1. Introduction
Environmental concerns have motivated the continuing development of new green energy sources, reduction of hazardous chemical compounds, and more intelligent uses of natural resources. There is a continuing need to devise new technological approaches in this direction. In the case of the petrochemical industry, replacing petrol fuels with hydrogen involves two main difficulties: storage and transport [
1,
2,
3]. Hydrogen storage methods can be divided into physical methods (compression, liquefaction, or cryocompression) and chemical methods, such as adsorption, conversion into metallic hydrides, complex hydrides and liquid organic carriers [
3]. A variety of materials have been examined for these purposes, such as metal organic frameworks, adsorption on many transition metal surfaces, or carbon-based materials. A recent literature report comparing carbon and boron nitride nanotubes indicated that carbon-based nanotubes are more energetically favorable [
4].
The work described below focuses on a newly developed carbon allotrope—cyclo[18]carbon (a polyyne-type molecule)—and how noncovalent interactions might enable trapping and storage of small molecule gases within its circular ring structure. Better understanding of these interactions might aid in finding the solutions to current problems such as hydrogen storage and the extraction of harmful gases from the atmosphere. Cyclo[n]carbons have been studied for more than 50 years now [
5]. Procedures for the formation of C
18 from precursors such as C
18Br
6 and C
18(CO)
6 have been developed and the reaction mechanism is also understood [
6,
7,
8,
9]. The C
18 carbon ring itself consists of 18 carbon atoms bonded with an alternating pattern of single and triple covalent bonds [
10]. It is widely recognized that C
n rings (where n = 4q + 2 (where q is an integer greater than 1)) containing at least 10 carbon atoms tend to be more stable than their linear chain analogues [
5].
Computational studies have shown that this particular kind of polyyne represents the global minimum; the cumulene structure with uniform C–C bond lengths represents the transition state for the bond transposition process from single to triple bonds, and vice versa. At the same time, a bonding structure consisting of carbons connected by double bonds is more aromatic than a polyyne ring due to more effective π orbitals overlapping [
11]. This interesting attribute of the cyclo[18]carbon molecule provoked several studies regarding this new carbon allotrope. The electron-accepting potential of the C
18 ring results from lower C–C bond saturation. Several studies investigated this property of the C
18 ring along with other cyclocarbons comprising larger or lesser numbers of carbon atoms (from 10 to 60). Research concerning the adsorption of selected gases (CO, NO, NH
3) has demonstrated that both chemisorption and physisorption are possible [
12]. It was also postulated that C
18 nanoclusters can be used as ultra-fast sensors for detecting CO and NO molecules.
Recent quantum calculations conducted by Hobza et al. proved cyclo[18]carbon’s ability to create dative bonds with piperidine molecules (from one to even four) that attacked from the outside of the C
18 ring [
10]. This interaction was accompanied via significant deformation of the carbon ring. This theme has been extended by the Nandi group [
13], which considered the effect of heavy-atom quantum mechanical tunneling on the transformation from a distant van der Waals complex into a more closely bound dative-bond complex at cryogenic temperatures. The energy barrier to this structural reshaping was measured as 2.2 kcal/mol [
13] compared to 3.6 kcal/mol in previously cited work [
10].
The history of study regarding the interaction between cyclo[n]carbons and piperidine shows that even C
60 can adsorb one or two piperidine molecules via a N–C dative bond, and the interaction energy of such adducts reaches up to 37 kcal/mol [
14]. Other research on this matter involved the examination of the ground and excited states of the C
18 ring and its complexes with certain electron-donating units such as tetrathiafulvalene, zinc porphyrin, or zinc phthalocyanine [
15]. Density functional calculations also explored combinations of cyclo[18]carbon complexes with the noble gases [
16], as well as with the XCN (X = H, F, Cl, Br, I) molecules [
17]. In the latter paper, the vdW potential map of the C
18 ring with the halogen probe revealed cylindrical (for F) or dumbbell-shaped (for other halogens) areas for the negative vdW potential, indicating that in this region attraction produced by dispersion forces dominates over exchange repulsion. These spherical forms grew in size from F to I, demonstrating the possibility of diverse objects circling close to C
18, interacting with this polyyne in various ways. The ability of the Si-substituted cyclo[18]carbon to bind with molecular N
2 was very recently investigated. The carbon ring had to be bent in order to create the resultant adduct. This reaction can serve as a potential precursor for the further conversion of C
17Si∙∙∙N
2 into ammonia [
18]. The cyclo[18]carbon species can be viewed as potential materials for hydrogen storage. Therefore, the binding energies of C
18∙∙∙H
2/(C
18)
2∙∙∙H
2 adducts might be compared to some other compounds reported earlier in the literature. The growing popularity of graphene motivated its study as a possible hydrogen storage unit. It has been shown that the physisorption energy of molecular hydrogen on flat carbon nanoparticles (graphitic platelets) and polycyclic aromatic hydrocarbons (PAHs) ranged from −0.84 to −1.72 kcal/mol, with the latter value assigned to graphene itself [
19]. Other systems potentially useful for hydrogen storage applications were pristine and functionalized GaS sheets, with the binding energies of molecular hydrogen ranging from −1.48 to −8.54 kcal/mol (for pristine GaS it was −1.78 kcal/mol) [
20]. Finally, for pristine and decorated GeC monolayers, the binding energies were reported as falling within the scope of −0.23 to −6.81 kcal/mol (the best were for Li and K atom doping) [
21].
