
Recent Advances in Molecule Synthesis Involving C-C Bond Cleavage of Ketoxime Esters

{kind=link}
{kind=link}
{kind=link}
{kind=link}
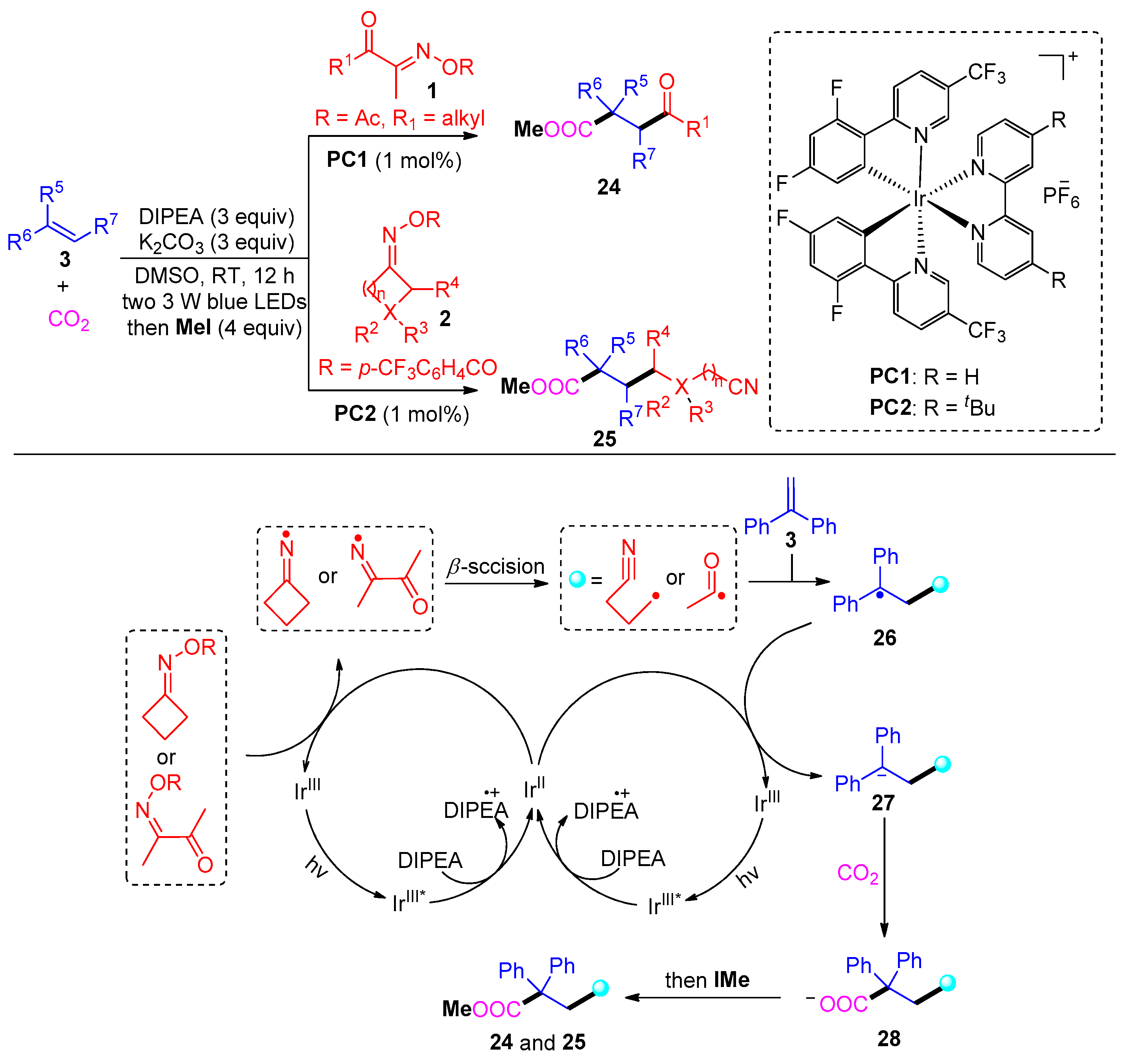{kind=link}
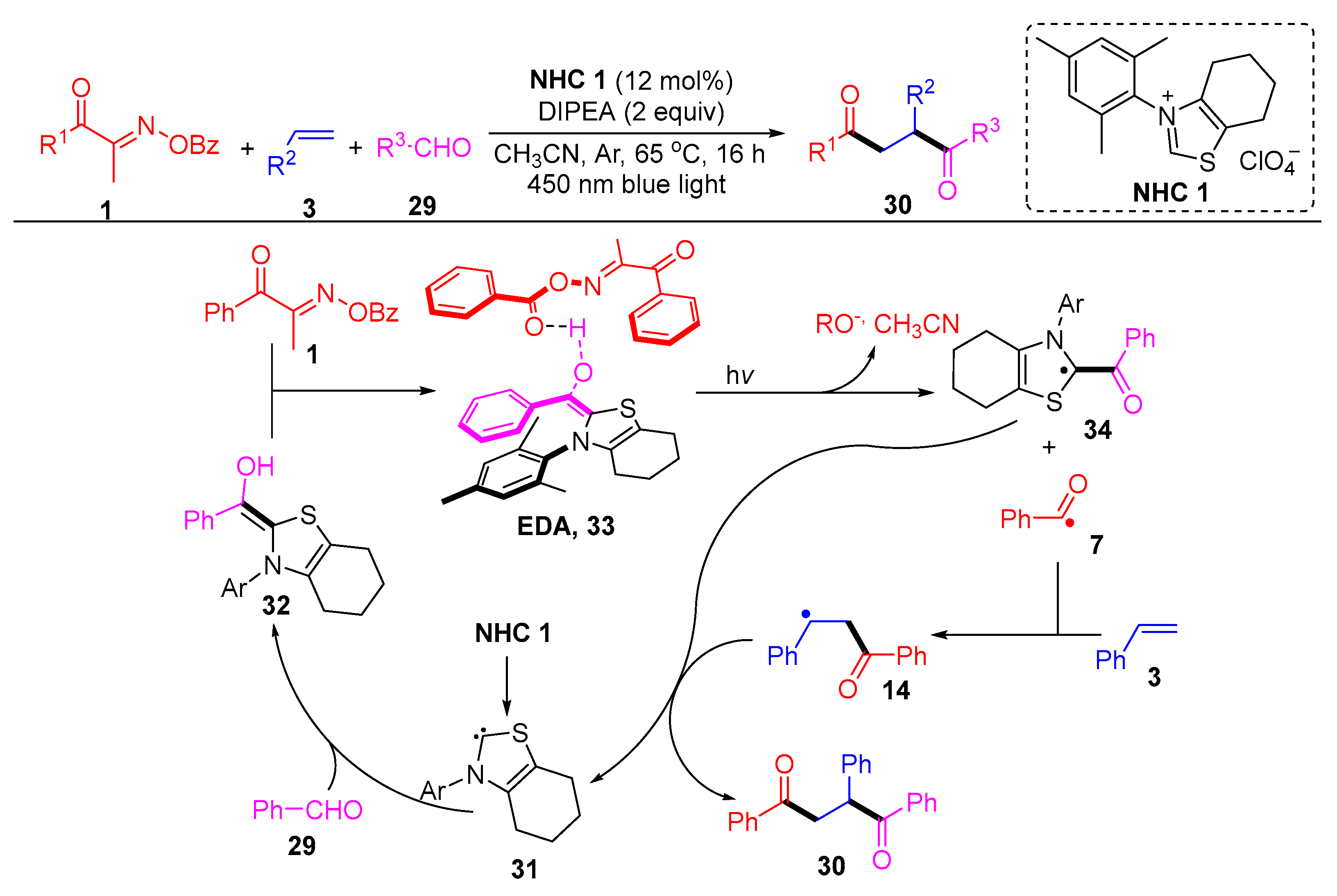{kind=link}
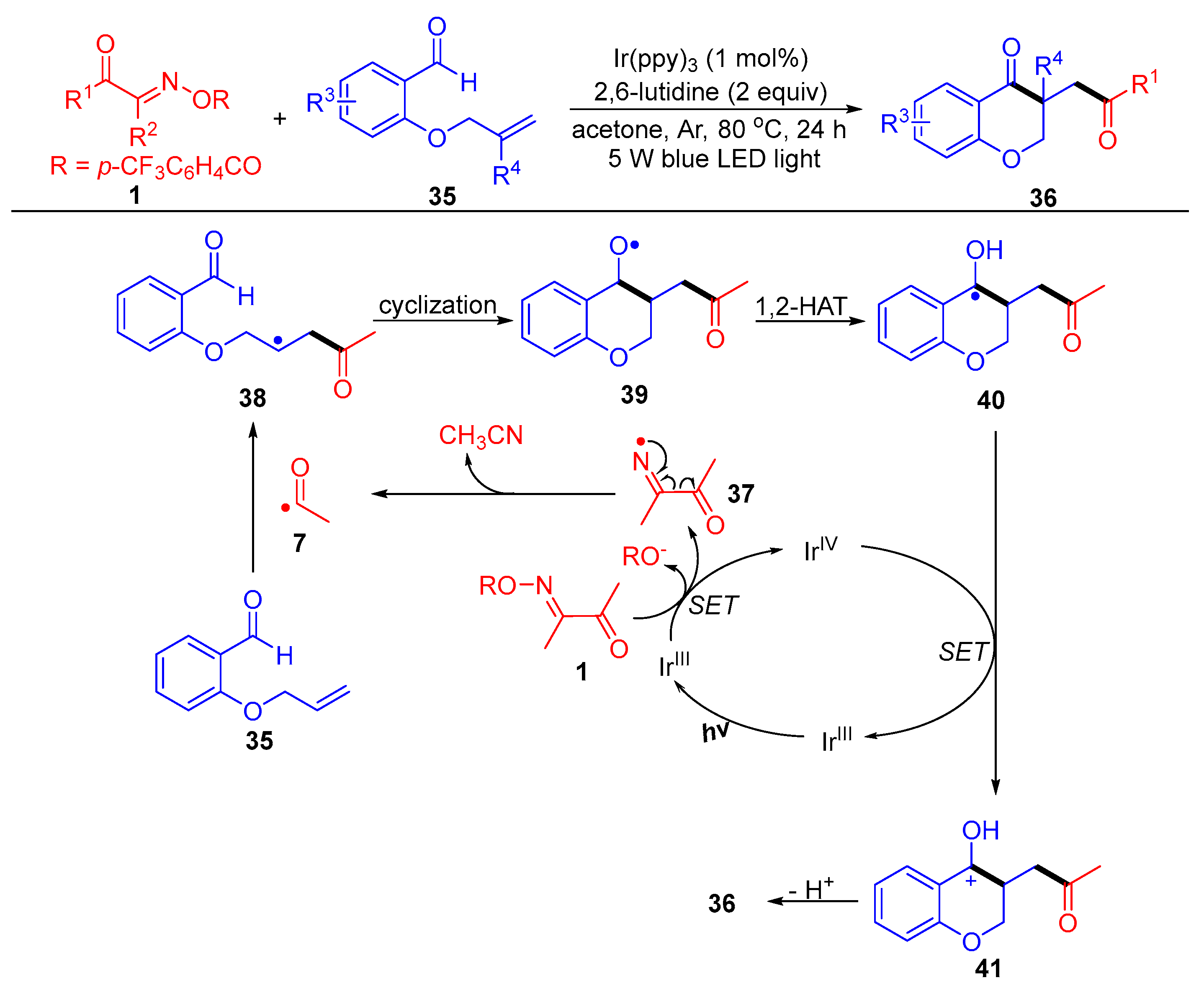{kind=link}
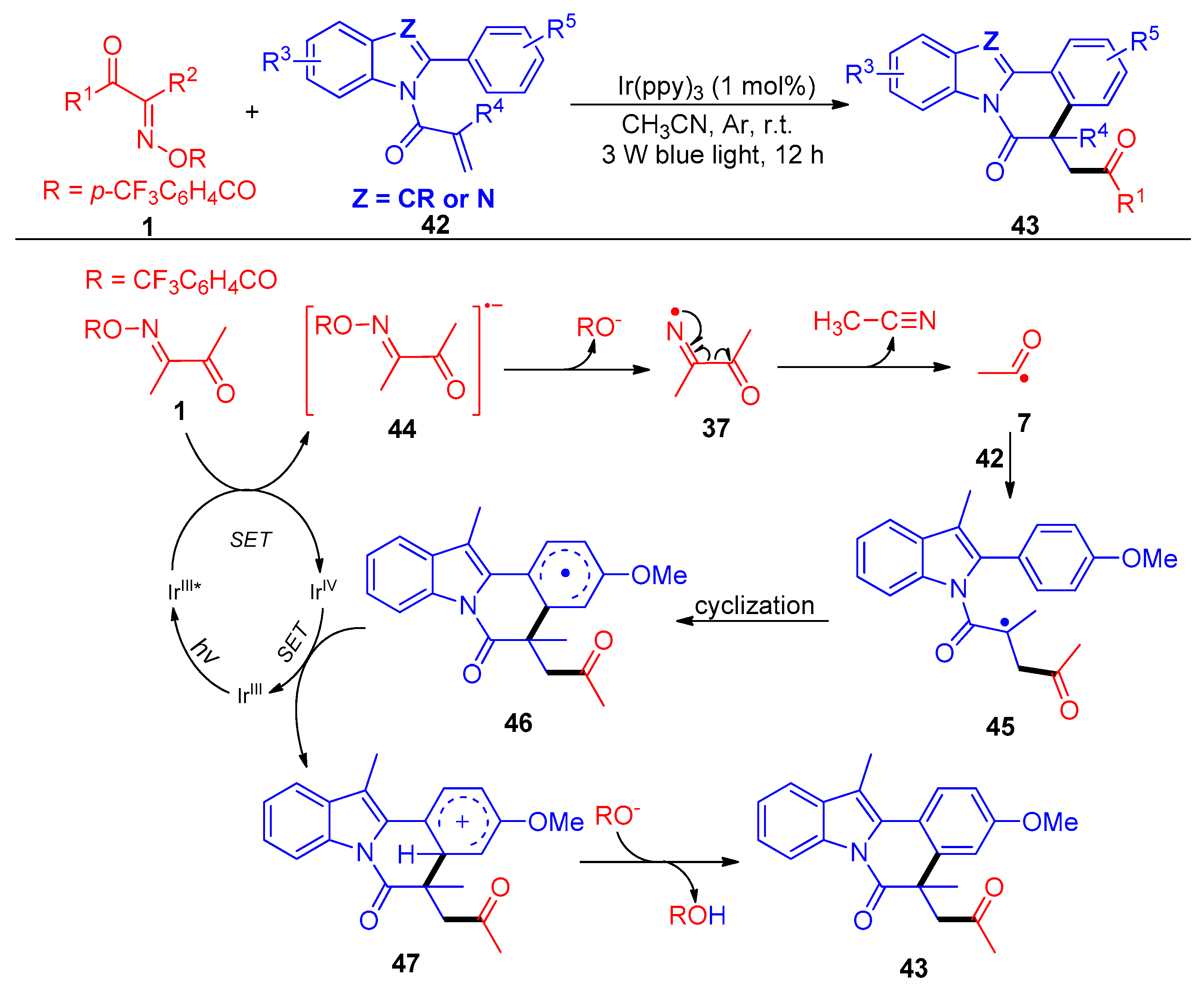{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
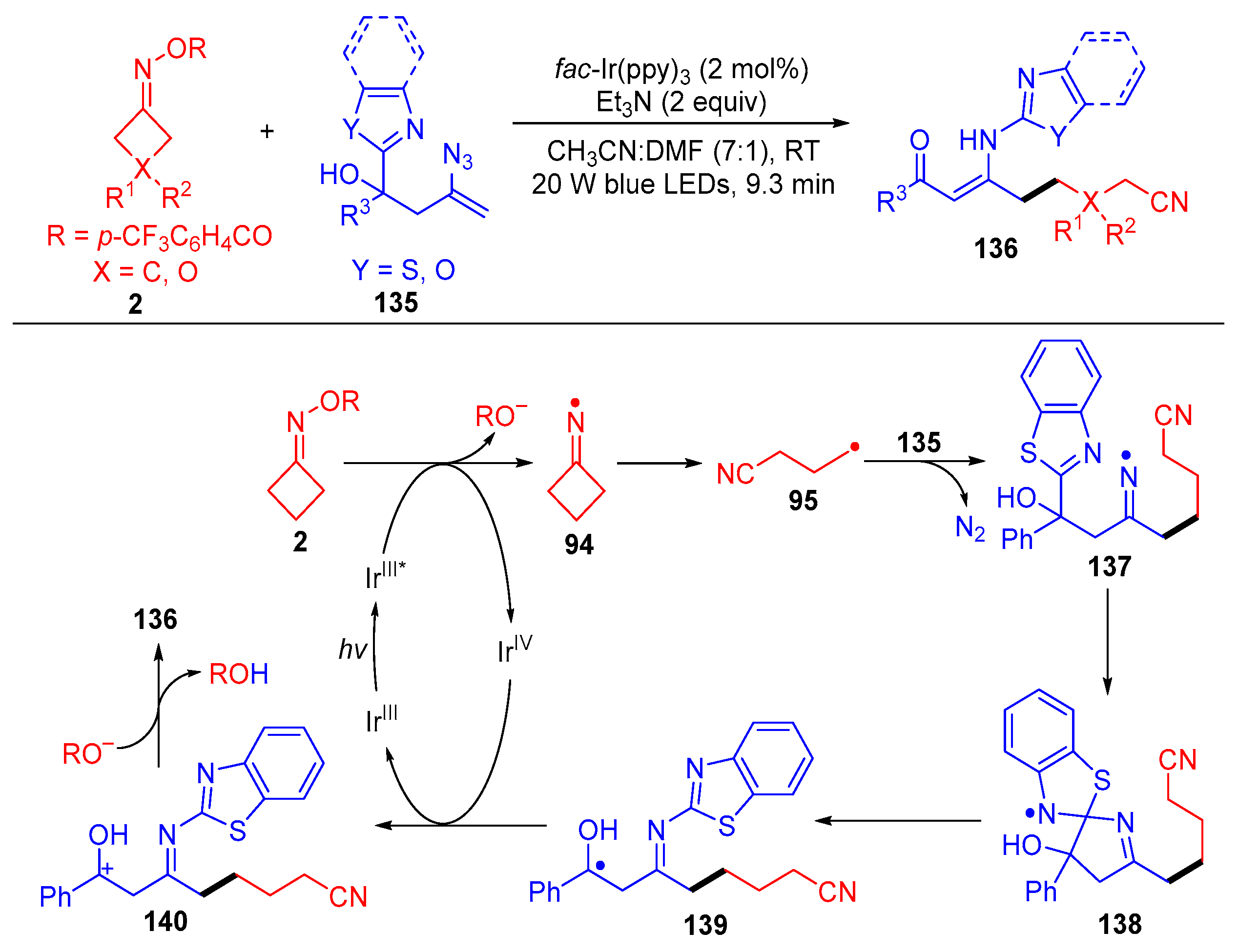{kind=link}
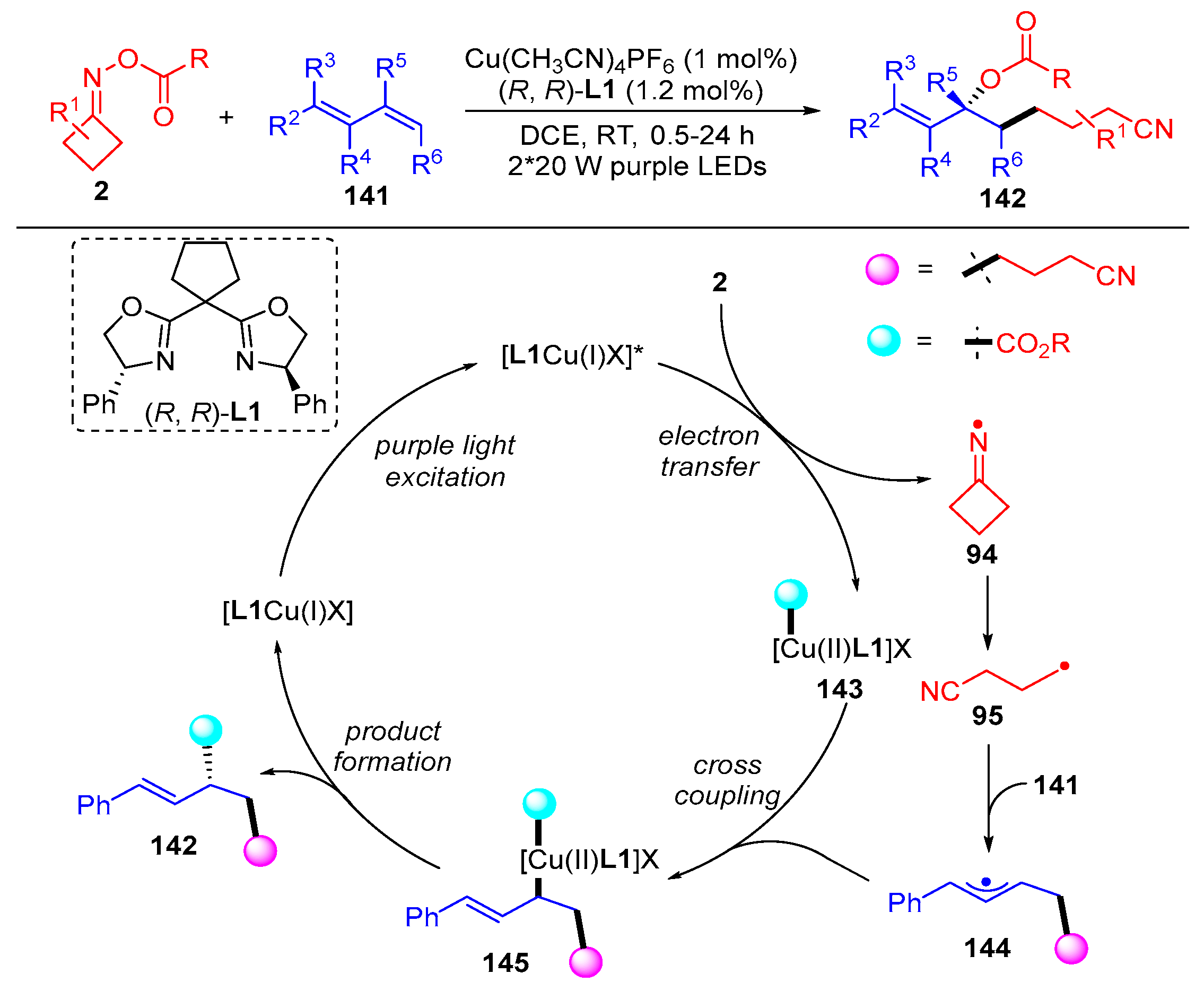{kind=link}
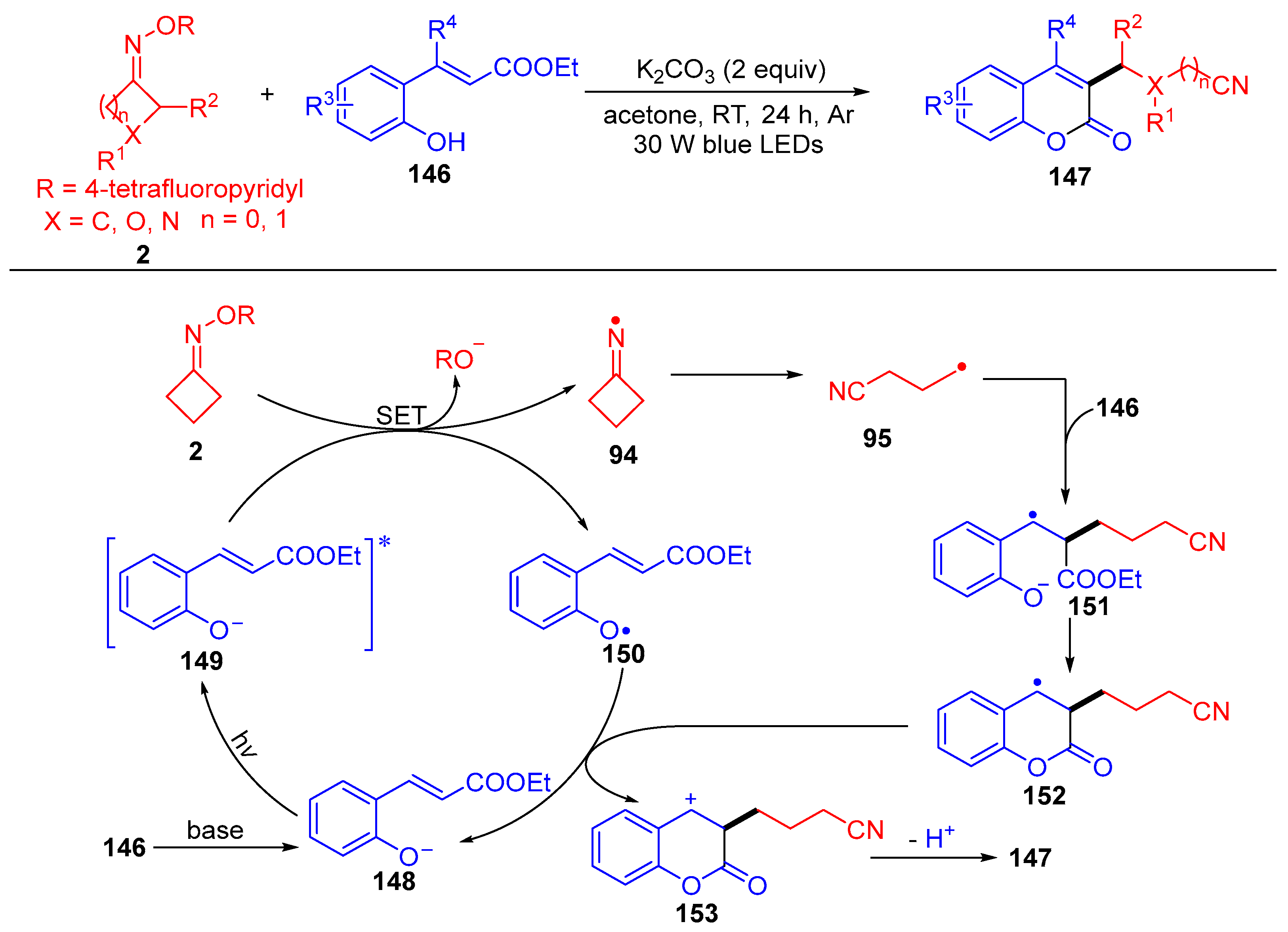{kind=link}
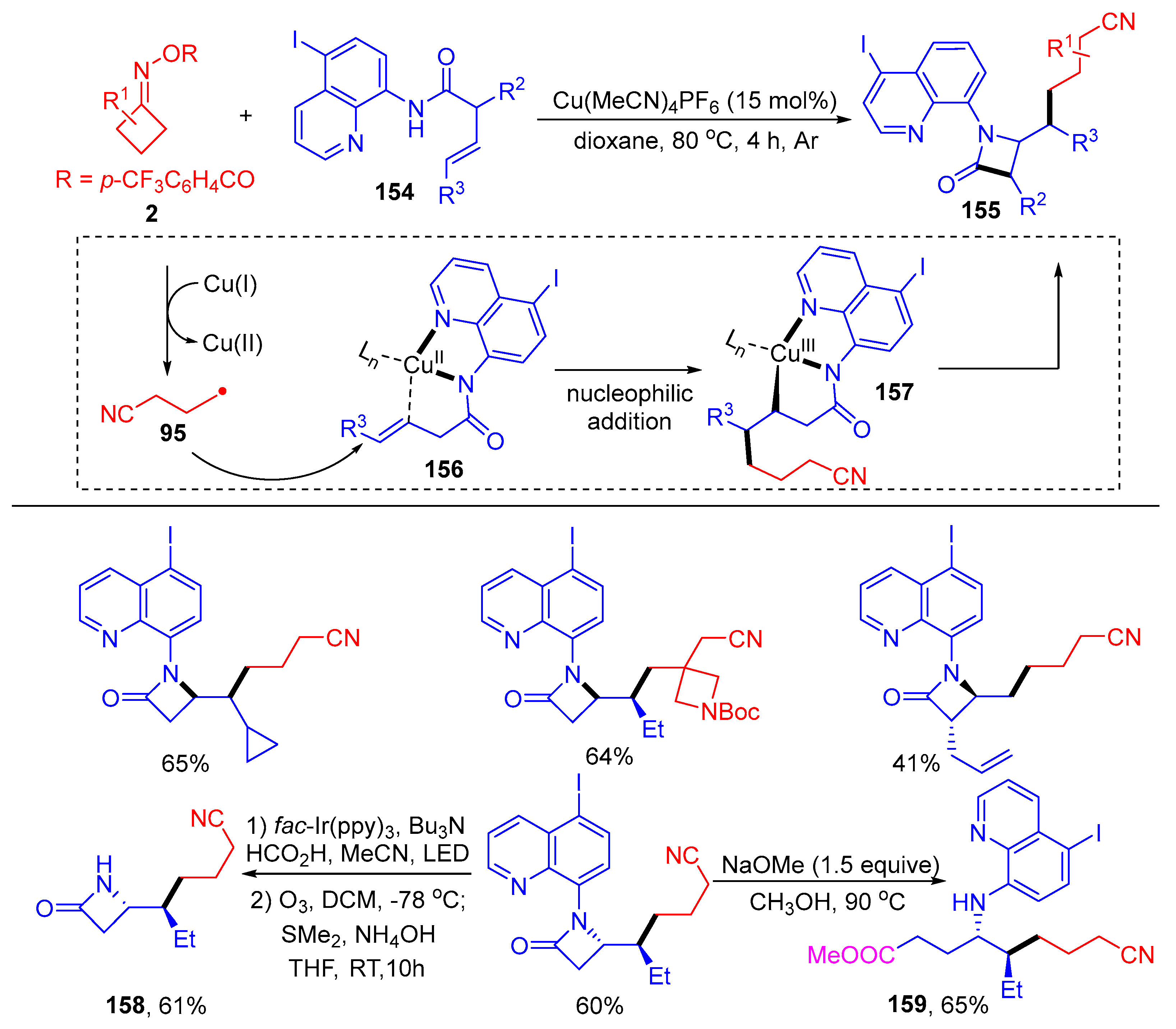{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
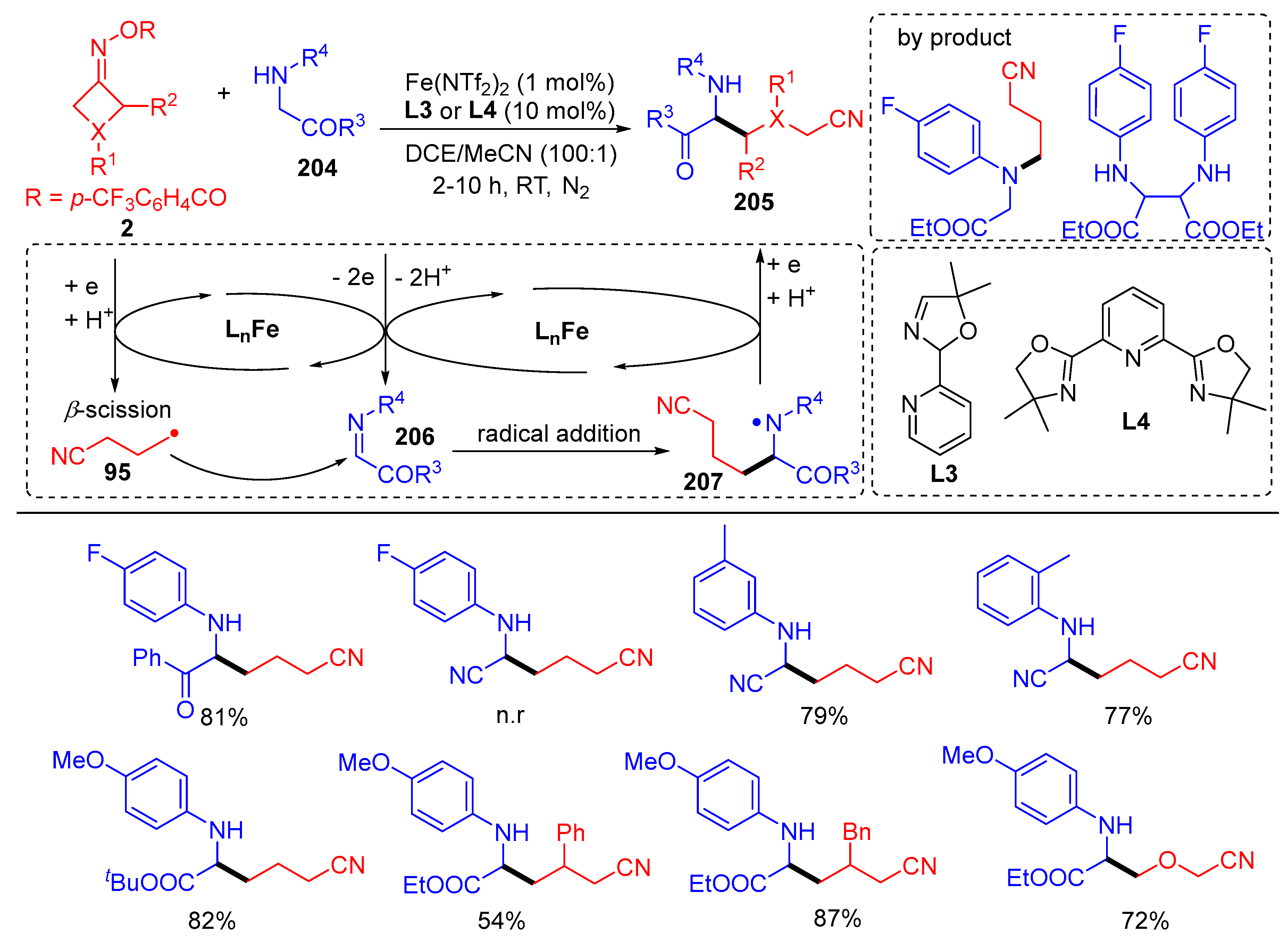{kind=link}
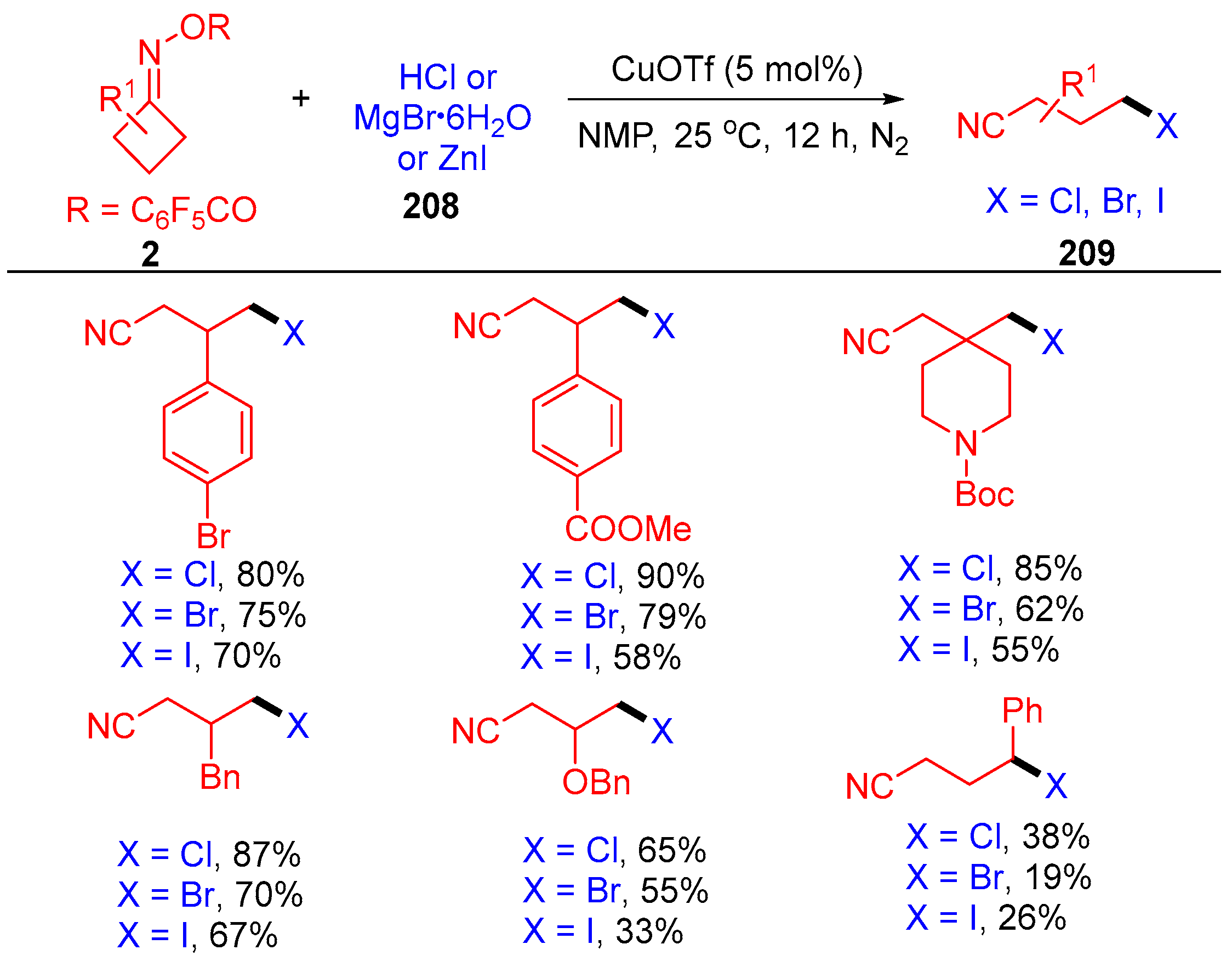{kind=link}
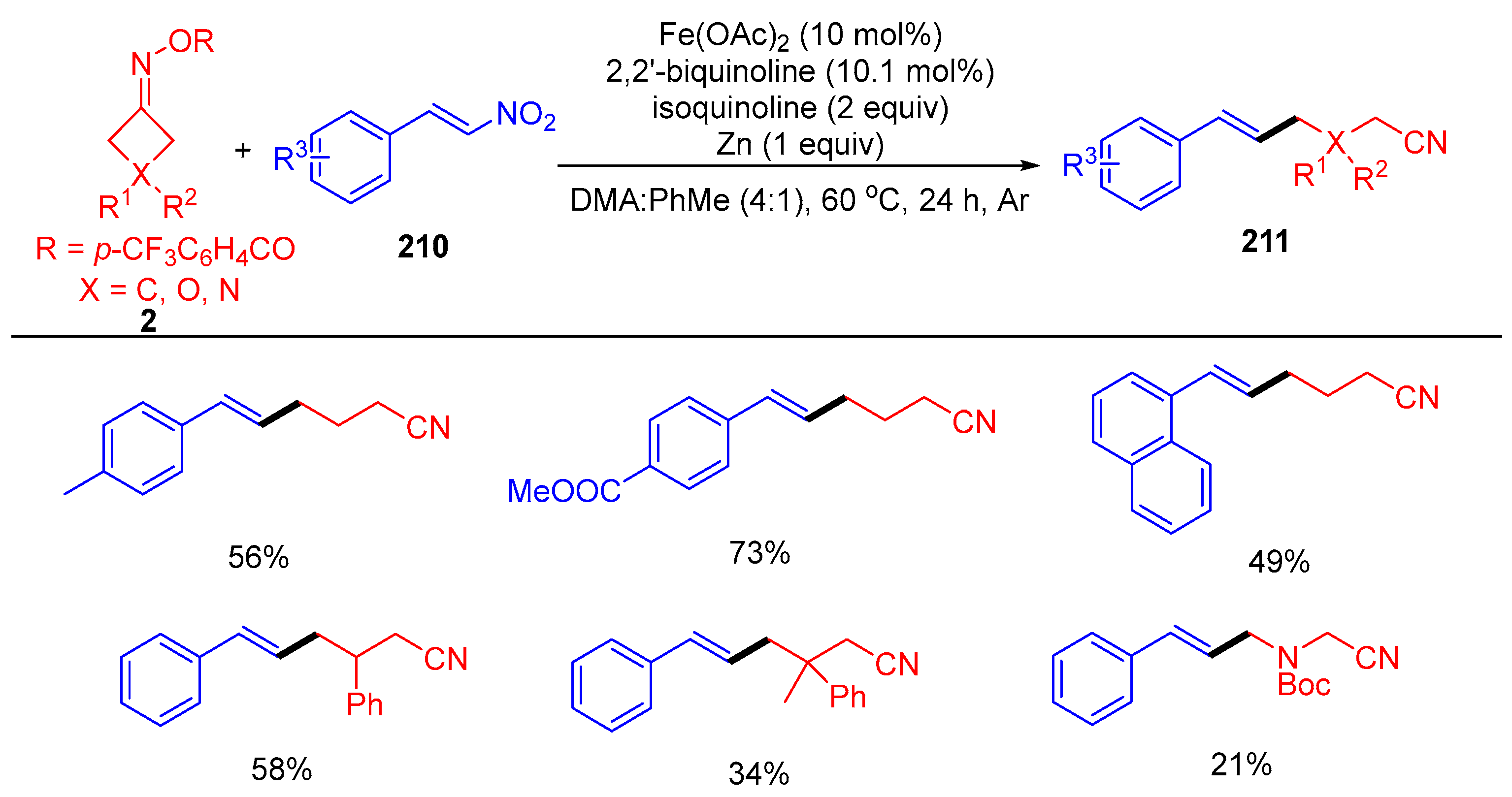{kind=link}
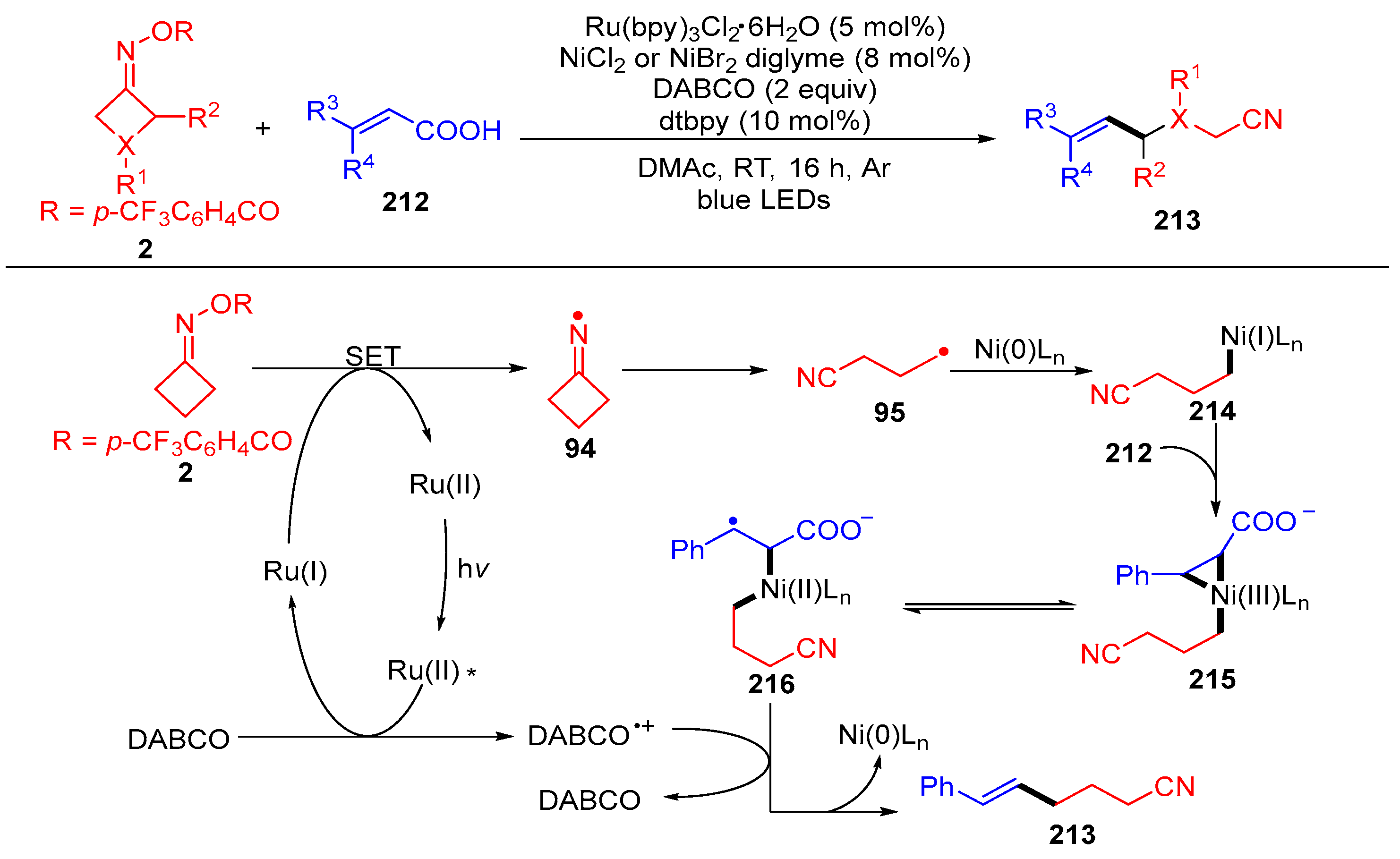{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Acyl Oxime Ester
2.1. Radical Addition
2.1.1. Acyl Addition to Alkenes
2.1.2. Acyl Addition to Alkynes
2.2. Radical Cross-Couple
3. Cyclic Ketoxime Esters
3.1. Radical Addition
3.1.1. Ring Opening and Addition to Alkenes
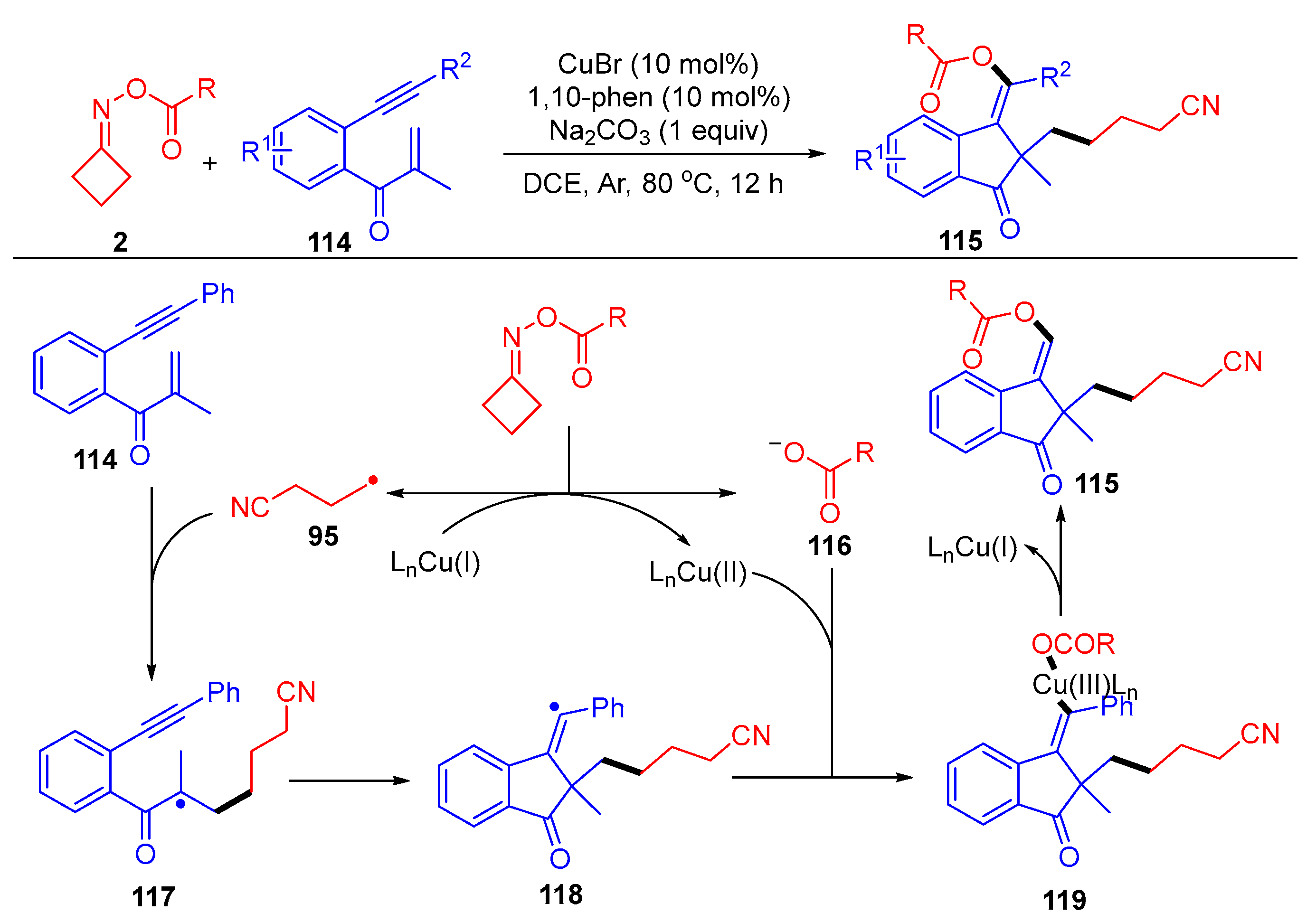
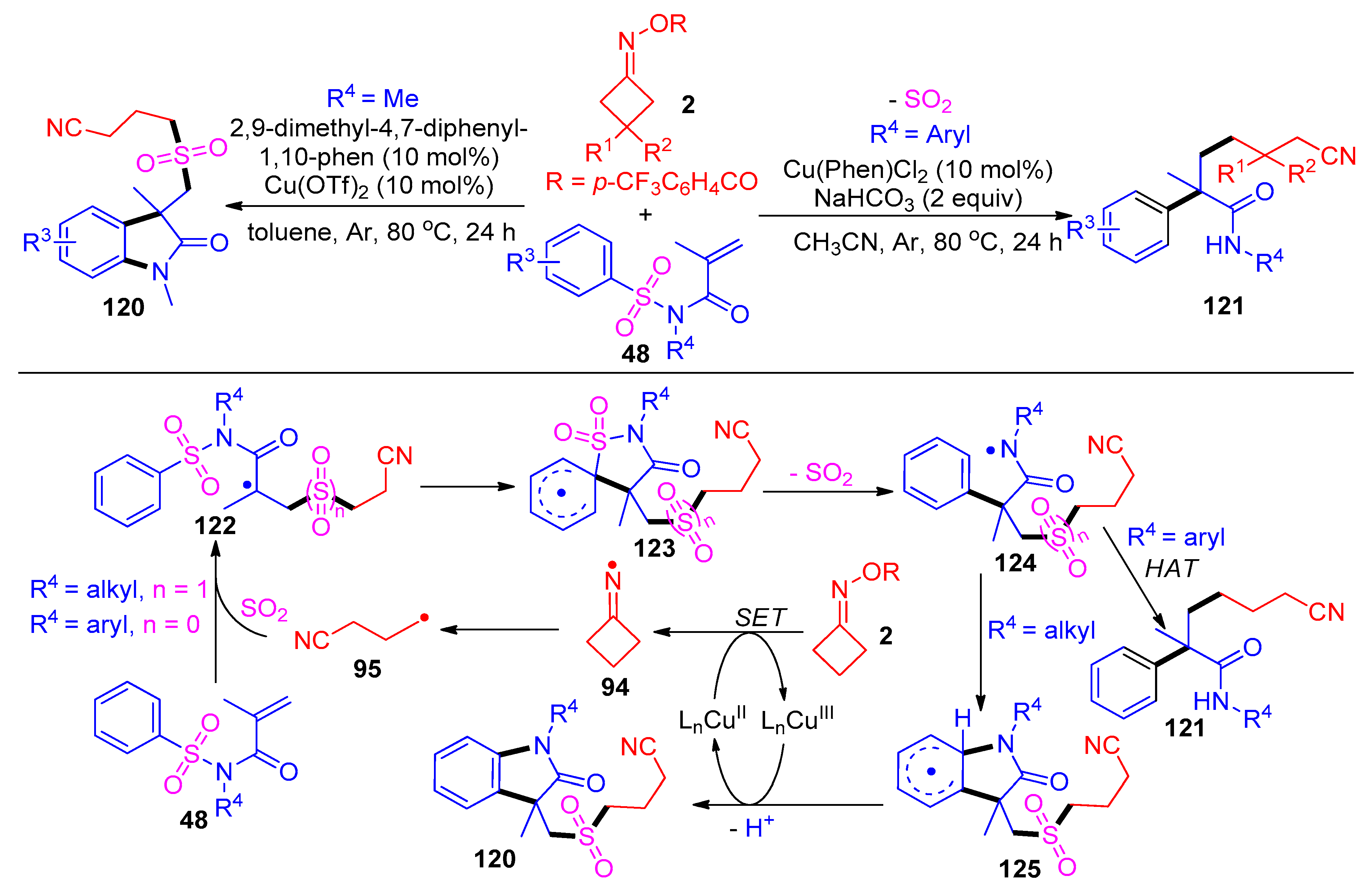
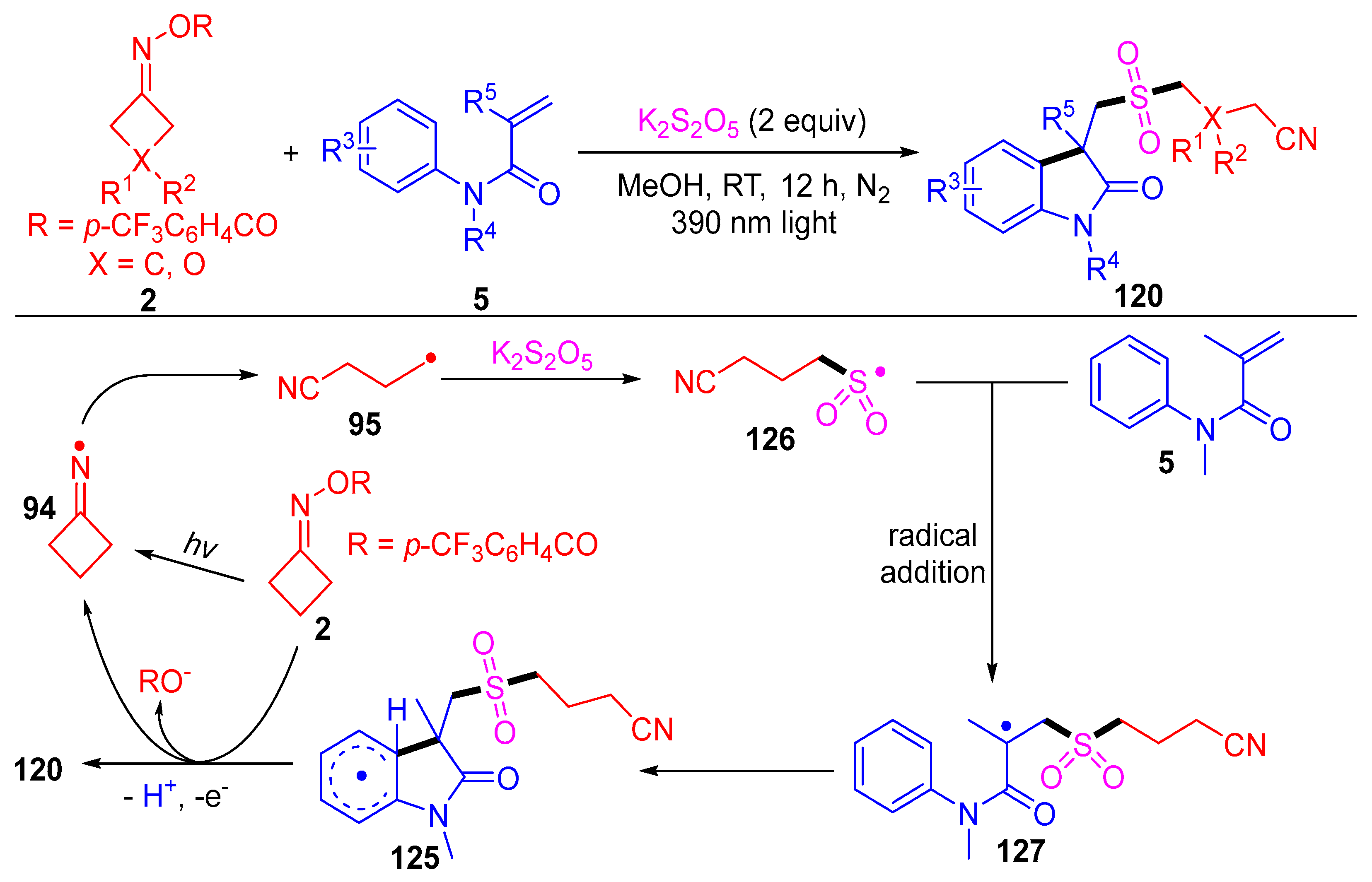
3.1.2. Ring Opening and Addition to Alkynes
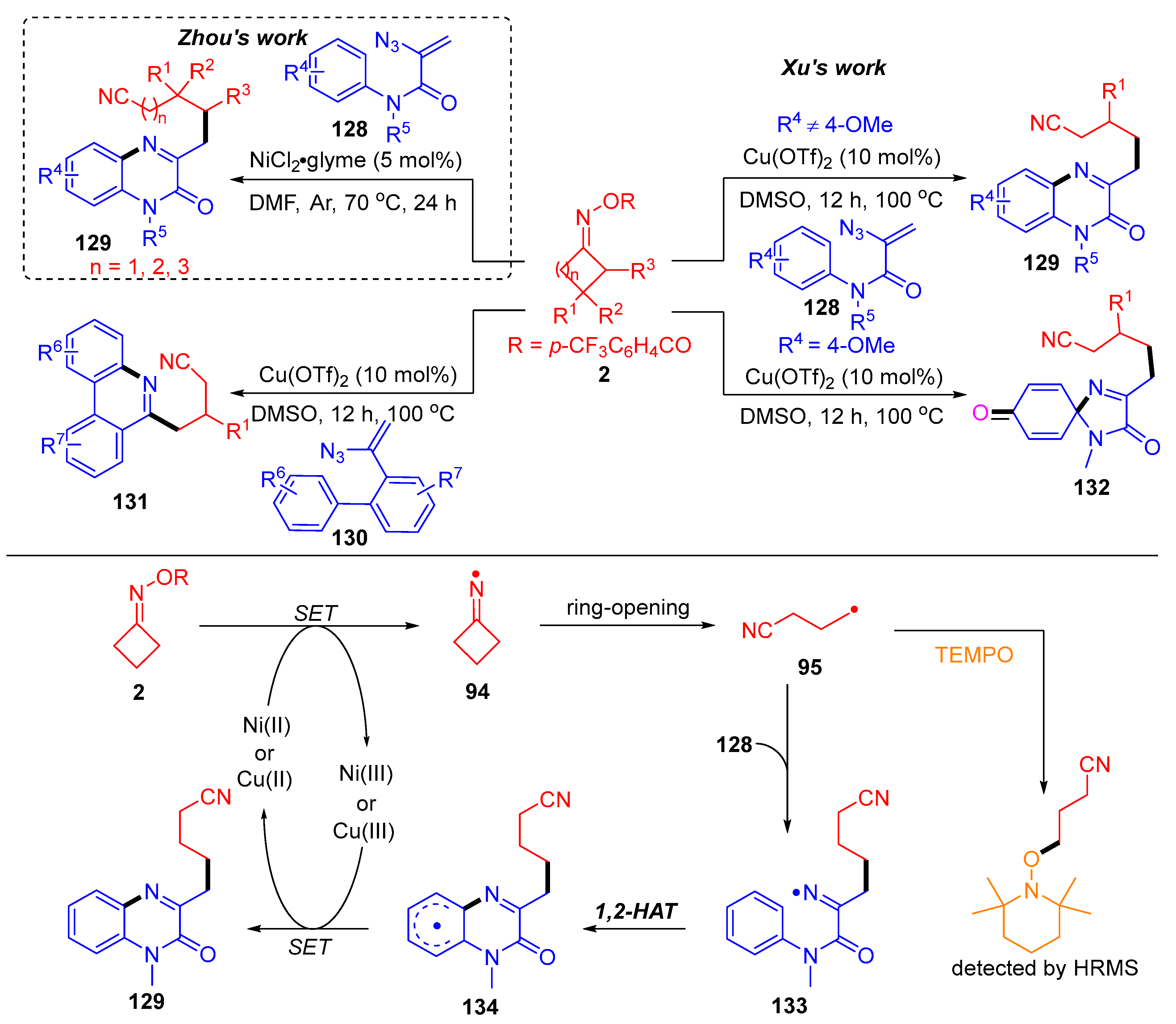
3.2. Radical Cross-Coupling
3.3. Radical Coupling
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Homer, J.A.; Sperry, J. Mushroom-Derived Indole Alkaloids. J. Nat. Prod. 2017, 80, 2178–2187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, C.; Zheng, Z.; Zhou, P.; Liu, W. Research Progress of Sulfoxonium Ylides in the Construction of Five/Six-Membered Nitrogen-Containing Heterocycles. Chin. J. Org. Chem. 2022, 42, 2745–2759. [Google Scholar] [CrossRef]
- Showalter, H.D.H. Progress in the Synthesis of Canthine Alkaloids and Ring-Truncated Congeners. J. Nat. Prod. 2013, 76, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Stepek, I.A.; Itami, K. Recent Advances in C–H Activation for the Synthesis of Π-Extended Materials. ACS Mater. Lett. 2020, 2, 951–974. [Google Scholar] [CrossRef]
- Qu, Z.; Wang, P.; Chen, X.; Deng, G.-J.; Huang, H. Visible-Light-Driven Cadogan Reaction. Chin. Chem. Lett. 2021, 32, 2582–2586. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, X.; Ji, X.; Huang, H. Visible Light-Induced Aerobic Oxidative Dehydrogenative Coupling of Thiophenols. Chin. J. Org. Chem. 2021, 41, 4704–4711. [Google Scholar] [CrossRef]
- Yang, S.; Song, J.; Dong, D.; Yang, H.; Zhou, M.; Zhang, H.; Wang, Z. Progress of N-Amino Pyridinium Salts as Nitrogen Radical Precursors in Visible Light Induced C-N Bond Formation Reactions. Chin. J. Org. Chem. 2022, 42, 4099–4110. [Google Scholar] [CrossRef]
- Zard, S.Z. Recent Progress in the Generation and Use of Nitrogen-Centred Radicals. Chem. Soc. Rev. 2008, 37, 1603–1618. [Google Scholar] [CrossRef]
- Xiong, T.; Zhang, Q. New Amination Strategies Based on Nitrogen-Centered Radical Chemistry. Chem. Soc. Rev. 2016, 45, 3069–3087. [Google Scholar] [CrossRef]
- Bolotin, D.S.; Bokach, N.A.; Demakova, M.Y.; Kukushkin, V.Y. Metal-Involving Synthesis and Reactions of Oximes. Chem. Rev. 2017, 117, 13039–13122. [Google Scholar] [CrossRef]
