

DNA Binding and Cleavage, Stopped-Flow Kinetic, Mechanistic, and Molecular Docking Studies of Cationic Ruthenium(II) Nitrosyl Complexes Containing “NS4” Core
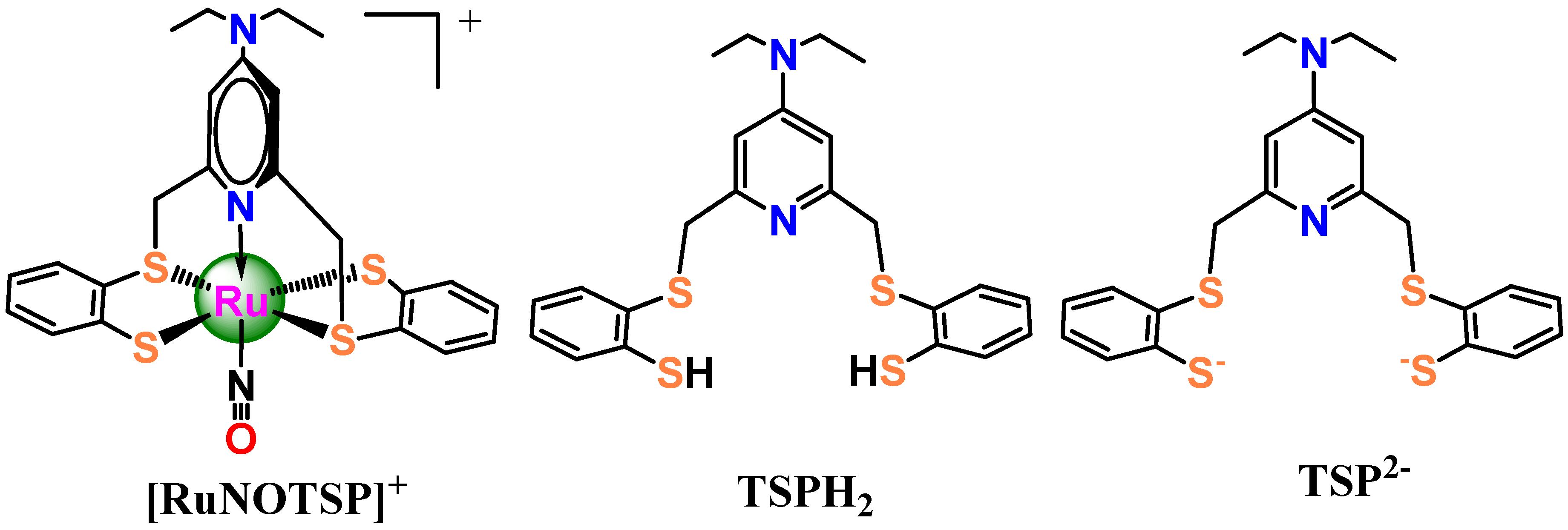
 ,
,  and
and
Abstract
:Highlights
- Theoretical studies were performed on [RuNOTSP]+, TSPH2, and its anion TSP2− using the DFT/B3LYP method.
- Cationic complex [RuNOTSP]+ and TSPH2 were investigated mechanistically for ctDNA interaction.
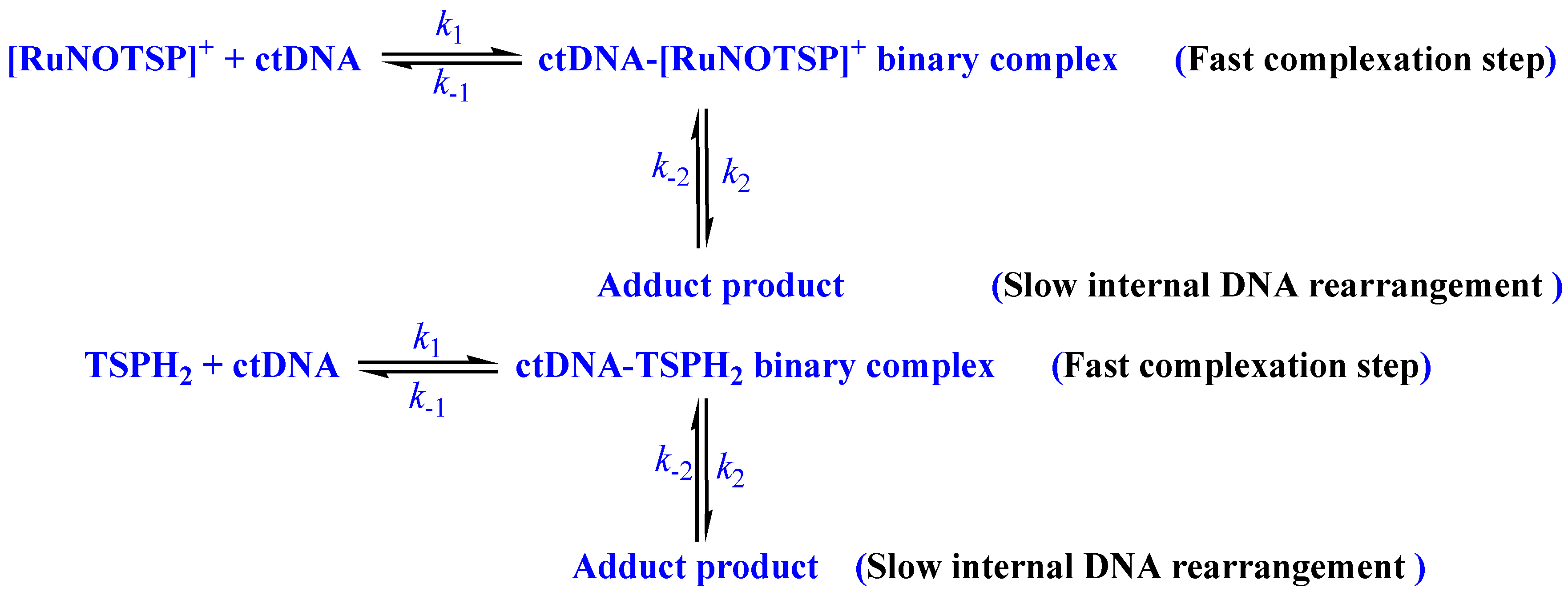
- Spontaneous ctDNA binding via a static mechanism with two steps was reported.
- Detailed kinetic data are reported and relative reactivity is [RuNOTSP]+/TSPH2 = 3/1.
- The ruthenium effect on affinity and mechanism is reported.
- The ruthenium center improves the reaction rate through coordination affinity, but does not change its mechanism.
- Molecular docking was used to predict the binding between [RuNOTSP]+ and TSPH2 and the receptors.
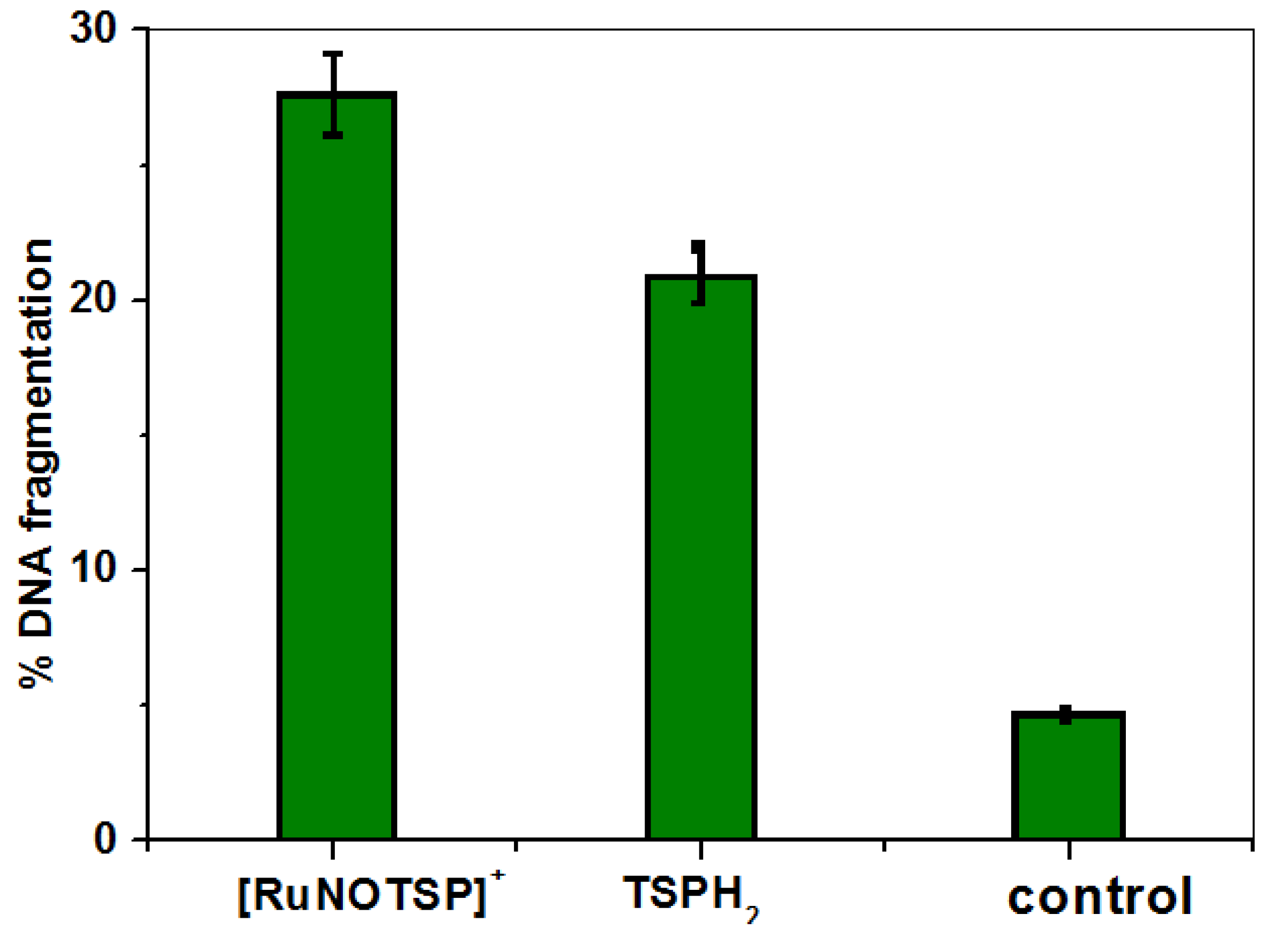
- DNA cleavage studies are correlated with kinetic data.