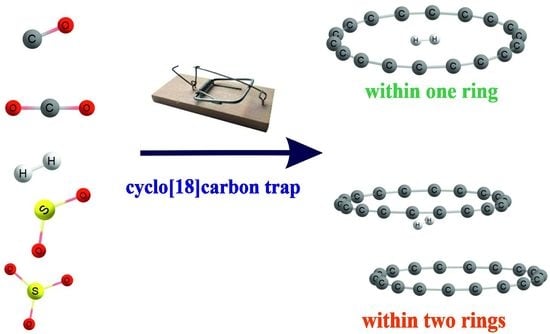
The purpose of the work described below was to investigate the nature of the interactions between cyclo[18]carbon (single or double) and small gas molecules such as H2, CO, CO2, SO2 and SO3, which can provide some fundamental information related to hydrogen storage and air pollution reduction. All the possible complexation routes were considered: both trapping the ligand inside the ring and adsorption from the outside of C18. How do these two interaction modes compare? Which gas molecules can be captured within the C18 ring? How large can it be? What is the exact nature of these interactions? Is this complexation driven by dispersion or other forces? What is the consequence of adding a second C18 ring? Does this addition enhance the binding or change it in a qualitative manner? Insights into the storage process that would occur in a condensed phase are predicated on a thorough understanding of these complexes in the gas phase, where each particular interaction can be studied individually and carefully.
2. Results
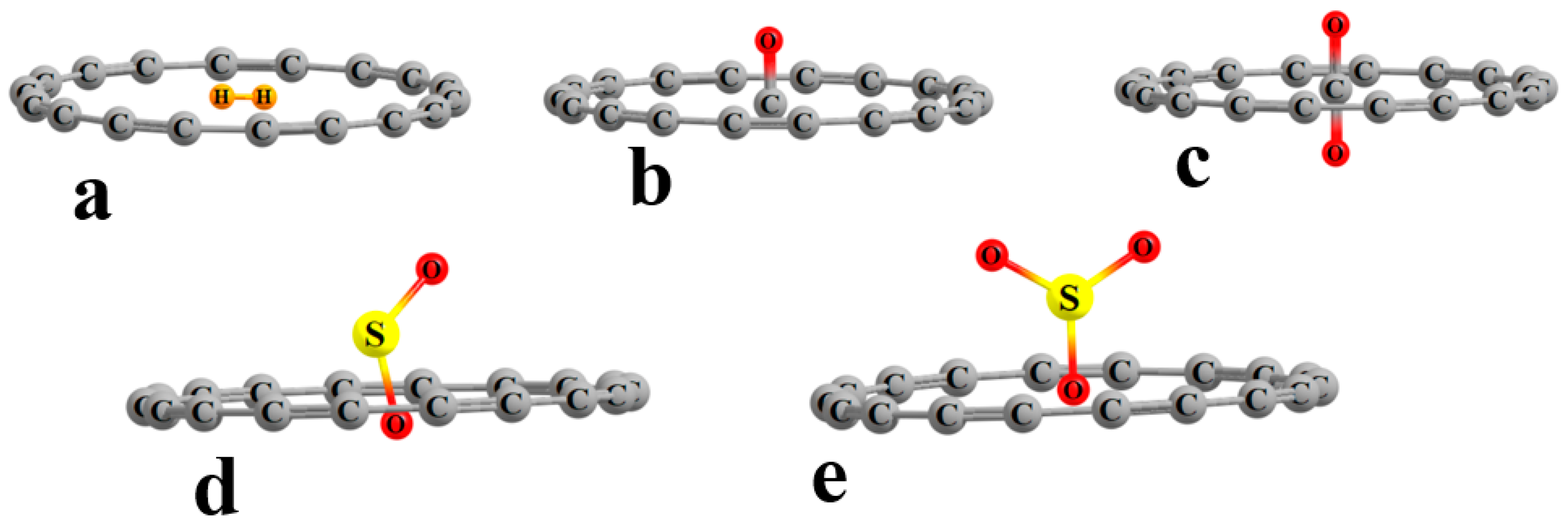
The structures of the various internal complexes with the ligand situated near the center of the C
18 ring are exhibited in
Figure 1. There are certain differences from one inclusion complex to the next. The H
2 molecule is small enough that its entirety can fit inside the circle, with both H atoms lying in the C
18 plane, as seen in
Figure 1a. It causes no deformation of the circular nature of the ring, with all the C atoms lying 3.693 Å from the ring center, as indicated in the first row of
Table 1. The H
2 lies slightly off center, with its midpoint displaced 0.062 Å from the ring center. The CO lies perpendicular to the ring plane, with the C within this plane, as may be seen in
Figure 1b. Its presence distorts the ring into a slight oval shape, with the maximum and minimum distances from the ring center being 3.695 and 3.689 Å. As indicated in the last column of
Table 1, the C atom lies some 0.086 Å from the ring center. The C atom of the CO
2 also lies in the ring plane, with one O above and the other below, as shown in
Figure 1c. The ring becomes more oval in the presence of SO
2 and SO
3, with differences between the long and short axes of 0.070 and 0.108 Å, respectively. In both cases, one of the O atoms lies closest to the ring center, with the remainder of the ligand lying above the ring, as shown
Figure 1d,e. In the case of SO
2, the O atom lies significantly below the plane, a full 0.406 Å from its center. Additional calculations for two complexes (C
18···H
2 and C
18···SO
