


Synthesis and Elimination Pathways of 1-Methanesulfonyl-1,2-dihydroquinoline Sulfonamides



Abstract
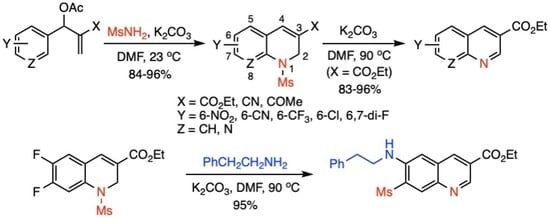
:
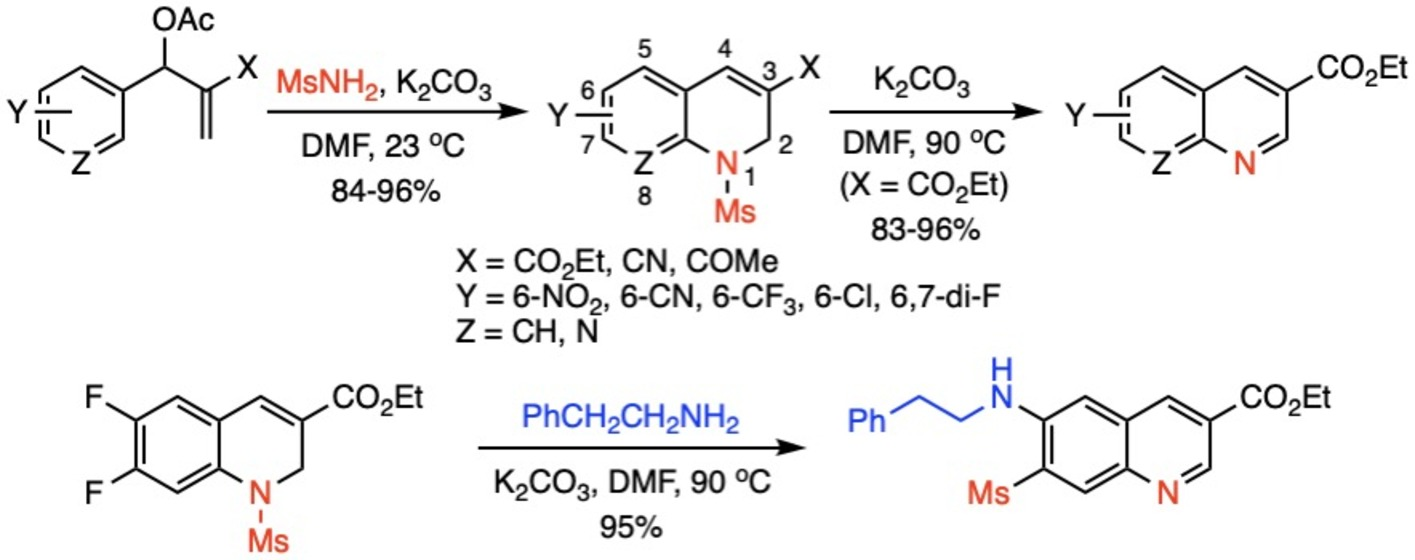
1. Introduction
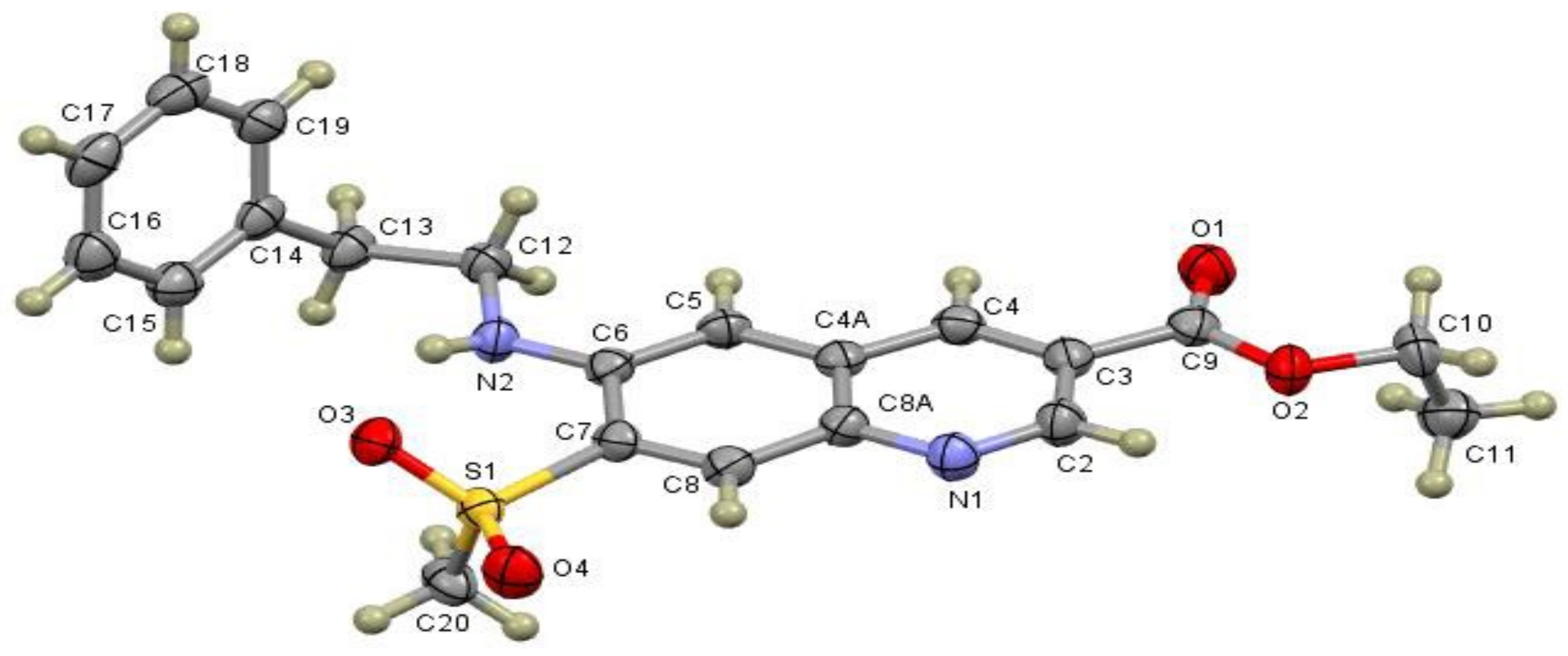
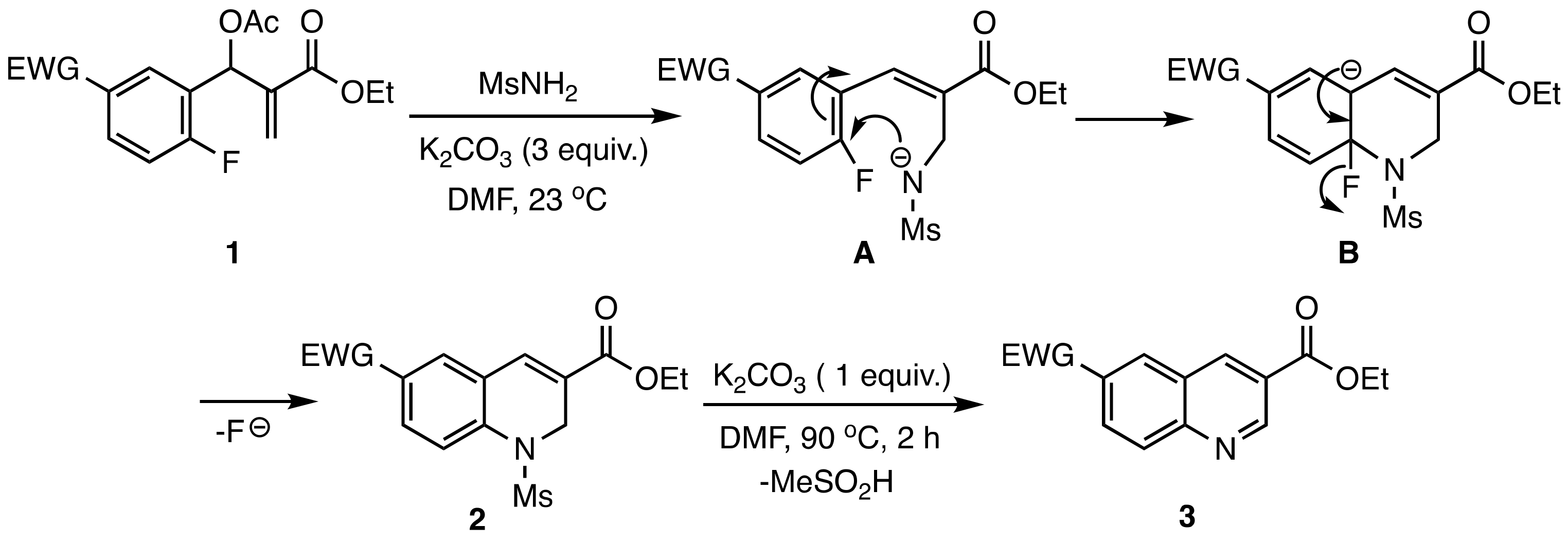
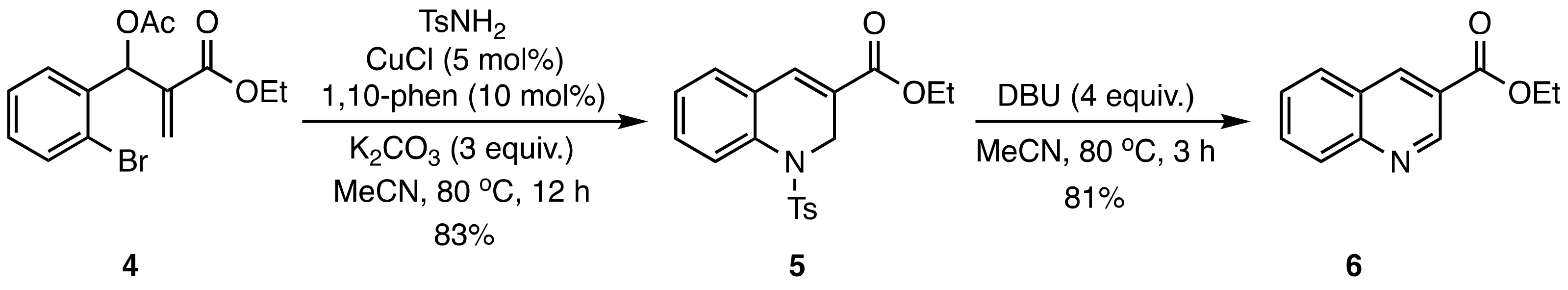
2. Results and Discussion
3. Methods and Materials
3.1. General Methods
3.2. General Procedure for the Preparation of MBH Acetates
3.2.1. (±)-Ethyl 2-(acetoxy(2-fluoro-5-(trifluoromethyl)phenyl)methyl)acrylate (15)