- Kitamura, M.; Narasaka, K. Synthesis of Aza-Heterocycles from Oximes by Amino-Heck Reaction. Chem. Rec. 2002, 2, 268–277. [Google Scholar] [CrossRef]
- Li, W.; Towne, T.B.; Liang, G.; Haight, A.R.; Barnes, D.M. Practical Large-Scale Synthesis of 6-Bromo-2-Naphthylmethanesulfonamide Using Semmler–Wolff Reaction. Org. Process Res. Dev. 2014, 18, 1696–1701. [Google Scholar] [CrossRef]
- Tabolin, A.A.; Ioffe, S.L. Rearrangement of N-Oxyenamines and Related Reactions. Chem. Rev. 2014, 114, 5426–5476. [Google Scholar] [CrossRef]
- Huang, H.; Cai, J.; Deng, G.-J. O-Acyl Oximes: Versatile Building Blocks for N-Heterocycle Formation in Recent Transition Metal Catalysis. Org. Biomol. Chem. 2016, 14, 1519–1530. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhao, J.; Shi, X.; Liu, L.; Zhu, Y.-P.; Sun, W.; Zhu, B. Recent Advances in Cyclization Reactions of Unsaturated Oxime Esters (Ethers): Synthesis of Versatile Functionalized Nitrogen-Containing Scaffolds. Org. Chem. Front. 2020, 7, 1948–1969. [Google Scholar] [CrossRef]
- Qu, Z.; Tian, T.; Deng, G.-J.; Huang, H. Diverse Catalytic Systems for Nitrogen-Heterocycle Formation from O-Acyl Ketoximes. Chin. Chem. Lett. 2023, 34, 107565. [Google Scholar] [CrossRef]
- Yi, H.; Zhang, G.; Wang, H.; Huang, Z.; Wang, J.; Singh, A.K.; Lei, A. Recent Advances in Radical C–H Activation/Radical Cross-Coupling. Chem. Rev. 2017, 117, 9016–9085. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-Y.; Chen, J.-R.; Wang, P.-Z.; Yang, M.-N.; Liang, D.; Xiao, W.-J. A Visible-Light-Driven Iminyl Radical-Mediated C−C Single Bond Cleavage/Radical Addition Cascade of Oxime Esters. Angew. Chem. Int. Ed. 2018, 57, 738–743. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Q.; Ji, X.; Deng, G.-J.; Huang, H. Bromide-Promoted Visible-Light-Induced Reductive Minisci Reaction with Aldehydes. ACS Catal. 2020, 10, 154–159. [Google Scholar] [CrossRef]
- Bhunia, A.; Studer, A. Recent Advances in Radical Chemistry Proceeding through Pro-Aromatic Radicals. Chem. 2021, 7, 2060–2100. [Google Scholar] [CrossRef]
- Wu, X.; Ma, Z.; Feng, T.; Zhu, C. Radical-Mediated Rearrangements: Past, Present, and Future. Chem. Soc. Rev. 2021, 50, 11577–11613. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Tian, T.; Tan, Y.; Ji, X.; Deng, G.-J.; Huang, H. Redox-Neutral Ketyl Radical Coupling/Cyclization of Carbonyls with N-Aryl Acrylamides through Consecutive Photoinduced Electron Transfer. Green Chem. 2022, 24, 7403–7409. [Google Scholar] [CrossRef]
- Ran, L.; Ren, Z.-H.; Wang, Y.-Y.; Guan, Z.-H. Copper-Catalyzed Homocoupling of Ketoxime Carboxylates for Synthesis of Symmetrical Pyrroles. Green Chem. 2014, 16, 112–115. [Google Scholar] [CrossRef]
- Ke, J.; Tang, Y.; Yi, H.; Li, Y.; Cheng, Y.; Liu, C.; Lei, A. Copper-Catalyzed Radical/Radical C-H/P-H Cross-Coupling: A-Phosphorylation of Aryl Ketone O-Acetyloximes. Angew. Chem. Int. Ed. 2015, 54, 6604–6607. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Booth, S.G.; Essafi, S.; Dryfe, R.A.W.; Leonori, D. Visible-Light-Mediated Generation of Nitrogen-Centered Radicals: Metal-Free Hydroimination and Iminohydroxylation Cyclization Reactions. Angew. Chem. Int. Ed. 2015, 54, 14017–14021. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Studer, A. Iminyl-Radicals by Oxidation of α-Imino-Oxy Acids: Photoredox-Neutral Alkene Carboimination for the Synthesis of Pyrrolines. Angew. Chem. Int. Ed. 2017, 56, 12273–12276. [Google Scholar] [CrossRef]