2) were carried out to consider the freedom of motion of each ligand within its bound environment. In both cases, the rotation barriers of the trapped molecules were estimated around an axis perpendicular to the plane of the ring passing through the center of the H–H bond or the sulfur atom, respectively. The barrier to rotation was found to be 0.58 and 1.21 kcal/mol, respectively. Thus, ligand rotation would require an additional energy equivalent to 38 and 21% of the binding energy for its optimal positioning.
The interaction energies between each ligand and the ring reported in
Table 2 indicate a weak to moderate strength. While the E
int is less than 2 kcal/mol for H
2, it rises to 2.8 kcal/mol for CO and exceeds 4 kcal/mol for the other three ligands. The deformation energies E
def resulting from the distortions of the two monomer units are quite small, 0.1 kcal/mol or less, as presented in the penultimate column of
Table 2, despite the deviations from circularity of some of these rings. Consequently, the binding energy E
b listed in
Table 2, which equates to the reaction energy of the formation of the inclusion complex from the two isolated monomers, is virtually identical to the E
int.
One can glean insight into the nature of the interaction via decomposition of the total E
int into components with physical meaning. The values of these components in
Table 3 suggest these interactions are largely dispersion-controlled, as the E
disp accounts for roughly 80% of the total attractive forces, with the electrostatic E
es and orbital interaction E
oi terms comprising some 10% each.
The AIM protocol is generally a useful tool for pinpointing specific interatomic bonding patterns. The bond paths elucidated for these inclusion complexes are shown in
Figure S1 and offer only partial understanding. For example, each of the H atoms of H
2 would appear to be bonded to a C–C midpoint, and only to those closest to these atoms, with little connection to other C atoms of the ring. The C atoms of CO and CO
2 each reach out to several ring atoms. The O atoms of SO
2 and SO
3 connect to the ring C atoms, despite being displaced above the ring. The weakness of any of these bonds is affirmed by the small densities of each bond critical point, which are in the 0.002–0.004 au range. The other AIM parameters of each complex, such as the ∇
2ρ and H, are all listed in
Table S1, the values of which all suggest a weak noncovalent bond in each case. Moreover, also displayed in
Table S1 are the bond critical point quantities for the internal C–C and C≡C bonds of the ring, which are consistent with an alternating weak/strong pattern.