3.2.2. (±)-Ethyl 2-(acetoxy(5-chloro-2-fluorophenyl)methyl)acrylate (16)
3.2.3. (±)-Ethyl 2-(acetoxy(2,4,5-trifluorophenyl)methyl)acrylate (17)
3.2.4. (±)-2-Cyano-1-(2-fluoro-5-(trifluoromethyl)phenyl)allyl Acetate (21)
3.2.5. (±)-1-(5-Chloro-2-fluorophenyl)-2-cyanoallyl Acetate (22)
3.2.6. (±)-1-(2-Fluoro-5-nitrophenyl)-2-methylene-3-oxobutyl Acetate (23)
3.2.7. (±)-1-(5-Cyano-2-fluorophenyl)-2-methylene-3-oxobutyl Acetate (24)
3.2.8. (±)-1-(2-Fluoro-5-(trifluoromethyl)phenyl)-2-methylene-3-oxobutyl Acetate (25)
3.2.9. (±)-2-Methylene-3-oxo-1-(2,4,5-trifluorophenyl)butyl Acetate (26)
3.2.10. (±)-1-(2-Fluoropyridin-3-yl)-2-methylene-3-oxobutyl Acetate (27)
3.3. General Procedure for the Preparation of 1-Methanesulfonyl-1,2-Dihydroquinolines from MBH Acetates
3.3.1. Ethyl 1-(Methylsulfonyl)-6-nitro-1,2-dihydroquinoline-3-carboxylate (28-Ms)
3.3.2. Ethyl 6-Nitro-1-tosyl-1,2-dihydroquinoline-3-carboxylate (28-Ts)
3.3.3. Ethyl 6-Cyano-1-(methylsulfonyl)-1,2-dihydroquinoline-3-carbonitrile (29)
3.3.4. Ethyl 1-(Methylsulfonyl)-6-(trifluoromethyl)-1,2-dihydroquinoline-3-carboxylate (30)