- Li, J.; Zhang, P.; Jiang, M.; Yang, H.; Zhao, Y.; Fu, H. Visible Light as a Sole Requirement for Intramolecular C(sp3)–H Imination. Org. Lett. 2017, 19, 1994–1997. [Google Scholar] [CrossRef]
- Shu, W.; Nevado, C. Visible-Light-Mediated Remote Aliphatic C−H Functionalizations through a 1,5-Hydrogen Transfer Cascade. Angew. Chem. Int. Ed. 2017, 56, 1881–1884. [Google Scholar] [CrossRef]
- Gu, Y.-R.; Duan, X.-H.; Chen, L.; Ma, Z.-Y.; Gao, P.; Guo, L.-N. Iminyl Radical-Triggered Intermolecular Distal C(sp3)–H Heteroarylation Via 1,5-Hydrogen-Atom Transfer (Hat) Cascade. Org. Lett. 2019, 21, 917–920. [Google Scholar] [CrossRef]
- Wu, X.; Zhu, C. Recent Advances in Ring-Opening Functionalization of Cycloalkanols by C–C σ-Bond Cleavage. Chem. Rec. 2018, 18, 587–598. [Google Scholar] [CrossRef]
- Xiao, T.; Huang, H.; Anand, D.; Zhou, L. Iminyl-Radical-Triggered C–C Bond Cleavage of Cycloketone Oxime Derivatives: Generation of Distal Cyano-Substituted Alkyl Radicals and Their Functionalization. Synthesis 2020, 52, 1585–1601. [Google Scholar] [CrossRef]
- Guin, S.; Majee, D.; Samanta, S. Recent Advances in Visible-Light-Driven Photocatalyzed Γ-Cyanoalkylation Reactions. Asian J. Org. Chem. 2021, 10, 1595–1618. [Google Scholar] [CrossRef]
- Yin, W.; Wang, X. Recent Advances in Iminyl Radical-Mediated Catalytic Cyclizations and Ring-Opening Reactions. New J. Chem. 2019, 43, 3254–3264. [Google Scholar] [CrossRef]
- Zhu, X.; Fu, H. Photocatalytic Cross-Couplings Via the Cleavage of N–O Bonds. Chem. Commun. 2021, 57, 9656–9671. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-Y.; Zhao, Q.-Q.; Chen, J.; Xiao, W.-J.; Chen, J.-R. When Light Meets Nitrogen-Centered Radicals: From Reagents to Catalysts. Acc. Chem. Res. 2020, 53, 1066–1083. [Google Scholar] [CrossRef] [PubMed]
- Xiong, P.; Xu, H.-C. Chemistry with Electrochemically Generated N-Centered Radicals. Acc. Chem. Res. 2019, 52, 3339–3350. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Xie, J.; Chen, Z.; Xiong, B.-Q.; Liu, Y.; Yang, C.-A.; Tang, K.-W. Visible-Light-Mediated Nitrogen-Centered Radical Strategy: Preparation of 3-Acylated Spiro[4,5]Trienones. Adv. Synth. Catal. 2021, 363, 4440–4446. [Google Scholar] [CrossRef]
- Fan, X.; Lei, T.; Liu, Z.; Yang, X.-L.; Cheng, Y.-Y.; Liang, G.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Benzyl C–O and C–N Bond Construction Via C–C Bond Dissociation of Oxime Ester under Visible Light Irradiation. Eur. J. Org. Chem. 2020, 2020, 1551–1558. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wu, S.; Wang, L.; Xia, Z.; Kuang, K.; Xu, Q.; Zhao, F.; Zhou, N. Visible-Light-Induced Cascade Cyclization of N-Propargyl Aromatic Amines and Acyl Oxime Esters: Rapid Access to 3-Acylated Quinolines. J. Org. Chem. 2022, 87, 10277–10284. [Google Scholar] [CrossRef]
- Fan, X.; Lei, T.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Photocatalytic C–C Bond Activation of Oxime Ester for Acyl Radical Generation and Application. Org. Lett. 2019, 21, 4153–4158. [Google Scholar] [CrossRef]