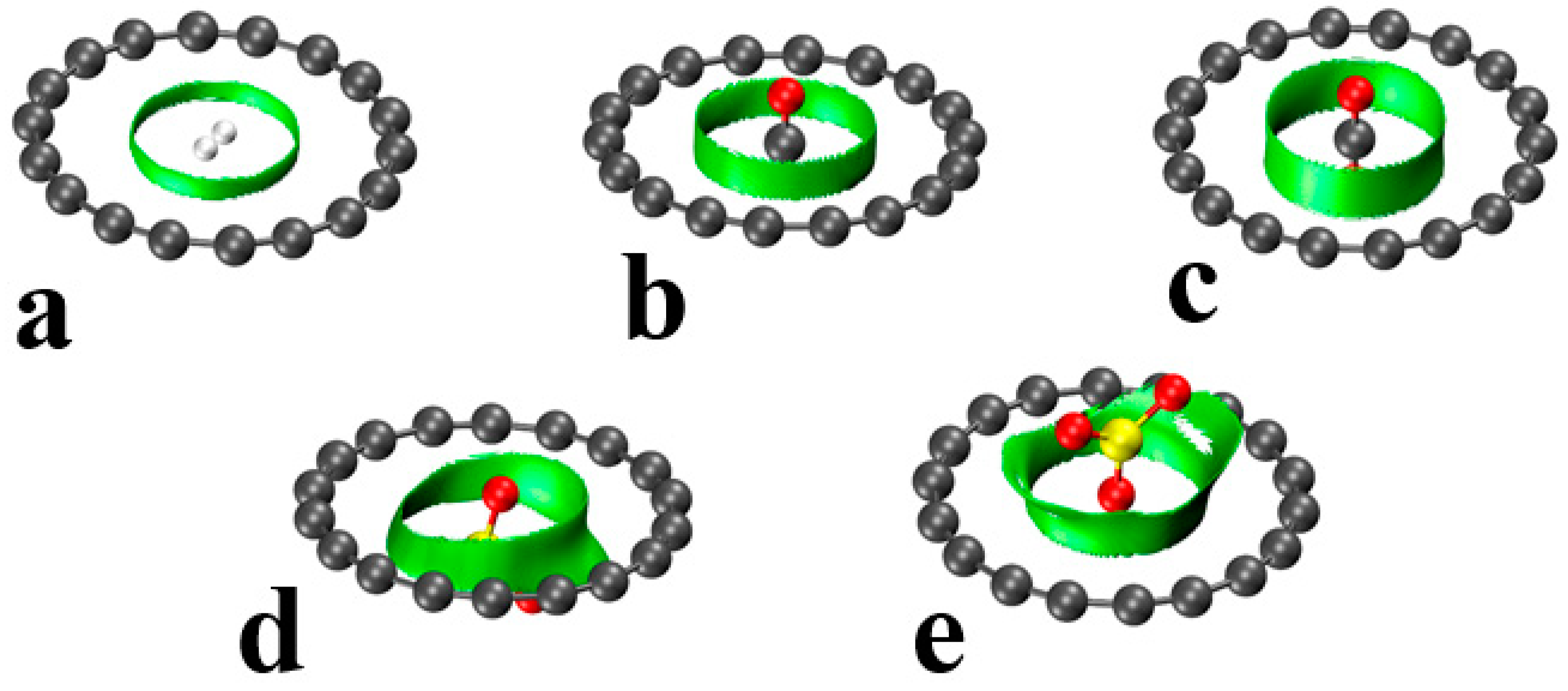
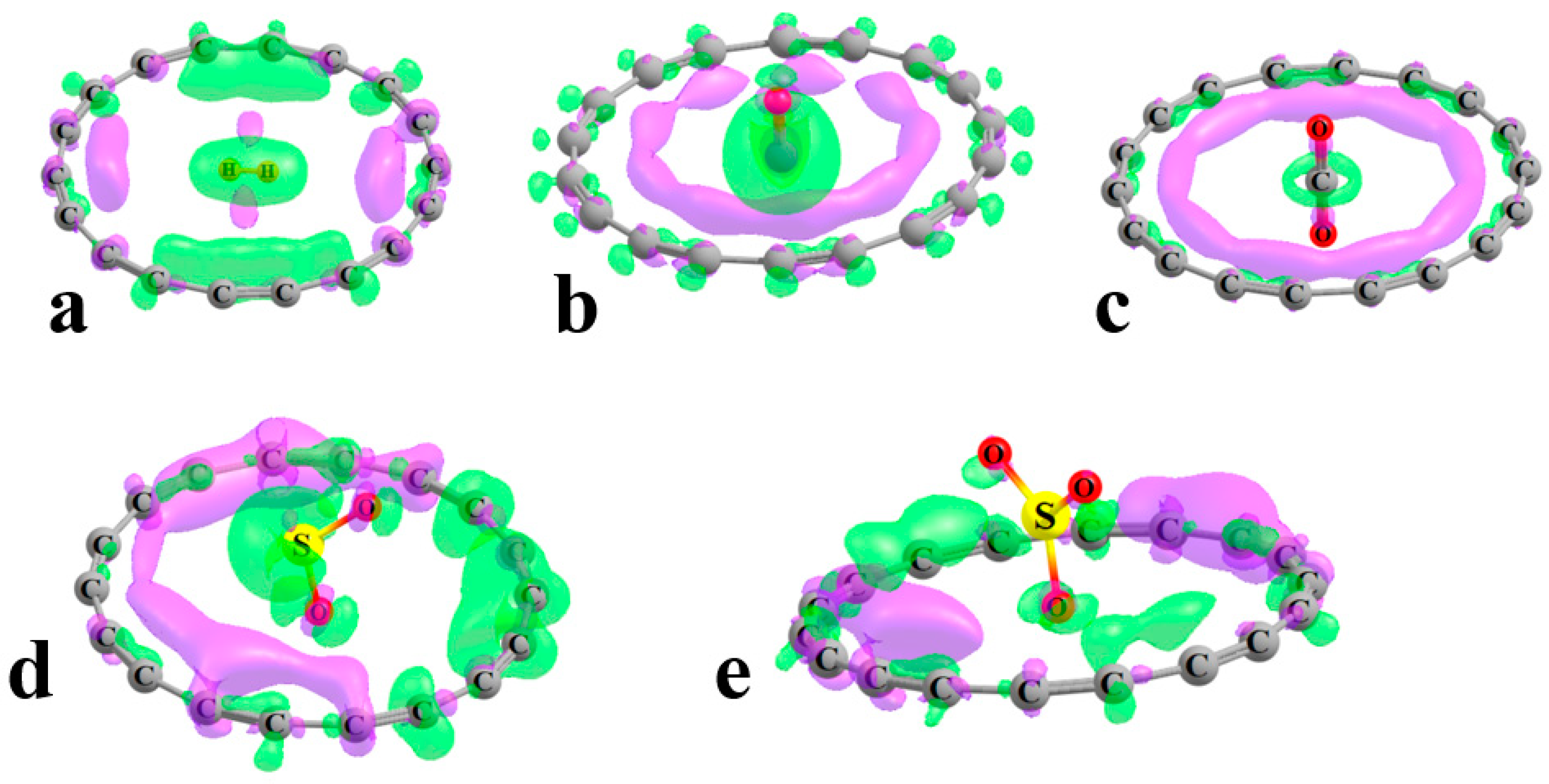
Noncovalent interaction analysis (NCI), also known as the reduced density gradient method, of these systems offers a perhaps more balanced view of the ligand–ring interactions. The green regions in
Figure 2, signaling a weak noncovalent interaction, are generally annular in shape, which suggests a less specific atom–atom bonding than might arise from a survey of the AIM diagrams. So, for example, while AIM proposes that each H atom of H
2 binds to one specific C ring atom, the NCI pattern in
Figure 2a provides a picture in which this bonding is delocalized over the entire ring. There is some asymmetry in the green regions, as in the SO
2 and SO
3 cases, which is based on the disposition of the ligand O atoms. Even in those cases, the green area fully encircles the ligand, which is again consistent with the idea that these complexes are dominated by nonspecific dispersive forces.
Another perspective on the binding arises from examination of how the complexation shifts the electron density around the entire system. The electron density shift patterns illustrated in
Figure 3 were derived by subtracting the sum of the unperturbed monomer densities from that of the full complex. Regions where the density has increased are designated by purple, while the green areas indicate loss. A different contour was used for each diagram in order to best display the shift patterns in each, although these contours represent small numbers, between 0.00005 and 0.0030 au, congruent with the lack of largescale shifts.
Figure 3a indicates that there is a shift from the H–H axis of the H