Abstract
1. Introduction
2. Results and Discussion
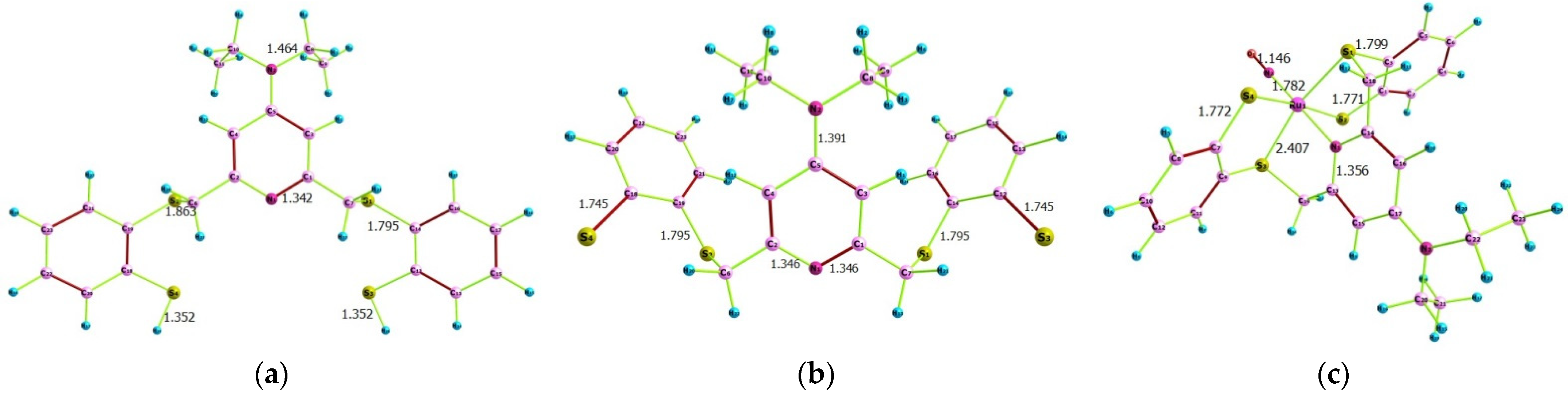
2.1. Computational Findings
2.1.1. Quantum Chemical Parameters
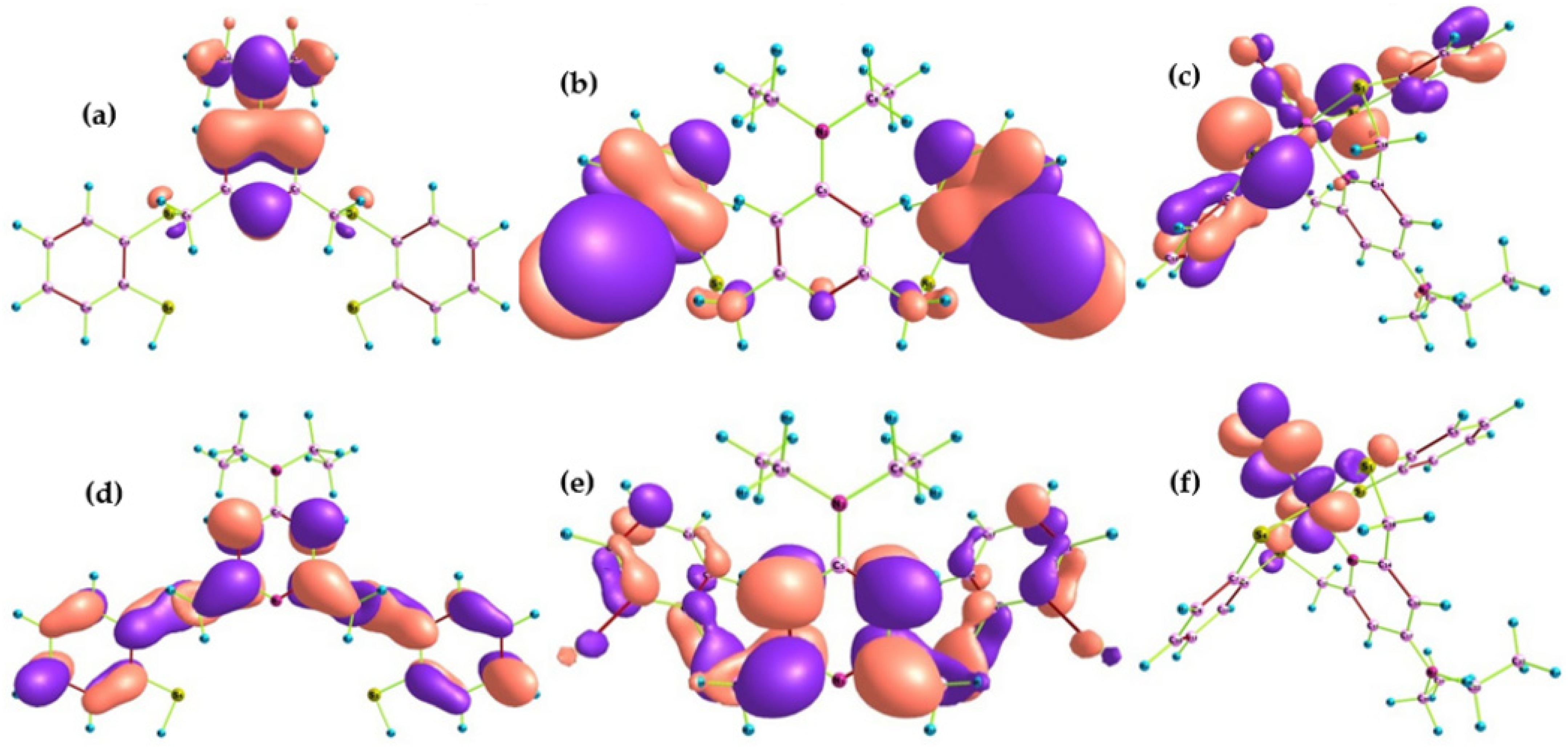
2.1.2. Frontier Molecular Orbitals and Chemical Reactivity
2.2. DNA Interaction Studies
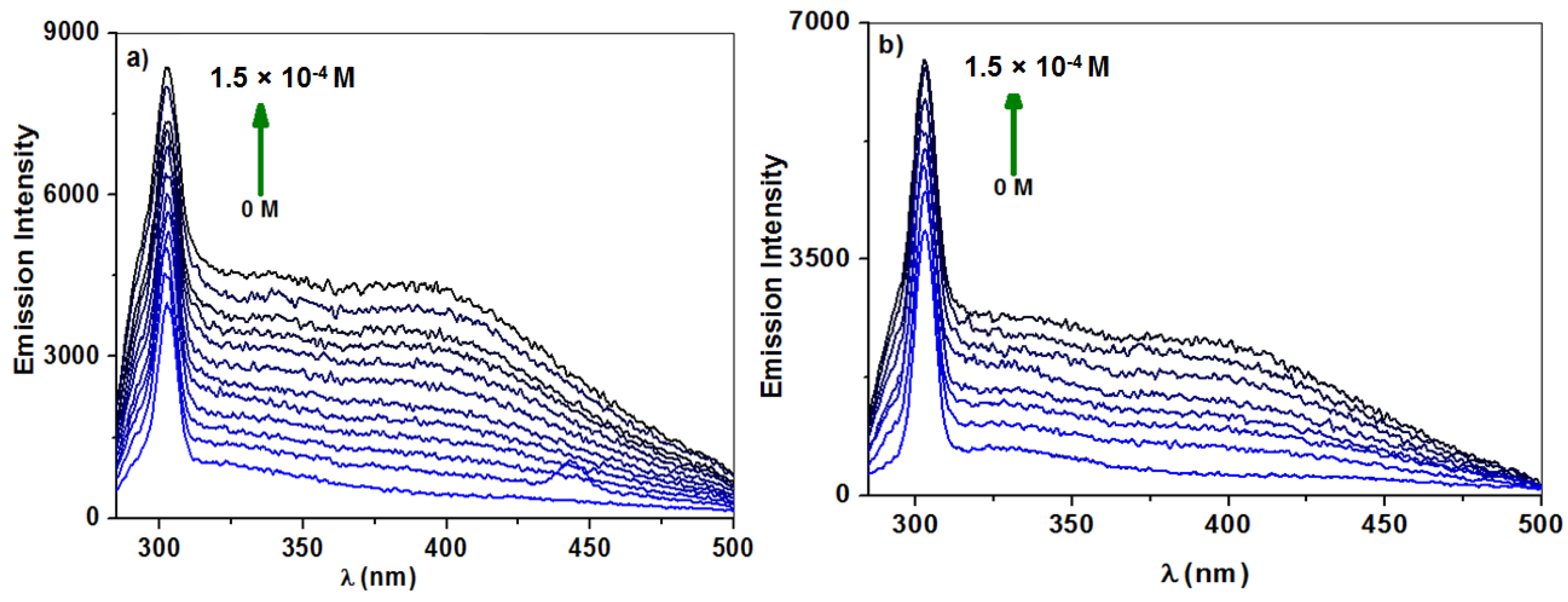
2.2.1. Fluorescence Spectroscopy
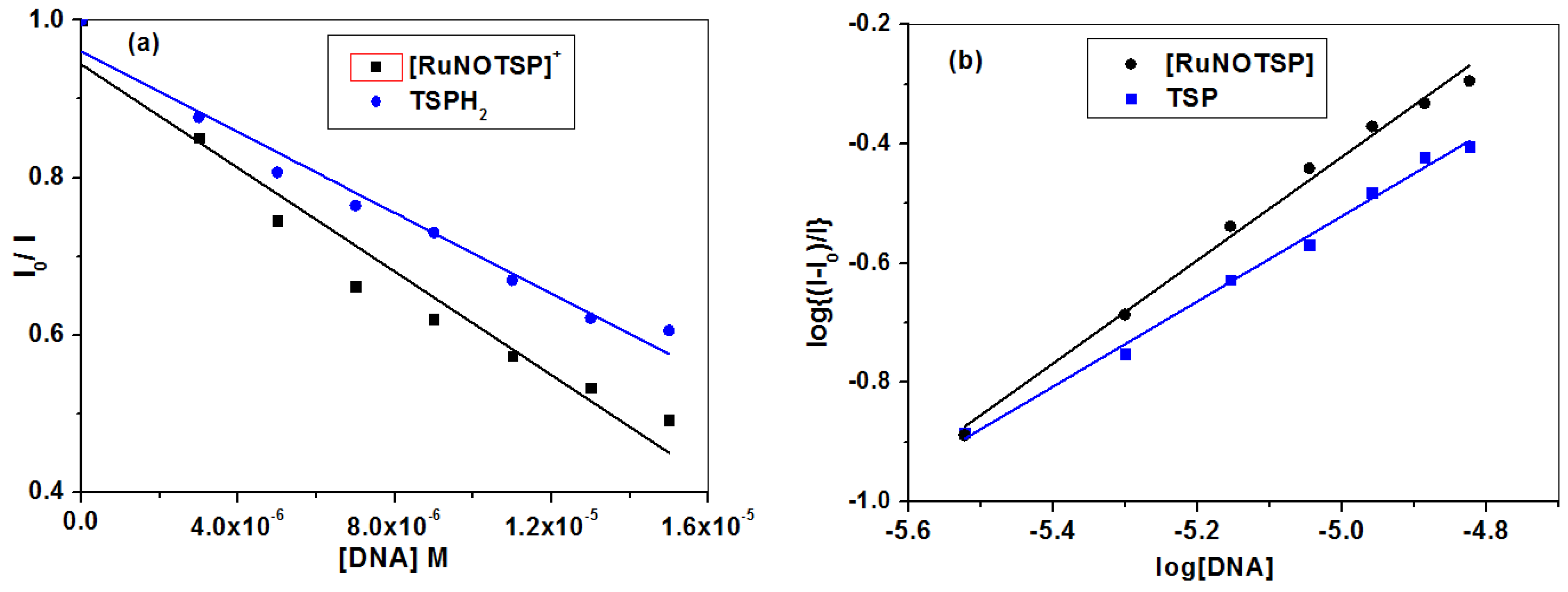
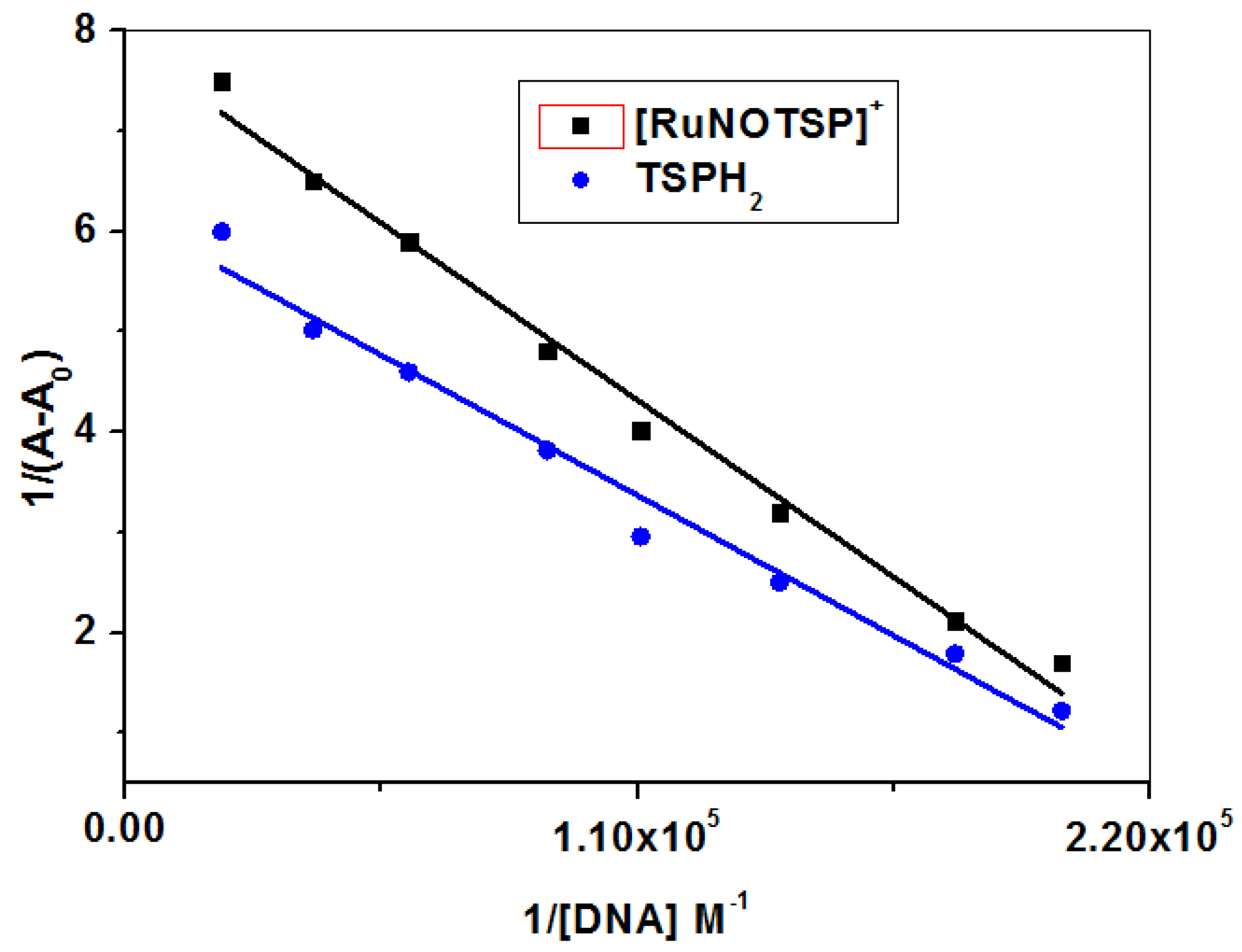
2.2.2. Equilibrium Binding Titration
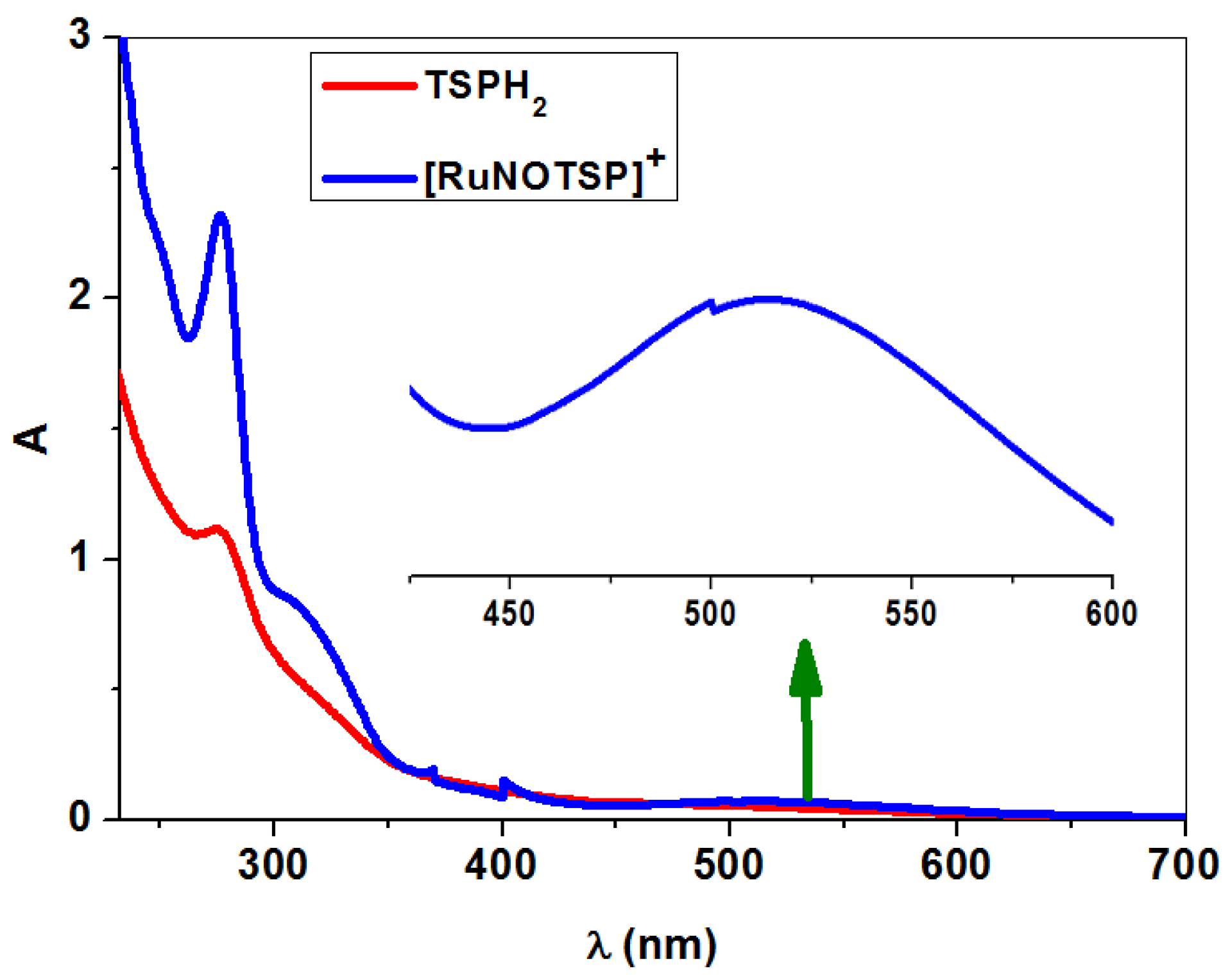
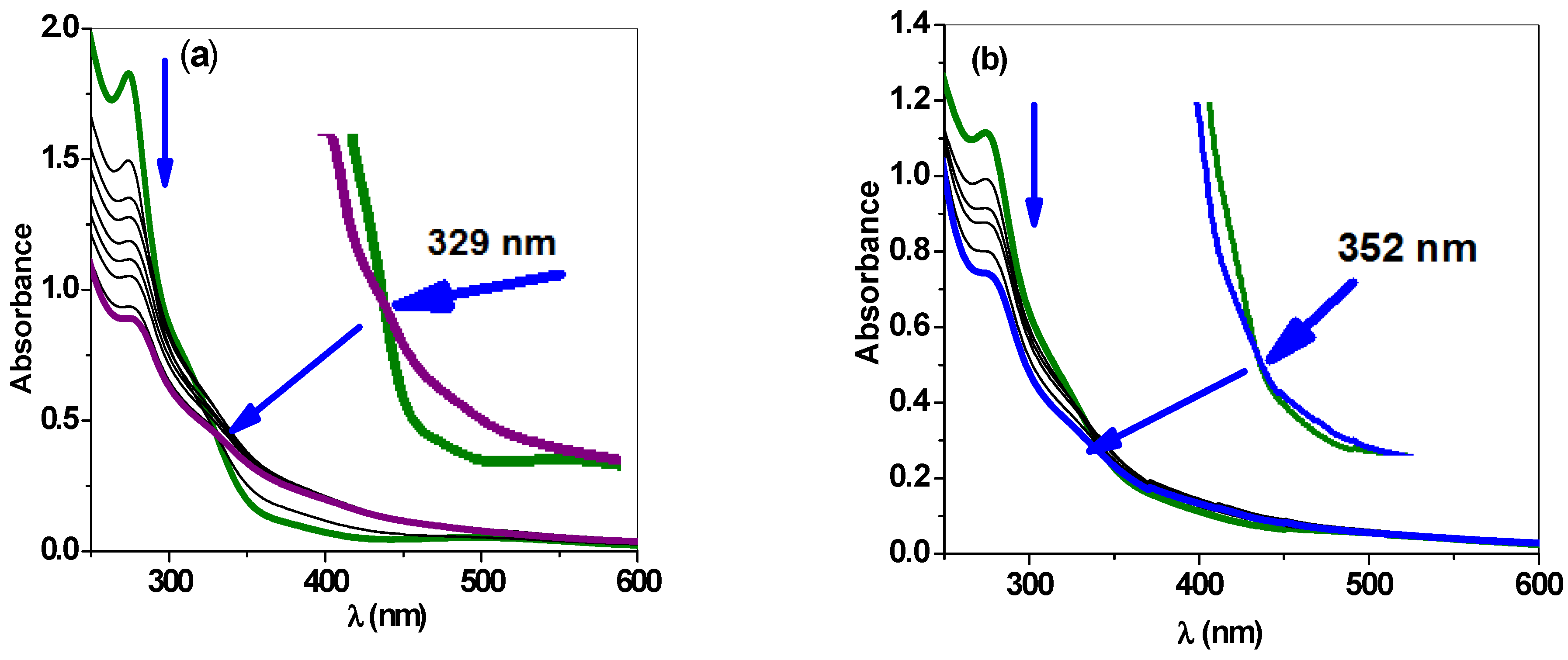
2.2.3. UV–Vis Spectroscopy
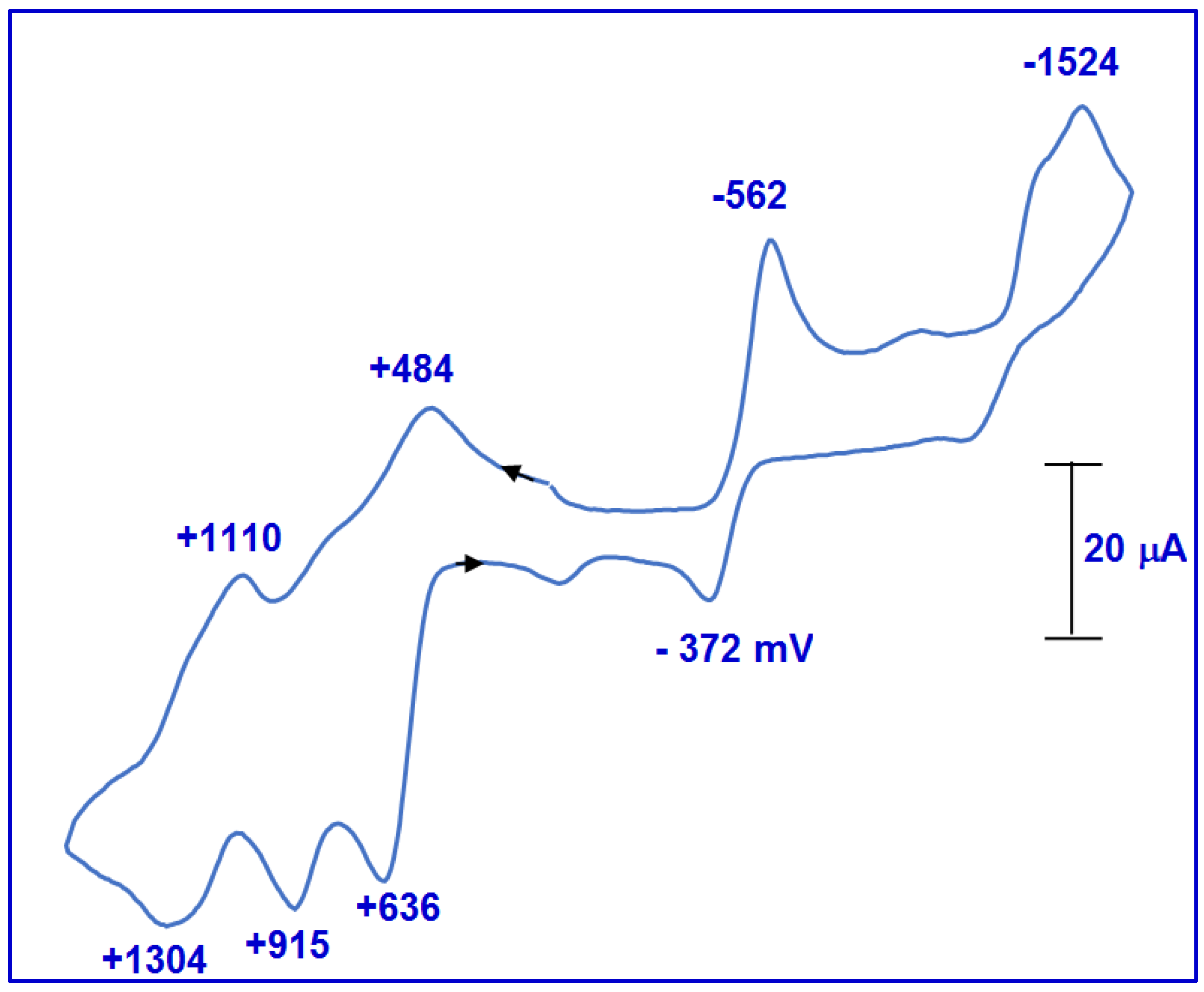
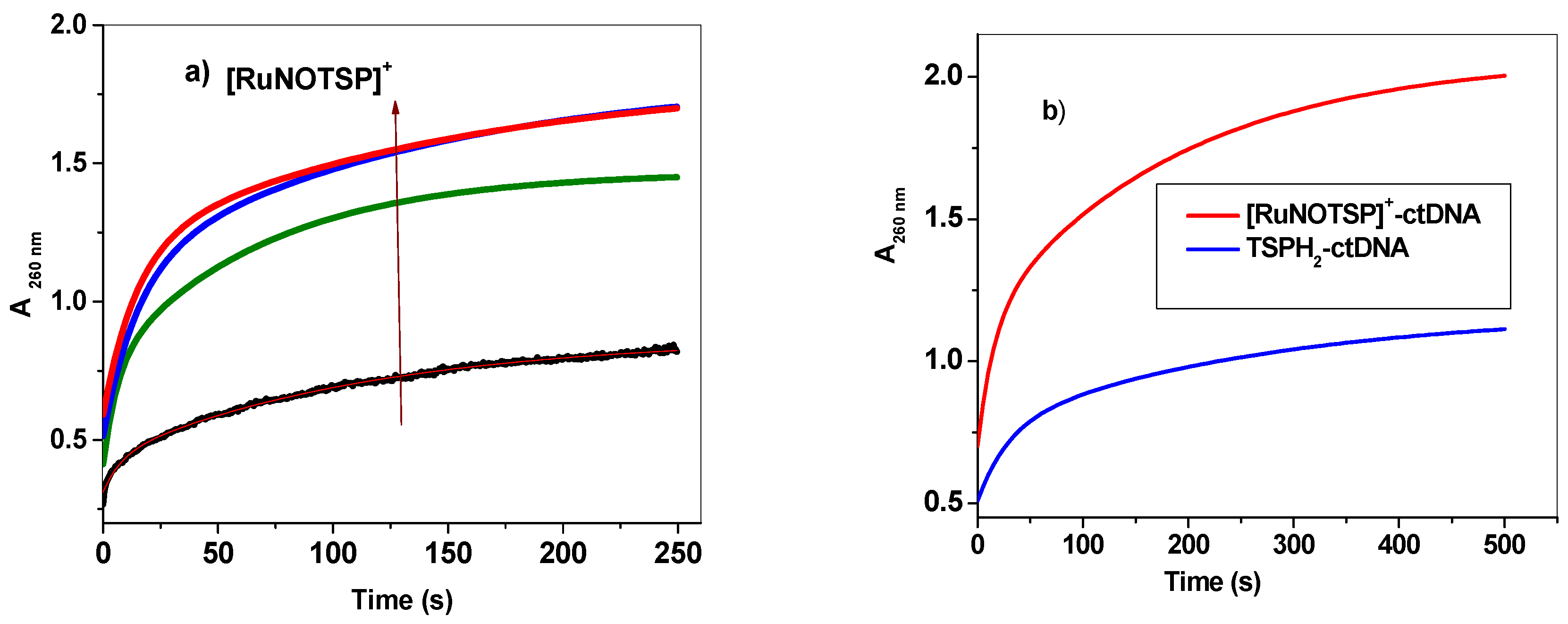
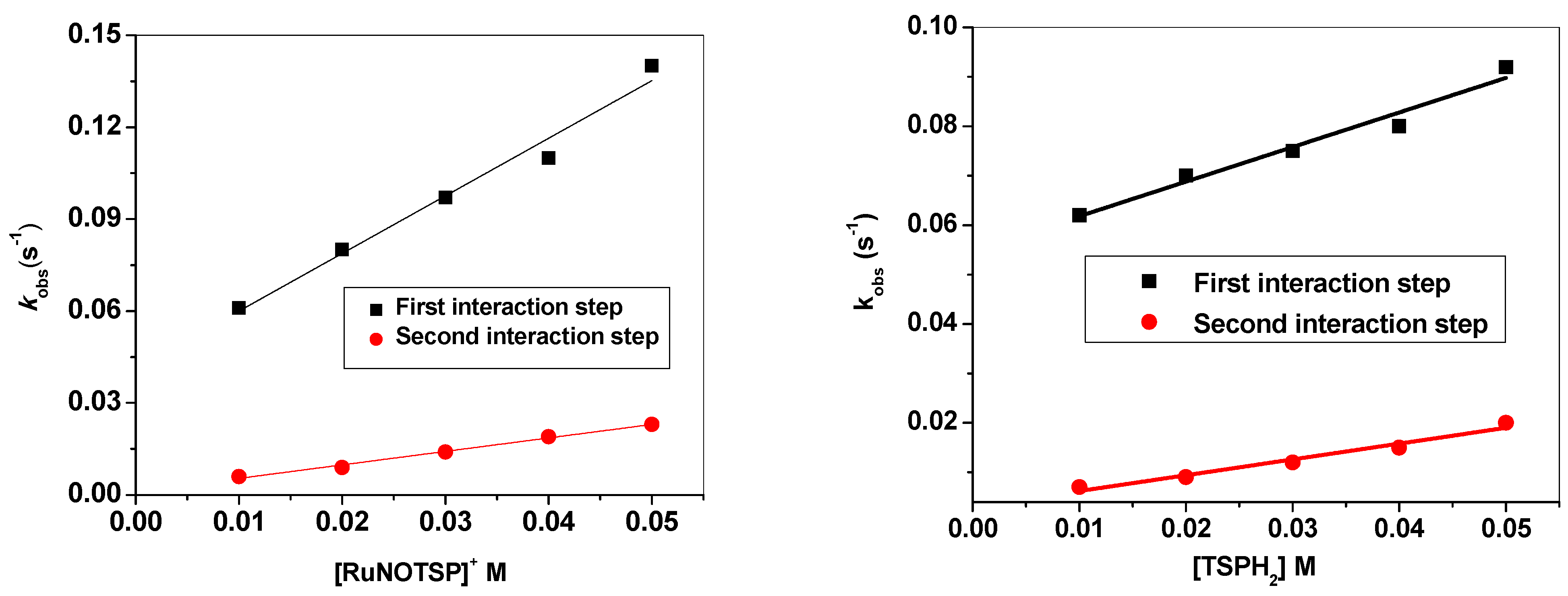
2.2.4. Stopped-Flow Spectroscopic Studies and Kinetic Investigation
2.3. DNA Cleavage Studies
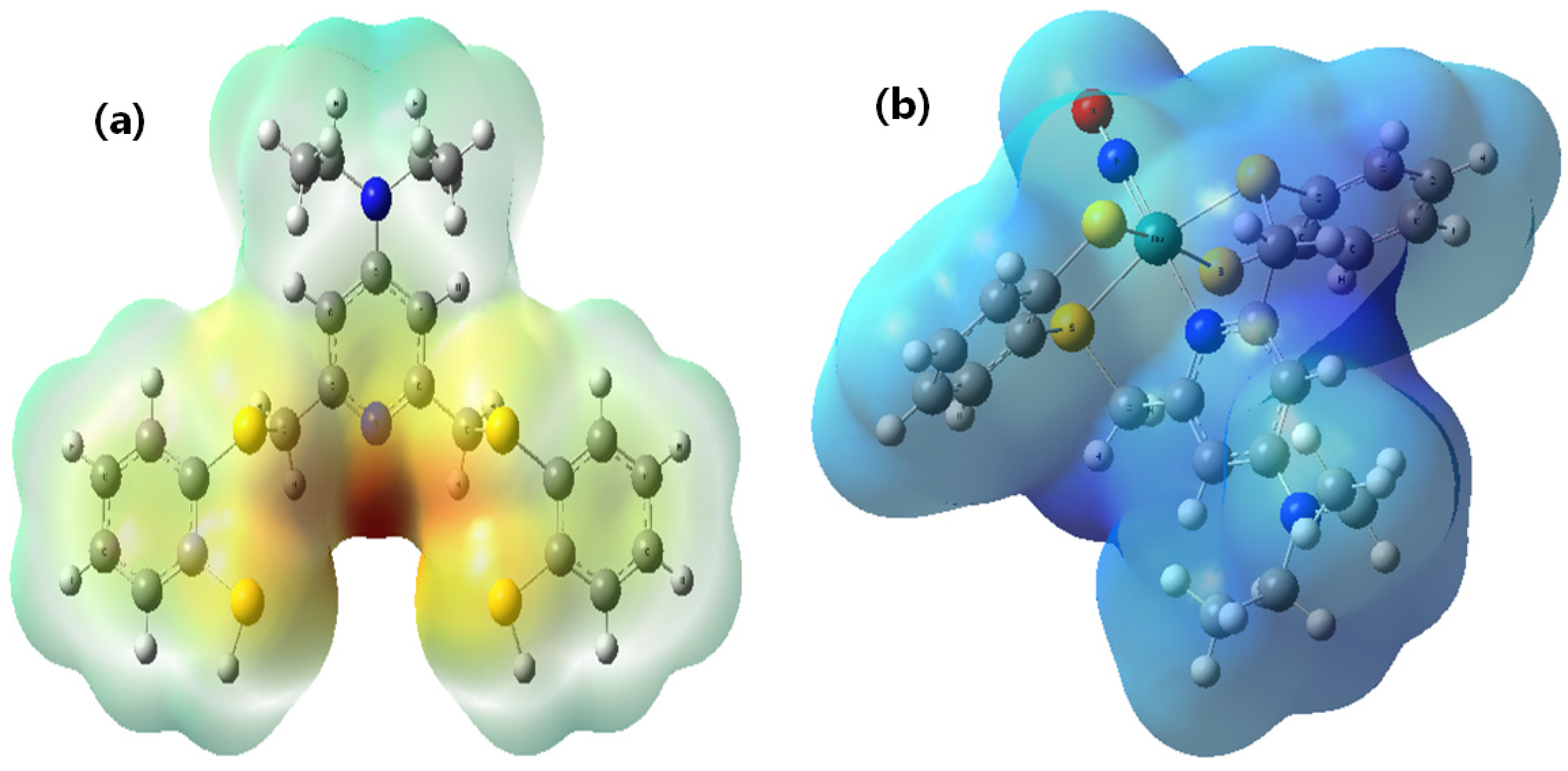
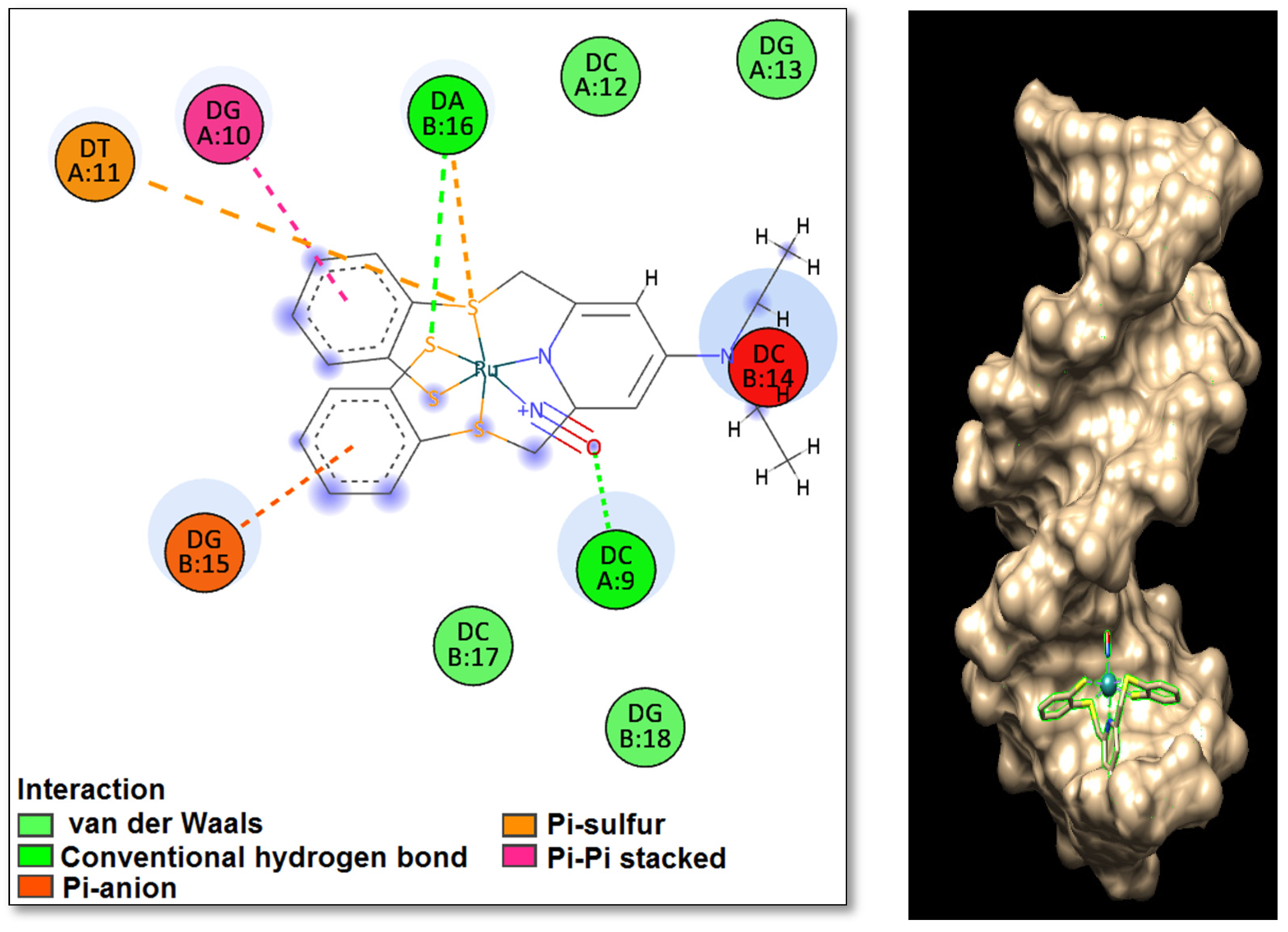
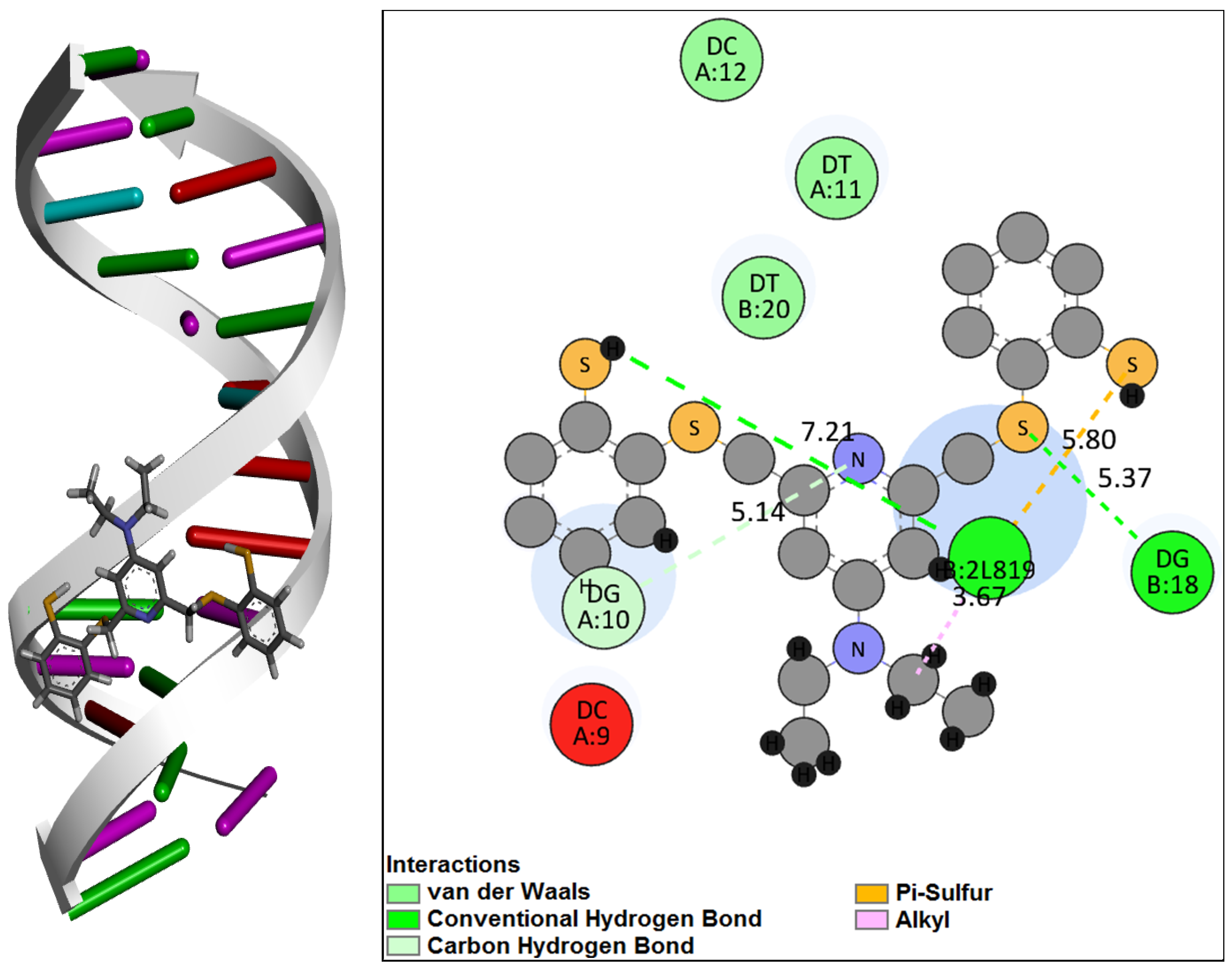
2.4. Molecular Docking of DNA with [RuNOTSP]+ and TSPH2
3. Conclusions
4. Experimental Section
4.1. Materials
4.2. Apparatus
4.3. Stopped-Flow Kinetic Studies
4.4. ctDNA Cleavage Activity
4.5. Computational Studies
4.6. Molecular Docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, C.; Xu, C.; Gao, X.; Yao, Q. Platinum-based drugs for cancer therapy and anti-tumor Strategies. Theranostics 2022, 12, 2115–2132. [Google Scholar] [CrossRef]
- Wheate, N.J.; Walker, S.; Craig, G.E.; Oun, R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010, 39, 8113–8127. [Google Scholar] [CrossRef] [Green Version]
- Rottenberg, S.; Disler, C.; Perego, P. The rediscovery of platinum-based cancer therapy. Nat. Rev. Cancer 2020, 21, 37–50. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Kim, C.Y.; Nam, T.-G. Ruthenium Complexes as Anticancer Agents: A Brief History and Perspectives. Drug Des. Dev. Ther. 2020, 14, 5375–5392. [Google Scholar] [CrossRef]
- Fabijańska, M.; Kasprzak, M.M.; Ochocki, J. Ruthenium(II) and Platinum(II) Complexes with Biologically Active Aminoflavone Ligands Exhibit In Vitro Anticancer Activity. Int. J. Mol. Sci. 2021, 22, 7568. [Google Scholar] [CrossRef]
- Sun, Q.; Li, Y.; Shi, H.; Wang, Y.; Zhang, J.; Zhang, Q. Ruthenium Complexes as Promising Candidates against Lung Cancer. Molecules 2021, 26, 4389. [Google Scholar] [CrossRef]
- Kanaoujiya, R.; Meenakshi; Srivastava, S.; Singh, R.; Mustafa, G. Recent advances and application of ruthenium complexes in tumor malignancy. Mater. Today Proc. 2023, 72, 2822–2827. [Google Scholar] [CrossRef]
- Singha, A.K.; Pandeyb, D.S.; Xua, Q.; Braunstein, P. Recent advances in supramolecular and biological aspects of arene ruthenium (II) complexes. Coord. Chem. Rev. 2014, 270, 31–56. [Google Scholar] [CrossRef]
- Bergamo, A.; Gaiddon, C.; Schellens, J.H.M.; Beijnen, J.H.; Sava, G.J. Approaching tumour therapy beyond platinum drugs: Status of the art and perspectives of ruthenium drug candidates. J. Inorg. Biochem. 2012, 106, 90–99. [Google Scholar] [CrossRef]
- Khamrang, T.; Kartikeyan, R.; Velusamy, M.; Rajendiran, V.; Dhivya, R.; Perumalsamy, B.; Akbarsha, M.A.; Palaniandavar, M. Synthesis, structures, and DNA and protein binding of ruthenium(ii)-p-cymene complexes of substituted pyridylimidazo[1,5-a]pyridine: Enhanced cytotoxicity of complexes of ligands appended with a carbazole moiety. RSC Adv. 2016, 6, 114143–114258. [Google Scholar] [CrossRef]