- Cheng, Y.-Y.; Lei, T.; Su, L.; Fan, X.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Visible Light Irradiation of Acyl Oxime Esters and Styrenes Efficiently Constructs β-Carbonyl Imides by a Scission and Four-Component Reassembly Process. Org. Lett. 2019, 21, 8789–8794. [Google Scholar] [CrossRef]
- Zheng, L.; Xia, P.-J.; Zhao, Q.-L.; Qian, Y.-E.; Jiang, W.-N.; Xiang, H.-Y.; Yang, H. Photocatalytic Hydroacylation of Alkenes by Directly Using Acyl Oximes. J. Org. Chem. 2020, 85, 11989–11996. [Google Scholar] [CrossRef]
- Bai, J.; Li, M.; Zhou, C.; Sha, Y.; Cheng, J.; Sun, J.; Sun, S. Visible-Light Photoredox-Catalyzed Dicarbofunctionalization of Styrenes with Oxime Esters and CO2: Multicomponent Reactions toward Cyanocarboxylic Acids and Γ-Keto Acids. Org. Lett. 2021, 23, 9654–9658. [Google Scholar] [CrossRef]
- Jin, S.; Sui, X.; Haug, G.C.; Nguyen, V.D.; Dang, H.T.; Arman, H.D.; Larionov, O.V. N-Heterocyclic Carbene-Photocatalyzed Tricomponent Regioselective 1,2-Diacylation of Alkenes Illuminates the Mechanistic Details of the Electron Donor–Acceptor Complex-Mediated Radical Relay Processes. ACS Catal. 2022, 12, 285–294. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Chen, P.; Li, X.-J.; Xiong, B.-Q.; Liu, Y.; Tang, K.-W.; Huang, P.-F. Visible-Light-Induced Dual Acylation of Alkenes for the Construction of 3-Substituted Chroman-4-Ones. J. Org. Chem. 2022, 87, 4263–4272. [Google Scholar] [CrossRef]
- Guo, Y.; Huang, P.-F.; Liu, Y.; He, B.-H. Visible-Light-Induced Acylation/Cyclization of Active Alkenes: Facile Access to Acylated Isoquinolinones. Org. Biomol. Chem. 2022, 20, 3767–3778. [Google Scholar] [CrossRef]
- Luo, Z.-T.; Fan, J.-H.; Xiong, B.-Q.; Liu, Y.; Tang, K.-W.; Huang, P.-F. Visible-Light-Induced Acylation/Arylation of Alkenes Via Aryl Migration/Desulfonylation. Eur. J. Org. Chem. 2022, 2022, e202200793. [Google Scholar] [CrossRef]
- Michael, J.P. Quinoline, Quinazoline and Acridone Alkaloids. Nat. Prod. Rep. 2002, 19, 742–760. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.; Tang, G.-L.; Pan, H.-X. Enzymatic Formation of Oxygen-Containing Heterocycles in Natural Product Biosynthesis. Chembiochem. 2018, 19, 2002–2022. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, R.; Stanek, F.; Stodulski, M. Recent Advances on Metal-Free, Visible-Light- Induced Catalysis for Assembling Nitrogen- and Oxygen-Based Heterocyclic Scaffolds. Molecules 2019, 24, 1533. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, Q.-L.; Zhou, C.-S.; Xiong, B.-Q.; Zhang, P.-L.; Yang, C.-A.; Tang, K.-W. Visible-Light-Mediated Ipso-Carboacylation of Alkynes: Synthesis of 3-Acylspiro[4,5]Trienones from N-(p-Methoxyaryl)Propiolamides and Acyl Chlorides. J. Org. Chem. 2018, 83, 2210–2218. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, Q.-L.; Chen, Z.; Zhou, Q.; Xiong, B.-Q.; Zhang, P.-L.; Tang, K.-W. Visible-Light Promoted One-Pot Synthesis of Sulfonated Spiro[4,5]Trienones from Propiolamides, Anilines and Sulfur Dioxide under Transition Metal-Free Conditions. Chem. Commun. 2019, 55, 12212–12215. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Fan, J.-H.; Yu, W.-Q.; Xiong, B.-Q.; Liu, Y.; Tang, K.-W.; Xie, J. Alkylation/Ipso-Cyclization of Active Alkynes Leading to 3-Alkylated Aza- and Oxa-Spiro[4,5]-Trienones. J. Org. Chem. 2022, 87, 5643–5659. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Sun, S.; Xia, M.; Gu, N.; Cheng, J. Copper-Catalyzed Radical 1,2-Cyclization of Indoles with Arylsulfonyl Hydrazides: Access to 2-Thiolated 3h-Pyrrolo[1,2-a]Indoles. Org. Chem. Front. 2017, 4, 2153–2155. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Q.-L.; Chen, Z.; Chen, P.; Tang, K.-W.; Zhou, Q.; Xie, J. Visible-Light-Induced Cascade Sulfonylation/Cyclization of N-Propargylindoles with Aryldiazonium Tetrafluoroborates via the Insertion of Sulfur Dioxide. Org. Biomol. Chem. 2019, 17, 10020–10029. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Z.; Wang, Q.-L.; Chen, P.; Xie, J.; Xiong, B.-Q.; Zhang, P.-L.; Tang, K.-W. Visible Light-Catalyzed Cascade Radical Cyclization of N-Propargylindoles with Acyl Chlorides for the Synthesis of 2-Acyl-9h-Pyrrolo[1,2-a]Indoles. J. Org. Chem. 2020, 85, 2385–2394. [Google Scholar] [CrossRef]
- Liu, T.; Ding, Q.; Zong, Q.; Qiu, G. Radical 5-exo Cyclization of Alkynoates with 2-Oxoacetic Acids for Synthesis of 3-Acylcoumarins. Org. Chem. Front. 2015, 2, 670–673. [Google Scholar] [CrossRef]
- Mi, X.; Wang, C.; Huang, M.; Wu, Y.; Wu, Y. Preparation of 3-Acyl-4-Arylcoumarins via Metal-Free Tandem Oxidative Acylation/Cyclization between Alkynoates with Aldehydes. J. Org. Chem. 2015, 80, 148–155. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, L.; Gao, Y.; Tang, G.; Zhao, Y. Synthesis of 3-Phosphinoylquinolines via a Phosphinoylation–Cyclization–Aromatization Process Mediated by tert-Butyl Hydroperoxide. RSC Adv. 2016, 6, 60922–60925. [Google Scholar] [CrossRef]
- Sun, D.; Yin, K.; Zhang, R. Visible-Light-Induced Multicomponent Cascade Cycloaddition Involving N-Propargyl Aromatic Amines, Diaryliodonium Salts and Sulfur Dioxide: Rapid Access to 3-Arylsulfonylquinolines. Chem. Commun. 2018, 54, 1335–1338. [Google Scholar] [CrossRef]
- Liu, J.; Wang, M.; Li, L.; Wang, L. Electrooxidative Tandem Cyclization of N-Propargylanilines with Sulfinic Acids for Rapid Access to 3-Arylsulfonylquinoline Derivatives. Green Chem. 2021, 23, 4733–4740. [Google Scholar] [CrossRef]
- Zhou, Q.; Xiong, F.-T.; Chen, P.; Xiong, B.-Q.; Tang, K.-W.; Liu, Y. The Visible-Light-Induced Acylation/Cyclization of Alkynoates with Acyl Oximes for the Construction of 3-Acylcoumarins. Org. Biomol. Chem. 2021, 19, 9012–9020. [Google Scholar] [CrossRef]
- Yu, W.-Q.; Xie, J.; Chen, Z.; Xiong, B.-Q.; Liu, Y.; Tang, K.-W. Visible-Light-Induced Transition-Metal-Free Nitrogen-Centered Radical Strategy for the Synthesis of 2-Acylated 9H-Pyrrolo[1,2-a]Indoles. J. Org. Chem. 2021, 86, 13720–13733. [Google Scholar] [CrossRef]
- He, B.-Q.; Gao, Y.; Wang, P.-Z.; Wu, H.; Zhou, H.-B.; Liu, X.-P.; Chen, J.-R. Dual Photoredox/Palladium-Catalyzed C–H Acylation of 2-Arylpyridines with Oxime Esters. Synlett 2021, 32, 373–377. [Google Scholar]
- Qian, H.; Chen, J.; Zhang, B.; Cheng, Y.; Xiao, W.-J.; Chen, J.-R. Visible-Light-Driven Photoredox-Catalyzed Three-Component Radical Cyanoalkylfluorination of Alkenes with Oxime Esters and a Fluoride Ion. Org. Lett. 2021, 23, 6987–6992. [Google Scholar] [CrossRef]
- Chen, L.; Jin, S.; Gao, J.; Liu, T.; Shao, Y.; Feng, J.; Wang, K.; Lu, T.; Du, D. N-Heterocyclic Carbene/Magnesium Cocatalyzed Radical Relay Assembly of Aliphatic Keto Nitriles. Org. Lett. 2021, 23, 394–399. [Google Scholar] [CrossRef]