2 ligand toward the ring atoms that lie generally along this axis. The shift is in the opposite direction, from the ring to H
2, in the direction perpendicular to the molecular axis. There is a minute shift of overall charge from the ring to H
2 (computed as the sum of the natural atom charges) of 0.0005 e. This shift is in the opposite direction, from the ligand to the ring for CO, in the amount of 0.0056 e. This shift is evident by the green area encompassing the ligand in
Figure 3b and the purple area closer to the C atoms of the ring. This pattern remains quite similar for CO
2 in
Figure 3c, although the larger contour masks a slight transfer to the central ligand of 0.0009 e. The noncylindrical shape of the SO
2 and SO
3 ligands introduces the asymmetry of the density shift patterns in
Figure 3d,e such that certain regions of the ring accrue additional charge and others lose density. In both cases, there is a small overall shift of charge from the ligand to ring, 0.0085 and 0.0013 e for SO
2 and SO
3, respectively, confirmed by the green regions surrounding these ligands.
Concerning the electrostatic portion of the interaction energies, the C
18 ring is nonpolar for all intents and purposes. The molecular electrostatic potential (MEP) surrounding this system is displayed in
Figure S2 on its 0.001 au isodensity surface. The generality of the green area reinforces its nonpolarity. The values of the maximum and minimum on the inside of the ring are respectively +1.8 and −1.4 kcal/mol, as indicated in
Table 4. These quantities are of such a small magnitude that they can interact only very weakly with the positive or negative regions of the ligands listed in the table, some of which can be sizable, and are displayed graphically in
Figure S2.