- Hu, P.C.; Wang, Y.; Zhang, Y.; Song, H.; Gao, F.F.; Lin, H.Y.; Wang, Z.H.; Wei, L.; Yang, F. Novel mononuclear ruthenium(ii) complexes as potent and low-toxicity antitumour agents: Synthesis, characterization, biological evaluation and mechanism of action. RSC Adv. 2016, 6, 29963–29976. [Google Scholar] [CrossRef]
- Kamatchi, T.S.; Kalaivani, P.F.; Fronczek, R.; Natarajan, K.; Prabhakaran, R. The effect of incorporating carboxylic acid functionalities into 2,2′-bipyridine on the biological activity of the complexes formed: Synthesis, structure, DNA/protein interaction, antioxidant activity and cytotoxicity. RSC Adv. 2017, 7, 46531–46547. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.M.; Peng, F.; Li, G.D.; Jie, X.M.; Cai, K.R.C.; Zhong, Y.; Zeng, H.; Li, W.; Zhang, Z.; Chen, J.C. The studies on the cytotoxicity in vitro, cellular uptake, cell cycle arrest and apoptosis-inducing properties of ruthenium methylimidazole complex [Ru(MeIm)4(p-cpip)](2.). J. Inorg. Biochem. 2016, 156, 64–74. [Google Scholar] [CrossRef]
- Chen, W.X.; Song, X.D.; He, S.F.; Sun, J.; Chen, J.X.; Wu, T.; Mao, Z.W. Ru(II) complexes bearing guanidinium ligands as potent anticancer agents. J. Inorg. Biochem. 2016, 164, 91–98. [Google Scholar] [CrossRef]
- Zhang, C.; Han, B.J.; Zeng, C.C.; Lai, S.H.; Li, W.; Tang, B.; Wan, D.; Jiang, G.B.; Liu, Y.J. Synthesis, characterization, in vitro cytotoxicity and anticancer effects of ruthenium (II) complexes on BEL-7402 cells. J. Inorg. Biochem. 2016, 157, 62–72. [Google Scholar] [CrossRef]
- Süss-Fink, G. Areneruthenium complexes as anticancer agents. Dalton Trans. 2010, 39, 1673–1688. [Google Scholar] [CrossRef]
- Bratsos, I.; Jedner, S.; Gianferrara, T.; Alessio, E. Ruthenium Anticancer Compounds: Challenges and Expectations. Chimia 2007, 61, 692–697. [Google Scholar] [CrossRef]
- Meng, X.; Leyva, M.L.; Jenny, M.; Gross, I.; Benosman, S.; Fricker, B.; Harlepp, S.; Ebraud, P.H.; Boos, A.; Wlosik, P.; et al. A Ruthenium-Containing Organometallic Compound Reduces Tumor Growth through Induction of the Endoplasmic Reticulum Stress Gene CHOP. Cancer Res. 2009, 69, 5458–5466. [Google Scholar] [CrossRef] [Green Version]
- Gupta, G.; Nowak-Sliwinska, P.; Dyson, N.P.J.; Therrien, B. Increasing the selectivity of biologically active tetranuclear arene ruthenium assemblies. Inorg. Chim. Acta 2014, 423, 524–529. [Google Scholar] [CrossRef]
- Sava, G.; Bergamo, A.; Dyson, P.J. Metal-based antitumour drugs in the post-genomic era: What comes next? Dalton Trans. 2011, 40, 9069–9075. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Metzler-Nolte, N.; Dyson, P.J. Challenges and Opportunities in the Development of Organometallic Anticancer Drugs. Organometallics 2012, 31, 5677–5685. [Google Scholar] [CrossRef]
- Kilpin, K.J.; Crot, S.; Riedel, T.; Kitchen, J.A.; Dyson, P.J. Ruthenium(ii) and osmium(ii) 1,2,3-triazolylidene organometallics: A preliminary investigation into the biological activity of ‘click’ carbene complexes. Dalton Trans. 2014, 43, 1443–1448. [Google Scholar] [CrossRef] [Green Version]
- Stepanenko, I.; Zalibera, M.; Schaniel, D.; Telser, J.; Arion, V.B. Ruthenium-nitrosyl complexes as NO-releasing molecules, potential anticancer drugs, and photoswitches based on linkage isomerism. Dalton Trans. 2022, 51, 5367–5393. [Google Scholar] [CrossRef]