3.3.5. Ethyl 6-Chloro-1-(methylsulfonyl)-1,2-dihydroquinoline-3-carboxylate (31)
3.3.6. Ethyl 6,7-Difluoro-1-(methylsulfonyl)-1,2-dihydroquinoline-3-carboxylate (32)
3.3.7. Ethyl 1-(Methylsulfonyl)-1,2-dihydro-1,8-naphthyridine-3-carboxylate (33)
3.3.8. 1-(Methylsulfonyl)-6-nitro-1,2-dihydroquinoline-3-carbonitrile (34)
3.3.9. 1-(1-(Methylsulfonyl)-6-nitro-1,2-dihydroquinolin-3-yl)ethan-1-one (38)
3.3.10. 3-Acetyl-1-(methylsulfonyl)-1,2-dihydroquinoline-6-carbonitrile (39)
3.3.11. 1-(1-(Methylsulfonyl)-6-(trifluoromethyl)-1,2-dihydroquinolin-3-yl)ethan-1-one (40)
3.3.12. 1-(6,7-Difluoro-1-(methylsulfonyl)-1,2-dihydroquinolin-3-yl)ethan-1-one (41)
3.3.13. 1-(1-(Methylsulfonyl)-1,2-dihydro-1,8-naphthyridin-3-yl)ethan-1-one (42)
3.4. General Procedure for Elimination of the Methylsulfonyl Group to Give Quinolines
3.4.1. Ethyl 6-Nitroquinoline-3-carboxylate (43)
3.4.2. Ethyl 6-Cyanoquinoline-3-carboxylate (44)
3.4.3. Ethyl 6-(Trifluoromethyl)quinoline-3-carboxylate (45)
3.4.4. Ethyl 6-Chloroquinoline-3-carboxylate (46)
3.4.5. 6-Nitroquinoline-3-carbonitrile (48)
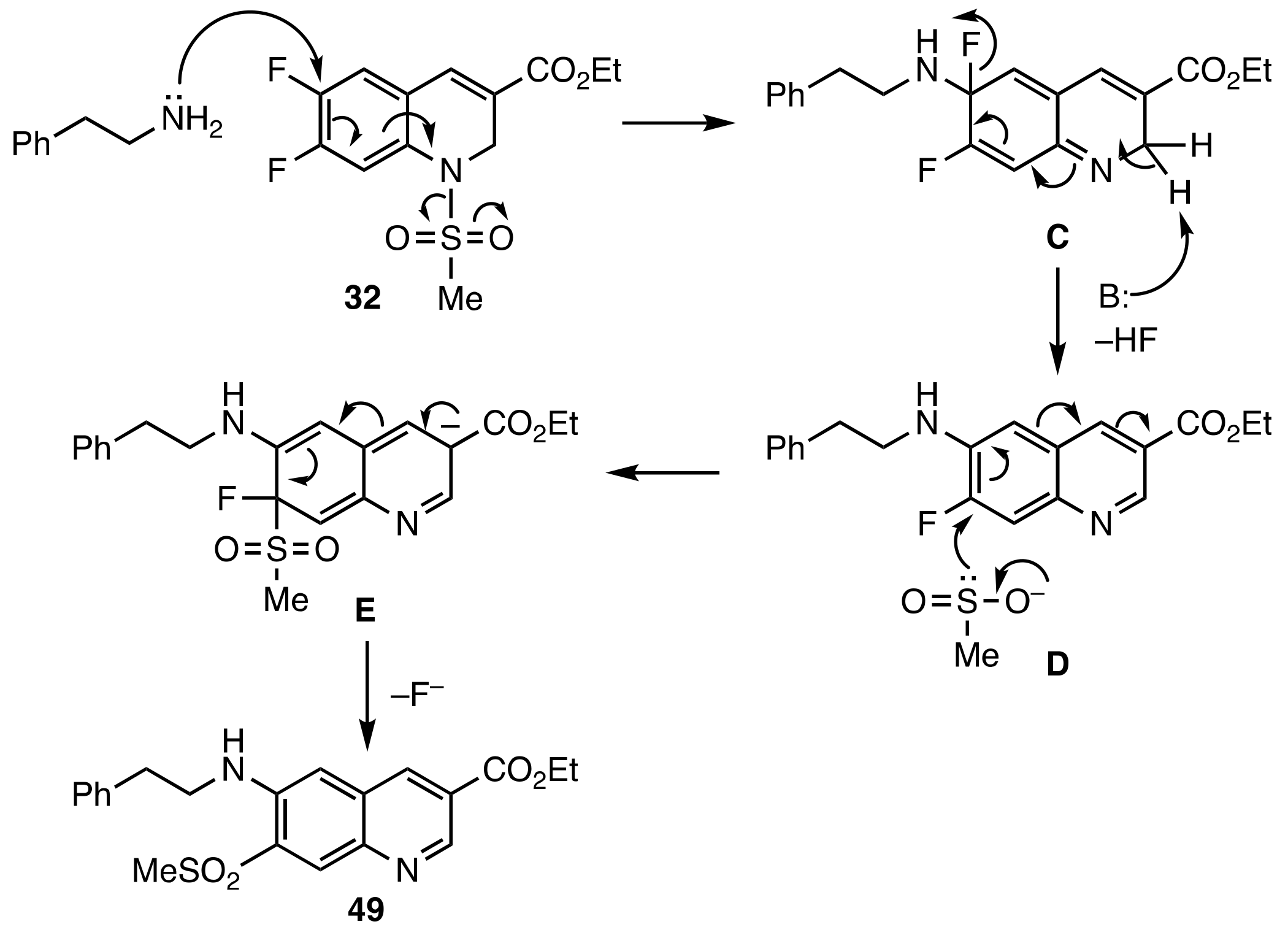