In addition to situating themselves in the middle of the C
18 ring, these ligands can also attach to the outside. The geometries depicted in
Figure S3 place the binding C or S atom near the center of a C–C bond of the ring. The C atom of CO, like one of the hydrogen atoms of H
2, approaches one of the longer C–C single bonds of the ring, whereas it is the shorter triple C≡C bond that attracts the C of CO
2 or the S of SO
2 and SO
3. These outer complexes are significantly weaker than the inner inclusion structures, by a factor of 2 to 6, with their binding energies listed in the first column of
Table S2. This weaker binding occurs despite the much larger MEP maximum on the outside of the C
18 ring, 8.0 vs. 1.8 kcal/mol for the interior maximum.
From this exterior position, it is possible for the ligands to approach the ring more closely so as to form a covalent dative bond with one or more C ring atoms. A certain exception here is the H
2 molecule, which dissociates before joining the ring, which results in a hydrogenated ring. These products are displayed in
Figure S4, along with some of the related energetic quantities. As may be expected, the deformation energies are quite high for both the ring and the erstwhile ligand. The second and third columns of
Table S2 summarize the binding energies of the noncovalent and covalent complexes, as well as the activation energy required to convert from the former into the latter. Even though some of the covalently bound complexes are more stable than their noncovalent counterparts, the activation energies are prohibitively high for their formation, in excess of 25 kcal/mol. The exception is SO
3, where the barrier is only 8 kcal/mol, which would enable the system to be stabilized by some 30 kcal/mol relative to the noncovalent complex.
Double Ring
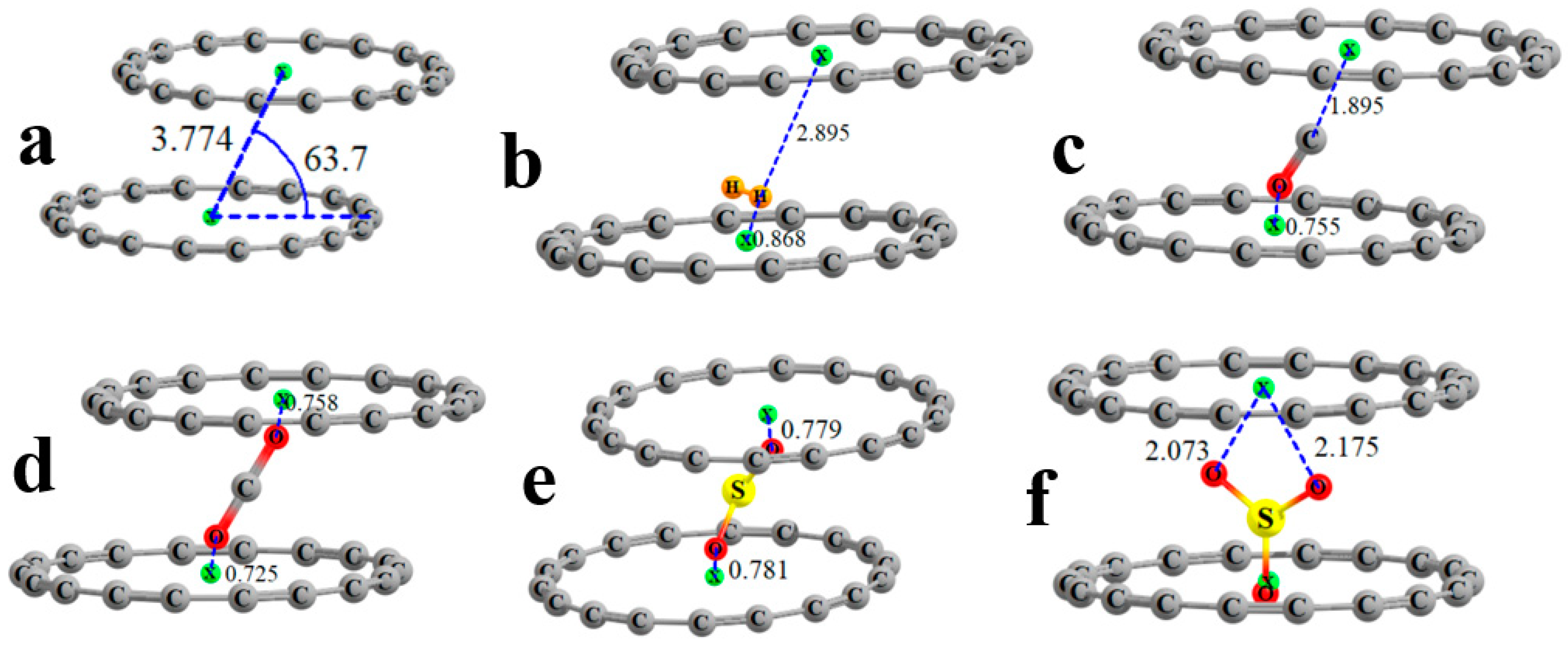
The flat shape of the C
18 ring begs the question of whether two such rings might dimerize. An optimization of such a dimer leads to the structure pictured in
Figure 4a, which can be categorized as a slipped parallel geometry, reminiscent of the favored geometry of the benzene dimer. The centers of the two rings, indicated by the small green balls, lie some 3.774 Å from one another, and the center-to-center axis makes an angle of 63.7° with the plane of each ring, which places the planes of the two units 3.38 Å apart, as indicated by the d quantity in the last column of
Table 5. The interaction energy between the two rings in this dimer amounts to −8.96 kcal/mol.