- Gallo, O.; Masini, E.; Morbidelli, L.; Franchi, A.; Fini-Storchi, I.; Vergari, W.A.; Ziche, M. Role of nitric oxide in angiogenesis and tumor progression in head and neck cancer. J. Natl. Cancer Inst. 1998, 90, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Ziche, M.; Morbidelli, L. Nitric oxide and angiogenesis. J. Neurooncol. 2000, 50, 139–148. [Google Scholar] [CrossRef]
- Gomes, M.G.; Davanzo, C.U.; Silva, S.C.; Lopes, L.G.F.; Santos, P.S.; Franco, D.W. Cis- and trans-nitrosyltetraammineruthenium(II). Spectral and electrochemical properties and reactivity. J. Chem. Soc. Dalton Trans. 1998, 601–608. [Google Scholar] [CrossRef]
- Enemark, J.H.; Feltham, R.D. Principles of structure, bonding, and reactivity for metal nitrosyl complexes. Coord. Chem. Rev. 1974, 13, 339–406. [Google Scholar] [CrossRef]
- Wolak, M.; van Eldik, R. To be or not to be NO in coordination chemistry? A mechanistic approach. Coord. Chem. Rev. 2002, 230, 263–282. [Google Scholar] [CrossRef]
- Karidi, K.; Garoufis, A.; Tsipis, A.; Hadjiliadis, N.; den Dulk, H.; Reedijk., J. Synthesis, characterization, in vitro antitumor activity, DNA-binding properties and electronic structure (DFT) of the new complex cis-(Cl,Cl)[RuIICl2(NO+)(terpy)]Cl. Dalton Trans. 2005, 1176–1187. [Google Scholar] [CrossRef]
- Sellmann, D.; Shaban, S.Y.; Heinemann, F.W. Syntheses, Structures and Reactivity of Electron-Rich Fe and Ru Complexes with the New Pentadentate Ligand Et2NpyS4−H2 {4-(Diethylamino)2,6-bis[(2-mercaptophenyl) thiomethyl] pyridine}. Eur. J. Inorg. Chem. 2004, 23, 4591–4601. [Google Scholar] [CrossRef]
- Sellmann, D.; Binder, H.; Häußinger, D.; Heinemann, F.W.; Sutter, J. Transition metal complexes with sulfur ligands: Part CXLIV. Square planar nickel complexes with NiS4 cores in three different oxidation states: Synthesis, X-ray structural and spectroscopic studies. Inorg. Chim. Acta 2000, 300, 829–836. [Google Scholar] [CrossRef]
- Sellmann, D.; Gottschalk-Gaudig, T.; Haussinger, D.; Heinemann, F.W.; Hess, B.A. Ru(HNO)(′pybuS4′)], the First HNO Complex Resulting from Hydride Addition to a NO Complex (′pybuS4′2−=2,6-Bis(2-mercapto-3,5-di-tert-butylphenylthio)dimethylpyridine(2−)). Chem. Eur. J. 2001, 7, 1874–1880. [Google Scholar] [CrossRef]
- Prakash, R.; Czaja, A.U.; Heinemann, F.W.; Sellmann, D. Visible Light Induced Reversible Extrusion of Nitric Oxide from a Ruthenium(II) Nitrosyl Complex: A Facile Delivery of Nitric Oxide. J. Am. Chem. Soc. 2005, 127, 13758–13759. [Google Scholar] [CrossRef]
- Dias, R.S.; Lindman, B. DNA Interactions with Polymers and Surfactants; Wiley and Sons: Hoboken, NJ, USA, 2008; pp. 1–432. [Google Scholar]
- Martinez, R.; Chacn-Garca, L. The search of DNA-intercalators as antitumoral drugs: What it worked and what did not work. Curr. Med. Chem. 2005, 12, 127–151. [Google Scholar] [CrossRef]
- Shereef, H.A.; Shaban, S.Y.; Moemen, Y.S.; El-Khouly, M.E.; El-Nahas, A.M. Biophysicochemical studies of a ruthenium(II) nitrosyl thioether-thiolate complex binding to BSA: Mechanistic information, molecular docking, and relationship to antibacterial and cytotoxic activities. Appl. Organomet. Chem. 2022, 34, e6583. [Google Scholar] [CrossRef]