- Gao, Y.; Quan, Y.; Li, Z.; Gao, L.; Zhang, Z.; Zou, X.; Yan, R.; Qu, Y.; Guo, K. Organocatalytic Three-Component 1,2-Cyanoalkylacylation of Alkenes Via Radical Relay. Org. Lett. 2021, 23, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lin, C.; Zhang, S.; Zhang, X.; Zhang, J.; Xing, L.; Guo, Y.; Feng, J.; Gao, J.; Du, D. 1,4-Alkylcarbonylation of 1,3-Enynes to Access Tetra-Substituted Allenyl Ketones Via an NHC-Catalyzed Radical Relay. ACS Catal. 2021, 11, 13363–13373. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, X.-P.; Duan, X.; Qin, L.-Z.; Yuan, X.; Wu, M.-Y.; Liu, J.; Zhu, S.-S.; Qiu, J.-K.; Guo, K. Photoinduced Merging with Copper- or Nickel-Catalyzed 1,4-Cyanoalkylarylation of 1,3-Enynes to Access Multiple Functionalizatized Allenes in Batch and Continuous Flow. Chin. J. Chem. 2022, 40, 1537–1545. [Google Scholar] [CrossRef]
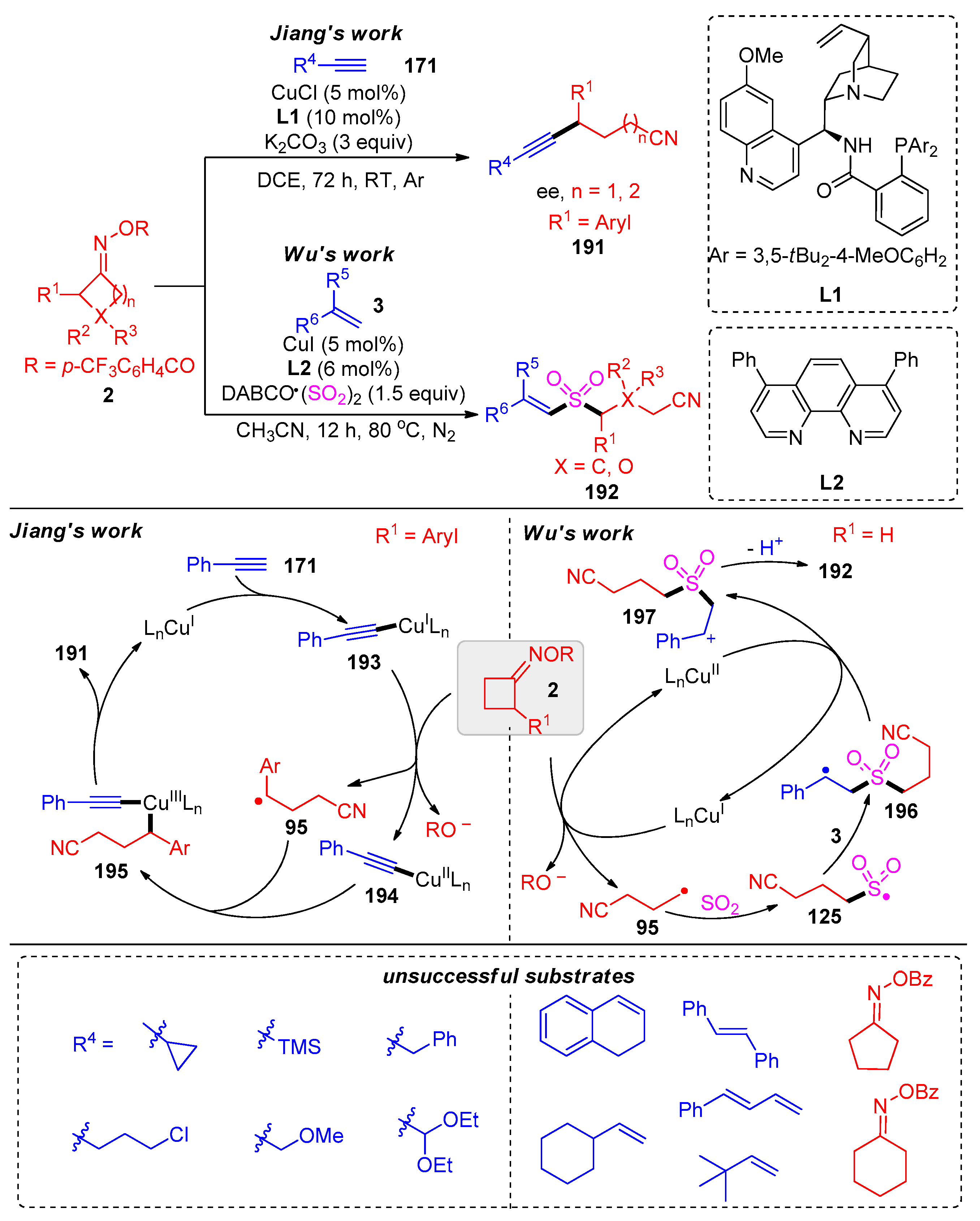
- Wang, S.-C.; Shen, Y.-T.; Zhang, T.-S.; Hao, W.-J.; Tu, S.-J.; Jiang, B. Cyclic Oxime Esters as Deconstructive Bifunctional Reagents for Cyanoalkyl Esterification of 1,6-Enynes. J. Org. Chem. 2021, 86, 15488–15497. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, C.-T.; Bao, Q.-F.; Qiu, Y.-F.; Wei, W.-X.; Li, X.-S.; Wang, Y.-Z.; Zhang, Z.; Wang, J.-L.; Liang, Y.-M. Copper-Catalyzed Radical Aryl Migration Approach for the Preparation of Cyanoalkylsulfonylated Oxindoles/Cyanoalkyl Amides. Org. Lett. 2021, 23, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Teng, F.; Du, J.; Xun, C.; Zhu, M.; Lu, Z.; Jiang, H.; Chen, Y.; Li, Y.; Gui, Q.-W. Photoinduced Efficient Synthesis of Cyanoalkylsulfonylated Oxindoles Via Sulfur Dioxide Insertion. Org. Biomol. Chem. 2021, 19, 8929–8933. [Google Scholar] [CrossRef] [PubMed]
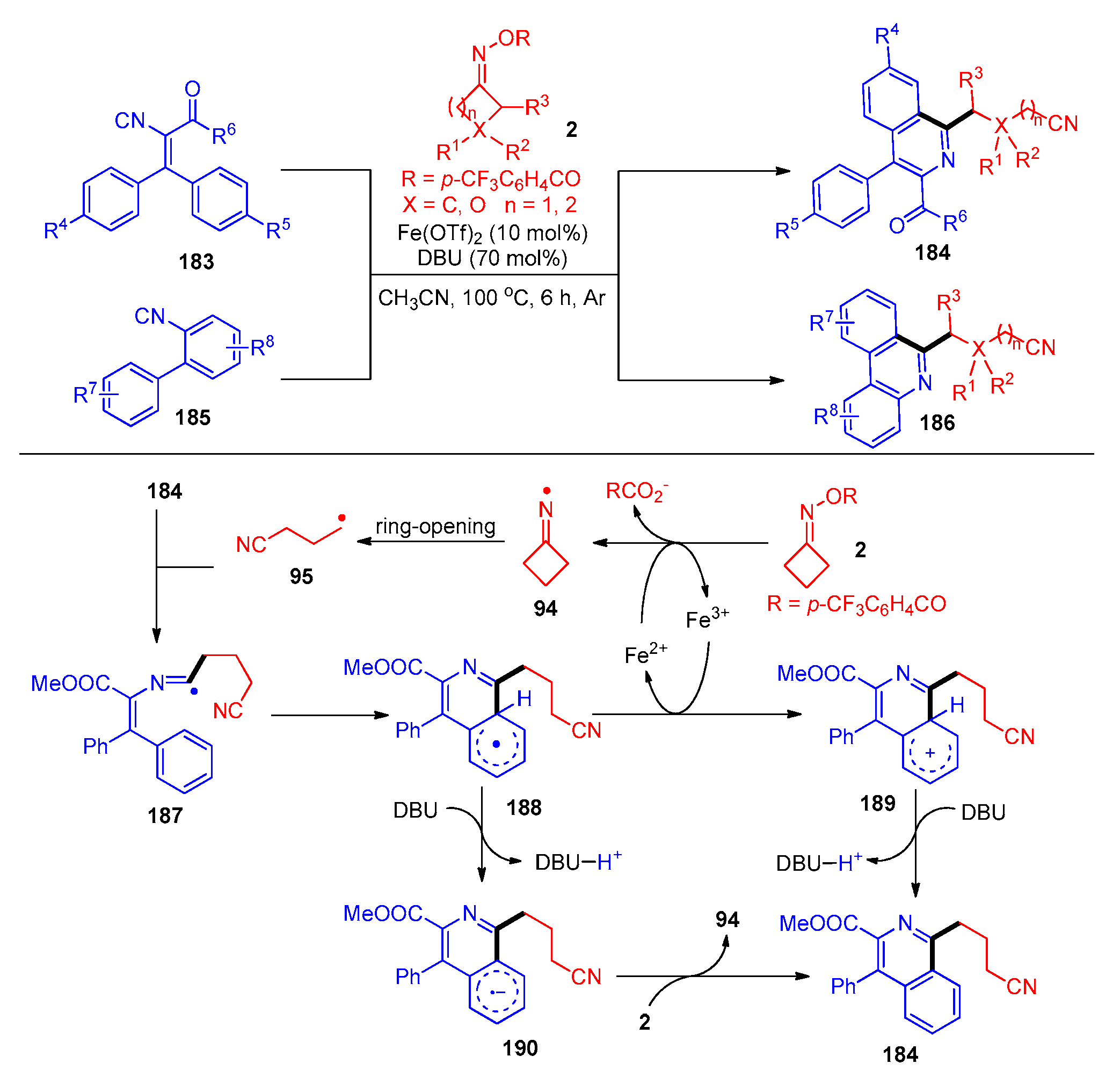
- Zhou, N.; Wu, S.; Kuang, K.; Wu, M.; Zhang, M. Ni-Catalyzed Radical Cyclization of Vinyl Azides with Cyclobutanone Oxime Esters to Access Cyanoalkyl Containing Quinoxalin-2(1H)-Ones. Org. Biomol. Chem. 2021, 19, 4697–4700. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Lin, L.; Li, G.; Kong, X.; Xu, B. Synthesis of Phenanthridine and Quinoxaline Derivatives via Copper-Catalyzed Radical Cyanoalkylation of Cyclobutanone Oxime Esters and Vinyl Azides. Chin. J. Chem. 2021, 39, 1948–1952. [Google Scholar] [CrossRef]
- Duan, X.; Sun, Q.; Yuan, X.; Qin, L.-Z.; Zhang, X.-P.; Liu, J.; Wu, M.-Y.; Zhu, S.-S.; Ma, C.-L.; Qiu, J.-K.; et al. Photoinduced Remote Heteroaryl Migration Accompanied by Cyanoalkylacylation in Continuous Flow. Green Chem. 2021, 23, 8916–8921. [Google Scholar] [CrossRef]
- Chen, J.; Liang, Y.-J.; Wang, P.-Z.; Li, G.-Q.; Zhang, B.; Qian, H.; Huan, X.-D.; Guan, W.; Xiao, W.-J.; Chen, J.-R. Photoinduced Copper-Catalyzed Asymmetric C–O Cross-Coupling. J. Am. Chem. Soc. 2021, 143, 13382–13392. [Google Scholar] [CrossRef]
- Hu, Y.-Z.; Ye, Z.-P.; Xia, P.-J.; Song, D.; Li, X.-J.; Liu, Z.-L.; Liu, F.; Chen, K.; Xiang, H.-Y.; Yang, H. Visible-Light-Driven, Photocatalyst-Free Cascade to Access 3-Cyanoalkyl Coumarins from Ortho-Hydroxycinnamic Esters. J. Org. Chem. 2021, 86, 4245–4253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lv, X.; Yu, H.; Bai, Z.; Chen, G.; He, G. Β-Lactam Synthesis Via Copper-Catalyzed Directed Aminoalkylation of Unactivated Alkenes with Cyclobutanone O-Benzoyloximes. Org. Lett. 2021, 23, 3620–3625. [Google Scholar] [CrossRef]