The geometries of the complexes in which each ligand is inserted into the C
18 dimer systems are represented in
Figure 4b–f. In the cases of H
2, CO, and SO
3, the ligand is asymmetrically positioned, being much closer to one ring than the other. For example, one H atom of H
2 lies 0.9 Å from the center of the lower ring but nearly 3 Å from the upper. By contrast, the terminal O atoms of CO
2 are each some 0.7 Å from the upper and lower rings, respectively, with similar distances noted in SO
2. In most cases, the ligand adopts a very different position with the triad than with the single ring. H
2 is pulled out of the plane of the lower ring, as are CO and CO
2, which are also tilted away from the vertical. SO
2 and especially SO
3 seem to be less affected by the presence of the upper ring.
The energetics of adding each ligand to the double ring compiled in
Table 5 correspond to the interaction energy between the ligand and the pair of rings, all in the geometry they adopt within the triad. These quantities span the range between 2.2 kcal/mol for H
2 and 7.4 kcal/mol for SO
3. These triad interaction energies represent a roughly 50% increase when compared to the single-ring complexes in
Table 2. The deformation energies in
Table 5 are somewhat larger than those in
Table 2, signaling that the inclusion of the ligand has a larger effect on the double ring geometry than it does on the single ring. A good part of this distortion has to do with the pushing of the two rings apart by the ligand. The distance between the ring planes reported in the last column of
Table 5 can be compared with the 3.38 Å in the unliganded ring dimer, which is closely related to the deformation energy of the system.
An energy decomposition of each of these triads, as presented in
Table 6, shows that like the single ring systems, so too are the larger triads dominated by dispersion, which accounts for 70–80% of the total attractive energy. The AIM diagrams of these complexes are exhibited in
Figure S5 and show certain similarities to those of the single ring dyads. Bond path densities connecting ligand atoms to those of the ring lie in the range between 0.003 and 0.005 au, and again one sees an alternating pattern of ring C–C and C≡C bonds with densities of 0.33 and 0.41 au, respectively. The NCI plots in
Figure S6 are again characterized by delocalized bonding involving the entire ring, rather than any one specific C atom.
3. Discussion
There has been a bit of controversy regarding the bonding nature of cyclo[18]carbon, in that some calculations confirmed a polyyne structure [
10,
11] with alternating short and long C–C bonds, as found experimentally [
22], while others suggested a cumulene geometry with uniform bond lengths [
11,
23]. While our own ωB97XD/Def2TZVPP computations fell in line with the experimental polyyne idea, which has been further quantified as being 10 kcal/mol more favorable than the cumulene [
11], our pilot calculations with the PBE0 functional falsely suggested the alternate. It would seem that the bonding pattern within this system is rather sensitive to the particular level of calculation.
The electron-acceptor properties of C
18 were studied in the work of Hobza et al. in C
18 complexes with piperidine [
10], which also bolsters confidence in the level of theory applied here. After applying this same ωB97XD/Def2TZVPP level [
10] in their work on piperidine complexes with different carbon ring systems, these authors also performed single-point energy calculations with the coupled cluster single and double CCSD/cc-pVTZ level and concluded that “The DFT values are in reasonable agreement with the more accurate CCSD ones”. In addition to the vdW complex for which the binding energy was −3.6 kcal/mol, a complex stabilized by a coordination bond (dative bond) was also examined, for which the E
b was −16.2 kcal/mol. Our own calculations were consistent in that it was found that a noncovalently bound ligand on the outside of the C
18 ring could approach more closely and engage in short covalent bonds with two C atoms. The energetics of this process presented in
Table S2 show that this process can be an exothermic one, for example, as much as −33.6 kcal/mol in the case of SO
3. However, this conversion from a noncovalent to a covalent complex must overcome an energy barrier. This barrier ranges from 8.1 kcal/mol for SO
3 up to nearly 50 kcal/mol for CO
2 and even 135 kcal/mol for H
2, which, as mentioned before, dissociates before joining the ring. This sort of high barrier contrasts with the previously published barrier of only 3.6 kcal/mol for piperidine. This earlier set of computations did not consider complexes with the piperidine located within the ring, probably due to the fact that piperidine is too large to fit inside the C
18 ring. It was found here that a similar restriction already applies even to benzene. When placed initially in the center of the ring, benzene was quickly expelled in an exothermic process.