- Shaban, N.Z.; Aboelsaad, A.M.; Shoueir, K.R.; Abdulmalek, S.A.; Awad, D.; Shaban, S.Y.; Mansour, H. Chitosan-based dithiophenolato nanoparticles: Preparation, mechanistic information of DNA binding, antibacterial and cytotoxic activities. J. Mol. Liq. 2020, 318, 114252. [Google Scholar] [CrossRef]
- Elshami, F.I.; Ramadan, A.E.M.M.; Ibrahim, M.M.; El-Mehasseb, I.M.; Al-Juaid,, S.; Shaban, S.Y. Metformin Containing Nickel (II) Complexes: Synthesis, Structural Characterization, Binding and Kinetic Interactions with BSA, Antibacterial and in-vitro Cytotoxicity Studies. Appl. Organomet. Chem. 2020, 34, e5437. [Google Scholar] [CrossRef]
- Shaban, N.Z.; Yehia, S.A.; Shoueir, K.R.; Saleh, S.R.; Awad, D.; Shaban, S.Y. Design, DNA binding and kinetic studies, antibacterial and cytotoxic activities of stable dithiophenolato titanium(IV)-chitosan Nanocomposite. J. Mol. Liq. 2019, 287, 111002. [Google Scholar] [CrossRef]
- Ramzy, E.; Ibrahim, M.M.; El-Mehasseb, I.M.; Ramadan, A.E.M.M.; Elshami, F.I.; Shaban, Y.S.; van Eldik, R. Synthesis, Biophysical Interaction of DNA/BSA, Equilibrium and Stopped-Flow Kinetic Studies, and Biological Evaluation of bis(2-Picolyl)amine-Based Nickel(II) Complex. Biomimetics 2022, 7, 172. [Google Scholar] [CrossRef]
- Sellmann, D.; Engel, K.; Heinemann, F.W. Inert and Labile [Ru(L)(‘pyS4′)] Complexes with Rigid [RuNS4] Cores and trans-Thiolate Donors [L = PPh3, PEt3, DMSO, CO, NO+, N2H4; ‘pyS4′2– = 2,6-Bis(2-mercaptophenylthio)dimethylpyridine(2–)]. Eur. J. Inorg. Chem. 2000, 3, 423–429. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F.J. Natural population analysis. Chem. Phys. 1985, 83, 735. [Google Scholar] [CrossRef]
- Reed, A.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899. [Google Scholar] [CrossRef]
- Kwon, O.; Sevin, F.; McKee, M.L. Density Functional Calculations of Methyllithium, t-Butyllithium, and Phenyllithium Oligomers: Effect of Hyperconjugation on Conformation. J. Phys. Chem. A 2001, 105, 913–922. [Google Scholar] [CrossRef]
- Masoud, M.S.; Ali, A.E.; Shaker, M.A.; Elasala, G.S. Synthesis, computational, spectroscopic, thermal and antimicrobial activity studies on some metal-urate complexes. Spectrochim. Acta A 2012, 90, 93–108. [Google Scholar] [CrossRef]
- Zhao, C.; Lin, H.; Zhu, S.; Sun, H.; Chen, Y. Dinuclear palladium (II) complexes containing two monofunctional [Pd(en)(pyridine)Cl]+ units bridged by Se or S. Synthesis, characterization, cytotoxicity and kinetic studies of DNA-binding. J. Inorg. Biochem. 1998, 70, 219–226. [Google Scholar] [CrossRef]
- Lwebuga-Mukasa, J.S.; Harper, E. Taylor Palmer. Collagenase enzymes from Clostridium: Characterization of individual enzymes. Biochemistry 1976, 15, 4736–4741. [Google Scholar]
- Tabassum, S.; Zaki, M.; Arjmand, F.; Ahmad, I. Synthesis of heterobimetallic complexes: In vitro DNA binding, cleavage and antimicrobial studies. J. Photochem. Photobiol. B. Biol. 2012, 114, 108–118. [Google Scholar] [CrossRef]
- Tabassum, S.; Parveen, M.; Ali, A.; Alam, M.; Ahmad, A.; Khan, A.U.; Khan, R.A. Synthesis of Aryl-1, 2, 4, 5-tetrazinane-3-thiones, in vitro DNA binding studies, nuclease activity and its antimicrobial activity. J. Mol. Struct. 2012, 1020, 33–40. [Google Scholar] [CrossRef]