- Qiu, G.; Lai, L.; Cheng, J.; Wu, J. Recent Advances in the Sulfonylation of Alkenes with the Insertion of Sulfur Dioxide Via Radical Reactions. Chem. Commun. 2018, 54, 10405–10414. [Google Scholar] [CrossRef]
- Qiu, G.; Zhou, K.; Wu, J. Recent Advances in the Sulfonylation of C–H Bonds with the Insertion of Sulfur Dioxide. Chem. Commun. 2018, 54, 12561–12569. [Google Scholar] [CrossRef]
- Chen, P.; Chen, Z.; Xiong, B.-Q.; Liang, Y.; Tang, K.-W.; Xie, J.; Liu, Y. Visible-Light-Mediated Cascade Cyanoalkylsulfonylation/Cyclization of Alkynoates Leading to Coumarins Via SO2 Insertion. Org. Biomol. Chem. 2021, 19, 3181–3190. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xia, Y.; Tong, J.; Ouyang, L.; Lai, Y.; Luo, R.; Liao, J. Photoinduced Radical Cascade Cyclization of Acetylenic Acid Esters with Oxime Esters to Access Cyanalkylated Coumarins. Org. Biomol. Chem. 2022, 20, 5239–5244. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Xia, Z.; Wu, S.; Kuang, K.; Xu, Q.; Zhang, M. Visible-Light-Induced Multicomponent Cascade Cycloaddition of N-Propargyl Aromatic Amines, Cyclobutanone Oxime Esters, and K2S2O5: Access to Cyanoalkylsulfonylated Quinolines. J. Org. Chem. 2021, 86, 15253–15262. [Google Scholar] [CrossRef]
- He, F.-S.; Yao, Y.; Tang, Z.; Qiu, Y.; Xie, W.; Wu, J. Synthesis of β-Cyanoalkylsulfonylated Vinyl Selenides through a Four-Component Reaction. Chem. Commun. 2021, 57, 12603–12606. [Google Scholar] [CrossRef]
- Zhou, N.; Xu, Q.; Xia, Z.; Kuang, K.; Wu, S.; Li, W.; Zhang, M. Palladium-Catalyzed Radical Cascade Cyanoalkylsulfonylation/Cyclization of 3-Arylethynyl-[1,1′-Biphenyl]-2-Carbonitriles with Cyclobutanone Oxime Esters and Dabso. Chem. Commun. 2022, 58, 2335–2338. [Google Scholar] [CrossRef]
- Xue, D.; Liu, R.; Zhang, D.; Li, N.; Xue, Y.; Ge, Q.; Shao, L. Iron-Catalyzed Radical Cascade Cyclization of Oxime Esters with Isocyanides: Synthesis of 1-Cyanoalkyl Isoquinolines and 6-Cyanoalkyl Phenanthridines. Org. Biomol. Chem. 2021, 19, 8597–8606. [Google Scholar] [CrossRef]
- Zuo, H.-D.; Zhu, S.-S.; Hao, W.-J.; Wang, S.-C.; Tu, S.-J.; Jiang, B. Copper-Catalyzed Asymmetric Deconstructive Alkynylation of Cyclic Oximes. ACS Catal. 2021, 11, 6010–6019. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Zeng, L.-H.; Zhao, Y.; Zhu, T.; Wu, J. A Copper-Catalyzed Three-Component Reaction of Alkenes, Cycloketone Oximes and DABCO·(SO2)2: Direct C(sp2)-H Cyanoalkylsulfonylation. Chin. Chem. Lett. 2022, 33, 2383–2386. [Google Scholar] [CrossRef]
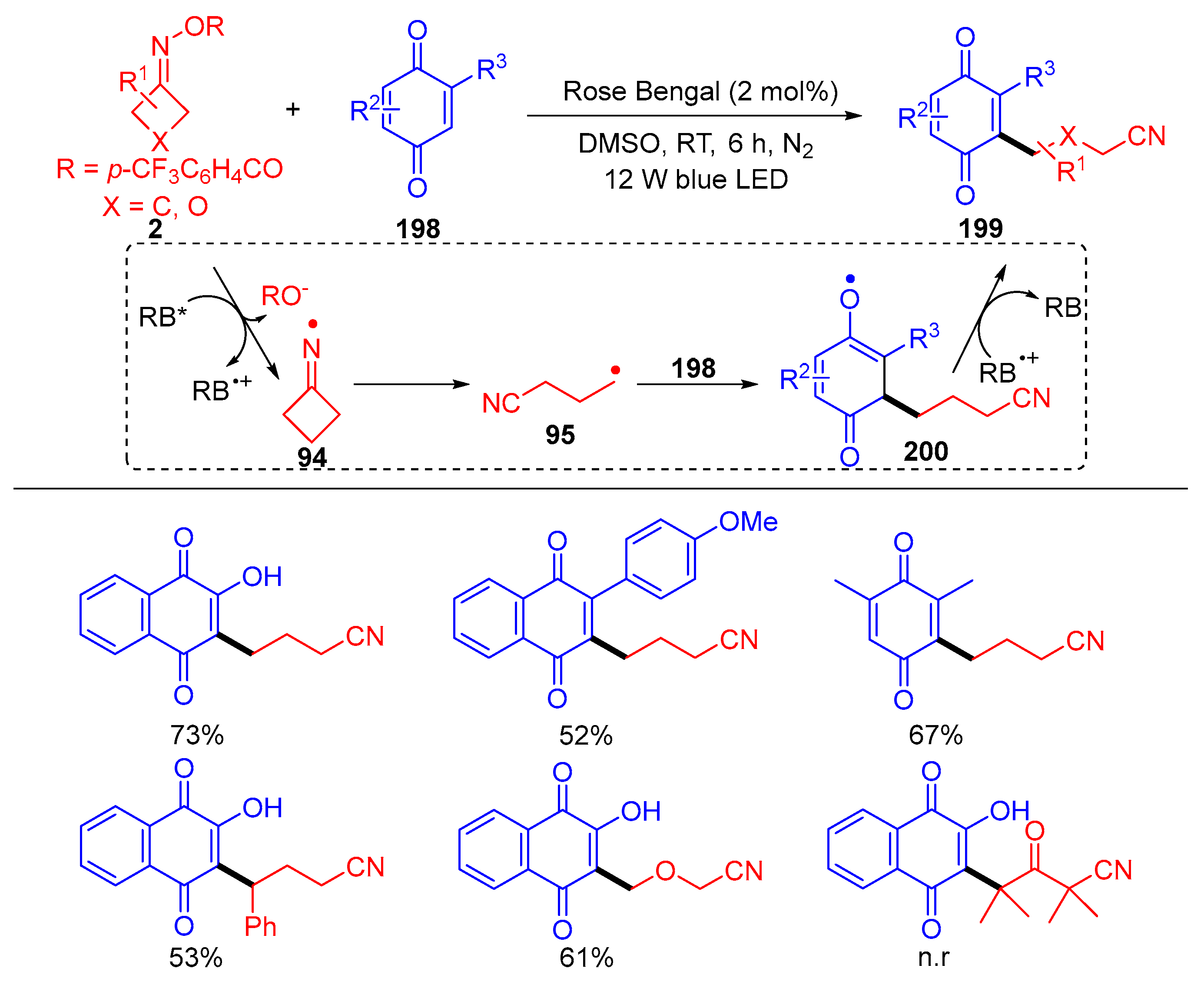
- Kulthe, A.-D.; Jaiswal, S.; Golagani, D.; Mainkar, P.-S.; Akondi, S.-M. Organophotoredox-Catalyzed Cyanoalkylation of 1,4-Quinones. Org. Biomol. Chem. 2022, 20, 4534–4538. [Google Scholar] [CrossRef]
- Ackermann, L. Metalla-Electrocatalyzed C–H Activation by Earth-Abundant 3d Metals and Beyond. Acc. Chem. Res. 2020, 53, 84–104. [Google Scholar] [CrossRef]
- Yuan, Y.; Yang, J.; Lei, A. Recent Advances in Electrochemical Oxidative Cross-Coupling with Hydrogen Evolution Involving Radicals. Chem. Soc. Rev. 2021, 50, 10058–10086. [Google Scholar] [CrossRef] [PubMed]