Nandi et al. confirmed that the connection of piperidine to the C
18 ring requires overcoming an energy barrier, this time estimated at 2.2 kcal/mol [
13]. Their work also showed that among the dative bond complexes of various polyynes with piperidine, the one with a 14-member carbon ring was the most stable, while the most stable vdW complex was that with C
16. In a paper by Vadalkar et al., the dative bond complexes between CO and the C
18 ring were discussed in detail [
12]. The results obtained at the ωB97XD/6-311++G(d, p) level for the C
18∙∙∙CO adduct were essentially the same as those obtained by the current work (E
b = −7.1 kcal/mol).
Mazumder et al. studied the ability of a double C
18 ring to encapsulate diatomic noble gas molecules at the M06-2X/def2-TZVP level [
16]. The binding energies ranged from −2.4 to −6.4 kcal/mol, indicating the most stable adduct for Kr
2, and this quantity increased in parallel with the size of the noble gas. The energetics of these dimers are similar to the small molecules examined in the current work, with the E
b being between −2.2 and −8.7 kcal/mol. The nature of the ring∙∙∙ligand complexes revealed by an SAPT2 decomposition scheme was also consistent with our findings in that the dispersion forces greatly outweighed the electrostatics and induction, with the latter two combining for less than 30% of the total attractive forces.
Different binding modes connecting C
18 to HCN were examined by Vadalkar et al. [
24]. The interaction energy between HCN and the bare ring was roughly −5 kcal/mol and the N∙∙∙C binding distance equal to 3.01 Å. This adsorption energy was found to rise to nearly −14 kcal/mol when these nanoclusters were doped with Al, Si, and P atoms. Similar clusters were examined by Zhao [
17], extending the set of ligands to XCN molecules. Both inside and outside binding modes were confirmed for ClCN, BrCN and ICN, while HCN and FCN were limited to only inside the carbon ring. The inner complexes were more stable than the outside ones, much like in our data. While the interaction energies did not surpass −2.4 kcal/mol for the latter, these quantities ranged from −5 to −8 kcal/mol. SAPT analysis confirmed that these dimers are primarily dominated by dispersion. Green rims indicative of the noncovalent interactions typical of vdW complexes, comparable to those seen in
Figure 2, were detected via NCI analysis.
As mentioned in the Introduction, the physisorption energy of molecular hydrogen on flat carbon nanoparticles ranged from −0.84 to −1.72 kcal/mol [
19]. Our computed H
2 binding energy by a single ring of −1.51 kcal/mol fits into this range, while the −2.22 kcal/mol for the double ring is superior.
The binding energies can be connected with complexation constants using the approximate expression that K = exp(−Eb/RT). Since all the Eb are negative, these association constants will all exceed unity. Assuming a temperature of 298 K, these constants with the single ring will vary from a minimum of 13 for H2 up to 1.6 × 104 for SO2. These values increase for the double-ring complexes, with a range of between 42 and 2.3 × 106. K rises markedly as the temperature is diminished. Taking 100 K as an example, this quantity varies between 2.0 × 103 and 3.2 × 1012 for the single-ring complexes. These large constants, in tandem with the lack of any energy barrier to their formation, suggests the spontaneity of the association reactions.
It might be noted from
Table S3 that the zero-point vibrational energy corrections to the interaction energies are small, in the order of 1 kcal/mol, and so have little effect on the conclusions outlined above. Another issue relates to the low-frequency vibrational modes of some of these systems. Taking the single C
18 ring as an example, the mode with the smallest frequency of 59 cm
−1 corresponds to an in-plane deformation that takes the circular shape toward an oval. The next lowest frequency of 76 cm
−1 represents an out-of-plane vibration. With regard to the double ring, there is a very low-frequency mode of 5 cm
−1 that can be described as a synchronized swinging of two rings and a second of 5 cm
−1 involving opposite twists.



 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}