- Ahmedi, F.; Alizadeh, A.A.; Shahabadi, N.; Rhimi-Nasrbadi, M. Study bonding of Al-Curcumin complex to ds-DNA, monitoring by multispectroscopic and voltammetric techniques. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2011, 79, 1466–1474, Erratum in Spectrochim. Acta A Mol. Biomol. Spectrosc. 2012, 90, 230. [Google Scholar] [CrossRef]
- Shahabadi, N.; Mohamedi, S. Synthesis Characterization and DNA Interaction Studies of a New Zn(II) Complex Containing Different Dinitrogen Aromatic Ligands. Bioinorg. Chem. Appl. 2012, 2012, 571913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Korolev, S.; Waksman, G. Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: Structural basis for nucleotide incorporation. EMBO J. 1998, 17, 7514–7525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahabadi, N.; Fatahi, A. Multispectroscopic DNA binding studies of a tris-chelate nickel(II) complex containing 4,7-diphenyl 1,10-phenanthroline ligands. J. Mol. Struct. 2010, 970, 90–95. [Google Scholar] [CrossRef]
- Barton, J.K.; Danishefsky, A.T.; Goldberg, J.M. Tris(phenanthroline) ruthenium (II): Stereoselectivity in binding to DNA. J. Am. Chem. Soc. 1984, 106, 2172–2176. [Google Scholar] [CrossRef]
- Liu, X.W.; Li, J.; Deng, H.; Zheng, K.C.; Mao, Z.W.; Ji, L.N. Experimental and DFT studies on the DNA-binding trend and spectral properties of complexes [Ru(bpy)2L]2+ (L = dmdpq, dpq, and dcdpq). Inorg. Chim. Acta 2005, 358, 3311–3319. [Google Scholar] [CrossRef]
- Liu, X.W.; Li, J.; Li, H.; Zheng, K.C.; Chao, H.; Ji, L.N. Synthesis, characterization, DNA-binding and photocleavage of complexes [Ru(phen)2(6-OH-dppz)]2+ and [Ru(phen)2(6-NO2-dppz)]2+. J. Inorg. Biochem. 2005, 99, 2372–2380. [Google Scholar] [CrossRef] [PubMed]
- Barton, J.K.; Basil, L.A. A cobalt derivative of the restriction enzyme EcoRI. Inorg. Chim. Acta 1983, 79, 152–153. [Google Scholar] [CrossRef]
- Benesi, H.A.; Hildebrand, J.A. Spectrophotometric Investigation of the Interaction of Iodine with Aromatic Hydrocarbons. J. Am. Chem. Soc. 1949, 71, 2703–2707. [Google Scholar] [CrossRef]
- Kennedy, S.D.; Bryant, R.G. Manganese-deoxyribonucleic acid binding modes. Nuclear magnetic relaxation dispersion results. Biophys. J. 1986, 50, 669–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satyanarayana, S.; Dabrowiak, J.C.; Chaires, J.B. Neither delta- nor lambda tris(phenanthroline)ruthenium(II) binds to DNA by classical intercalation. Biochemistry 1992, 31, 9319–9324. [Google Scholar] [CrossRef] [PubMed]
- Dey, D.; Maiti, C.; Maitib, S.; Dhara, D. Interaction between calf thymus DNA and cationic bottle-brush copolymers: Equilibrium and stopped-flow kinetic studies. Phys.Chem.Chem.Phys. 2015, 17, 2366–2377. [Google Scholar] [CrossRef] [PubMed]
- Ilarduya, C.T.D.; Sun, Y.; Duezguenes, N. Gene Delivery by Lipoplexes and Polyplexes. Eur. J. Pharm. Sci. 2010, 40, 159–170. [Google Scholar]
- Godbey, W.T.; Mikos, A.G. Recent Progress in Gene Delivery Using Non-viral Transfer Complexes. J. Control. Release 2001, 72, 115–125. [Google Scholar] [CrossRef]
- Barreleiro, P.C.A.; Lindman, B. The Kinetics of DNA Cationic Vesicle Complex Formation. J. Phys. Chem. B 2003, 107, 6208–6213. [Google Scholar] [CrossRef]
- Dey, D.; Kumar, S.; Maiti, S.; Dhara, D. Stopped-Flow Kinetic Studies of Poly(amidoamine) Dendrimers-Calf Thymus DNA to Form Dendriplexes. J. Phys. Chem. B 2013, 117, 13767–13774. [Google Scholar] [CrossRef] [PubMed]