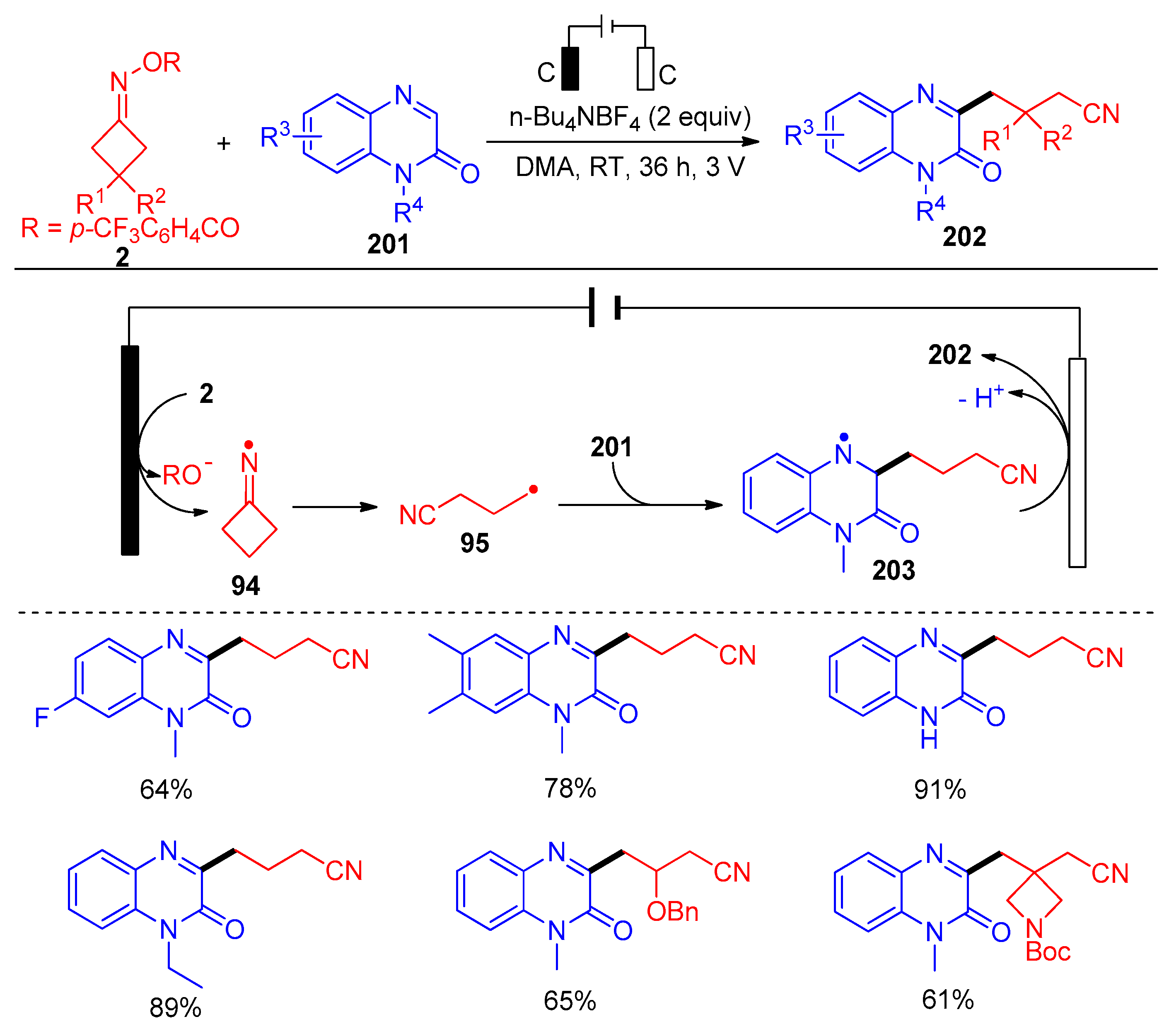
- Ding, L.; Niu, K.; Liu, Y.; Wang, Q. Electro-Reductive C-H Cyanoalkylation of Quinoxalin-2(1H)-Ones. Chin. Chem. Lett. 2022, 33, 4057–4060. [Google Scholar] [CrossRef]
- Lu, D.; Cui, J.; Yang, S.; Gong, Y. Iron-Catalyzed Cyanoalkylation of Glycine Derivatives Promoted by Pyridine-Oxazoline Ligands. ACS Catal. 2021, 11, 4288–4293. [Google Scholar] [CrossRef]
- Liu, S.; Bai, M.; Xu, P.-F.; Sun, Q.-X.; Duan, X.-H.; Guo, L.-N. Copper-Catalyzed Radical Ring-Opening Halogenation with HX. Chem. Commun. 2021, 57, 8652–8655. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Li, X.; Li, T.; Xiao, T.; Jiang, Y.; Qin, G. Fe-Catalyzed Denitrative Cyanoalkylation of Nitroalkenes with Cycloketone Oxime Esters Via Reductive C–C Bond Formation. Org. Chem. Front. 2022, 9, 6319–6324. [Google Scholar] [CrossRef]
- Lu, X.-Y.; Xia, Z.-J.; Gao, A.; Liu, Q.-L.; Jiang, R.-C.; Liu, C.-C. Dual Nickel/Ruthenium Strategy for Photoinduced Decarboxylative Cross-Coupling of α,β-Unsaturated Carboxylic Acids with Cycloketone Oxime Esters. J. Org. Chem. 2021, 86, 8829–8842. [Google Scholar] [CrossRef] [PubMed]
- Mugesh, G.; Du Mont, W.-W.; Sies, H. Chemistry of Biologically Important Synthetic Organoselenium Compounds. Chem. Rev. 2001, 101, 2125–2180. [Google Scholar] [CrossRef]
- Ji, L.; Qiao, J.; Liu, J.; Tian, M.; Lu, K.; Zhao, X. Metal-Free Chalcogenation of Cycloketone Oxime Esters with Dichalcogenides. Tetrahedron Lett. 2021, 75, 153202. [Google Scholar] [CrossRef]
- Zhao, X.; Ji, L.; Gao, Y.; Sun, T.; Qiao, J.; Li, A.; Lu, K. Visible-Light-Promoted Selenocyanation of Cyclobutanone Oxime Esters Using Potassium Selenocyanate. J. Org. Chem. 2021, 86, 11399–11406. [Google Scholar] [CrossRef]
- Ren, L.-Q.; Li, N.; Ke, J.; He, C. Recent Advances in Photo- and Electro-Enabled Radical Silylation. Org. Chem. Front. 2022, 9, 6400–6415. [Google Scholar] [CrossRef]
- Li, N.; Li, Y.; Wu, X.; Zhu, C.; Xie, J. Radical Deuteration. Chem. Soc. Rev. 2022, 51, 6291–6306. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cai, Z.; Warratz, S.; Ma, C.; Ackermann, L. Recent Advances in Electrooxidative Radical Transformations of Alkynes. Sci. Chin. Chem. 2022, 66, 703–724. [Google Scholar] [CrossRef]





































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, P.; Huang, H.; Tan, Q.; Ji, X.; Zhao, F. Recent Advances in Molecule Synthesis Involving C-C Bond Cleavage of Ketoxime Esters. Molecules 2023, 28, 2667. https://doi.org/10.3390/molecules28062667
Chen P, Huang H, Tan Q, Ji X, Zhao F. Recent Advances in Molecule Synthesis Involving C-C Bond Cleavage of Ketoxime Esters. Molecules. 2023; 28(6):2667. https://doi.org/10.3390/molecules28062667
Chicago/Turabian StyleChen, Pu, Huawen Huang, Qi Tan, Xiaochen Ji, and Feng Zhao. 2023. "Recent Advances in Molecule Synthesis Involving C-C Bond Cleavage of Ketoxime Esters" Molecules 28, no. 6: 2667. https://doi.org/10.3390/molecules28062667
APA StyleChen, P., Huang, H., Tan, Q., Ji, X., & Zhao, F. (2023). Recent Advances in Molecule Synthesis Involving C-C Bond Cleavage of Ketoxime Esters. Molecules, 28(6), 2667. https://doi.org/10.3390/molecules28062667