed]
- Barluenga, J.; Lonzi, G.; Tomas, M.; Lopez, L.A. Reactivity of stabilized vinyl diazo derivatives toward unsaturated hydrocarbons: Regioselective gold-catalyzed carbon-carbon bond formation. Chem. Eur. J. 2013, 19, 1573–1576. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, Z.; Luo, Z.; Zhang, J.; Liu, L.; Xia, F. Mechanistic Investigation of Aromatic C(sp2)-H and Alkyl C(sp3)-H Bond Insertion by Gold Carbenes. J. Phys. Chem. A 2016, 120, 1925–1932. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Chai, J.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Burns, L.A.; Vazquez-Mayagoitia, A.; Sumpter, B.G.; Sherrill, C.D. Density-functional approaches to noncovalent interactions: A comparison of dispersion corrections (DFT-D), exchange-hole dipole moment (XDM) theory, and specialized functionals. J. Chem. Phys. 2011, 134, 084107. [Google Scholar] [CrossRef]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the first row transition elements. J. Chem. Phys. 1987, 86, 866–872. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Fukui, K. A Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions-the IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Catalyst | Yield of 3 (%) 1 | Yield of 4 (%) 1 |
|---|---|---|
| CuCl | 76 | 0 |
| CuCl2 | 87 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, X.; Liu, X.; Xia, F.; Liu, L. Theoretical Study on the Copper-Catalyzed ortho-Selective C-H Functionalization of Naphthols with α-Phenyl-α-Diazoesters. Molecules 2023, 28, 1767. https://doi.org/10.3390/molecules28041767
Zhu X, Liu X, Xia F, Liu L. Theoretical Study on the Copper-Catalyzed ortho-Selective C-H Functionalization of Naphthols with α-Phenyl-α-Diazoesters. Molecules. 2023; 28(4):1767. https://doi.org/10.3390/molecules28041767
Chicago/Turabian StyleZhu, Xiaoli, Xunshen Liu, Fei Xia, and Lu Liu. 2023. "Theoretical Study on the Copper-Catalyzed ortho-Selective C-H Functionalization of Naphthols with α-Phenyl-α-Diazoesters" Molecules 28, no. 4: 1767. https://doi.org/10.3390/molecules28041767
APA StyleZhu, X., Liu, X., Xia, F., & Liu, L. (2023). Theoretical Study on the Copper-Catalyzed ortho-Selective C-H Functionalization of Naphthols with α-Phenyl-α-Diazoesters. Molecules, 28(4), 1767. https://doi.org/10.3390/molecules28041767