- Santhiya, D.; Dias, R.S.; Dutta, S.; Das, P.K.; Miguel, M.G.; Lindman, B.; Maiti, S. Kinetic Studies of Amino Acid- Based Surfactant Binding to DNA. J. Phys. Chem. B 2012, 116, 5831–5837. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-D.; Tian, J.-L.; Gu, W.; Liu, X.; Yan, S.-P. Synthesis X-ray crystal structures DNA binding and nuclease activities of two novel 124-triazole-based CuII complexes. Eur. J. Inorg. Chem. 2009, 2009, 5036–5045. [Google Scholar] [CrossRef]
- Hu, X.; Luo, Q.; Qin, Y.; Wu, Y.; Liu, X.-W. DNA Interaction, DNA Photocleavage, Photocytotoxicity In Vitro, and Molecular Docking of Naphthyl-Appended Ruthenium Complexes. Molecules 2022, 27, 3676. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Shen, F.; Wan, D.; Guo, B.-H.; Wang, Y.-J.; Yia, Q.-Y.; Liu, Y.-J. DNA-binding, molecular docking studies and biological activity studies of ruthenium(II) polypyridyl complexes. RSC Adv. 2017, 7, 34945–34958. [Google Scholar] [CrossRef] [Green Version]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucl. Acids. Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Truong, L.; Baker, Y.R.; El-Sagheer, A.H.; Brown, T. Synthesis, Affinity for Complementary RNA and DNA, and Enzymatic Stability of Triazole-Linked Locked Nucleic Acids (t-LNAs). ACS Omega 2018, 3, 6976–6987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallmann, A.; El-Sagheer, A.H.; Dehmel, L.; Mügge, C.; Griesinger, C.N.; Ernsting, P.; Brown, T. Structure and Dynamics of Triazole-Linked DNA: Biocompatibility Explained. Chem. Eur. J. 2011, 17, 14714–14717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, S.; Mujeeb, A.; Ameen, F.; Mubarak-Ishqi, H.; Ur-Rehman, S.; Tabish, M. Investigating the mechanism of binding of nalidixic acid with deoxyribonucleic acid and serum albumin: A biophysical and molecular docking approaches. J. Biomol. Struct. Dyn. 2020, 39, 570–585. [Google Scholar] [CrossRef] [PubMed]
- Reichmann, M.E.; Rice, S.A.; Thomas, C.A.; Doty, P. A Further Examination of the Molecular Weight and Size of Deoxypentose Nucleic Acid. J. Am. Chem. Soc. 1954, 76, 3047–3053. [Google Scholar] [CrossRef]
- Blumenthal, R.D. (Ed.) Methods in Molecular Medicine, Volume 111: Chemosensitivity: Volume 2: In Vivo Models, Imaging, and Molecular Regulators; Humana Press Inc.: Totowa, NJ, USA, 2005. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Parr, R.G.; Yang, W. Density-functional theory of atoms and molecules. Int. J. Quantum Chem. 1993, 47, 101. [Google Scholar]
- Ziegler, T. Approximate density functional theory as a practical tool in molecular energetics and dynamics. Chem. Rev. 1991, 91, 651–667. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Zhurko, G.A.; Zhurko, D.A. Chemcraft. Version 1.7 (Build 132). Available online: www.chemcraftprog.com (accessed on 16 January 2023).
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270. [Google Scholar] [CrossRef]
- Connolly, M.L. Solvent-accessible surfaces of proteins and nucleic acids. Science 1983, 221, 709–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, M.L. Analytical molecular surface calculation. J. Appl. Crystallogr. 1983, 16, 548–558. [Google Scholar] [CrossRef]
- Zhang, C.; Vasmatzis, G.; Cornette, J.L.; DeLisi, C. Determination of atomic desolvation energies from the structures of crystallized proteins. J. Mol. Biol. 1997, 267, 707–726. [Google Scholar] [CrossRef]
- Duhovny, D.; Nussinov, R.; Wolfson, H.J. Efficient Unbound Docking of Rigid Molecules. Algorithms Mol. Biol. 2002, 2452, 185–200. [Google Scholar]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom (No) | TSPH2 | TSP2− | Atom (No) | [RuNOTSP]+ |
|---|---|---|---|---|
| S (10) | 0.23735 | 0.21314 | S (11) | 0.68887 |
| S (26) | 0.23736 | 0.21314 | S (23) | 0.69449 |
| S (38) | 0.06633 | −0.47654 | S (12) | 0.12102 |
| S (48) | 0.06634 | −0.47655 | S (24) | 0.15628 |
| N (1) | −0.48666 | −0.51020 | N (26) | −0.43862 |
| N (11) | −0.44178 | −0.45570 | N (42) | −0.40769 |
| - | - | N (35) | 0.45495 | |
| - | - | Ru | −0.67395 |
| Parameter | TSPH2 | TSP2− | [RuNOTSP]+ |
|---|---|---|---|
| EHOMO (eV) | −5.56 | 1.04 | −8.09 |
| ELUMO (eV) | −0.72 | 4.70 | −5.87 |
| ELUMO – EHOMO (eV) | 4.84 | 3.66 | 2.22 |
| χ (eV) | 3.14 | −2.87 | 6.98 |
| η (eV) | 2.421 | 1.831 | 1.110 |
| Pi (eV) | −3.14 | 2.87 | −6.98 |
| σ (eV−1) | 0.413 | 0.456 | 0.901 |
| S (eV−1) | 0.206 | 0.273 | 0.451 |
| ω (eV) | 2.037 | 2.249 | 21.946 |
| Compound | KD (L mol−1) | KB (L mol−1 s−1) | n | Kf |
|---|---|---|---|---|
| [RuNOTSP]+ | (3.3 ± 0.3) × 104 | (3.3 ± 0.3) × 1012 | 0.9 | (0.81 ± 0.01) × 104 |
| TSPH2 | (2.6 ± 0.2) × 104 | (2.6 ± 0.2) × 1012 | 0.7 | (0.13 ± 0.10) × 104 |
| Activation Parameters | [RuNOTSP]+ | TSPH2 | |
|---|---|---|---|
| First interaction step | k1 [M−1 s−1] | 1.0 ± 0.2 | 0.7 ± 0.1 |
| k−1 [10−2 s−1] | 4.1 ± 0.0 | 5.5 ± 0.0 | |
| Ka1 [M−1] | 45.6 | 12.7 | |
| Kd1 [10−2 M] | 2.2 | 7.9 | |
| ΔG1# [kJ mol−1] | −17.1 | −6.1 | |
| Second interaction step | K2 [M−1 s−1] | 0.5 ± 0.0 | 0.2 ± 0.1 |
| k−2 [10−2 s−1] | 1.1 ± 0.0 | 0.3 ± 0.3 | |
| Ka2 [M−1] | 40 | 59 | |
| Kd2 [10−2 M] | 2.5 | 1.69 | |
| ΔG2# [kJ mol−1] | −19.48 | −13.17 | |
| Overall reaction data | Kd [10−2 M] | 2.28 | 66 |
| Ka [M] | 43.7 | 15.1 | |
| ΔG# [kJ mol−1] | −36.54 | −19.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shereef, H.A.; Moemen, Y.S.; Elshami, F.I.; El-Nahas, A.M.; Shaban, S.Y.; van Eldik, R. DNA Binding and Cleavage, Stopped-Flow Kinetic, Mechanistic, and Molecular Docking Studies of Cationic Ruthenium(II) Nitrosyl Complexes Containing “NS4” Core. Molecules 2023, 28, 3028. https://doi.org/10.3390/molecules28073028
Shereef HA, Moemen YS, Elshami FI, El-Nahas AM, Shaban SY, van Eldik R. DNA Binding and Cleavage, Stopped-Flow Kinetic, Mechanistic, and Molecular Docking Studies of Cationic Ruthenium(II) Nitrosyl Complexes Containing “NS4” Core. Molecules. 2023; 28(7):3028. https://doi.org/10.3390/molecules28073028
Chicago/Turabian StyleShereef, Hadeer A., Yasmine S. Moemen, Fawzia I. Elshami, Ahmed M. El-Nahas, Shaban Y. Shaban, and Rudi van Eldik. 2023. "DNA Binding and Cleavage, Stopped-Flow Kinetic, Mechanistic, and Molecular Docking Studies of Cationic Ruthenium(II) Nitrosyl Complexes Containing “NS4” Core" Molecules 28, no. 7: 3028. https://doi.org/10.3390/molecules28073028
APA StyleShereef, H. A., Moemen, Y. S., Elshami, F. I., El-Nahas, A. M., Shaban, S. Y., & van Eldik, R. (2023). DNA Binding and Cleavage, Stopped-Flow Kinetic, Mechanistic, and Molecular Docking Studies of Cationic Ruthenium(II) Nitrosyl Complexes Containing “NS4” Core. Molecules, 28(7), 3028. https://doi.org/10.3390/molecules28073028