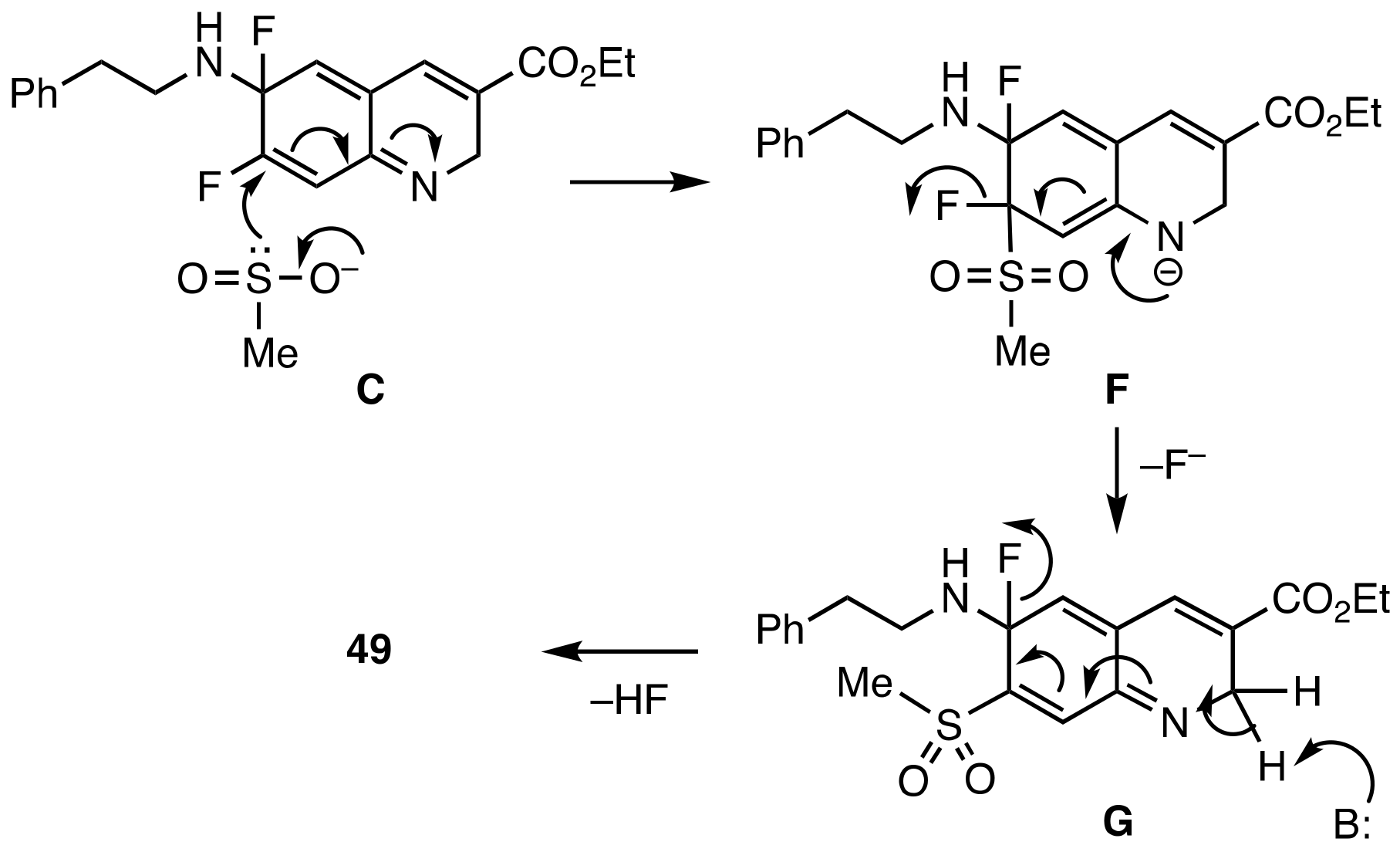
3.5. Base Promoted Sulfone Migration in 32 to Give Ethyl 7-(Methylsulfonyl)-6-(phenethyl-amino)quinoline-3-carboxylate (49)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Annor-Gyamfi, J.K.; Ametsetor, E.; Meraz, K.; Bunce, R.A. Dihydroquinolines, dihydronaphthyridines and quinolones by domino reactions of Morita-Baylis-Hillman acetates. Molecules 2021, 26, 890. [Google Scholar] [CrossRef] [PubMed]
- Annor-Gyamfi, J.K.; Ametsetor, E.; Meraz, K.; Bunce, R.A. Naphthalenes and Quinolines by Domino Reactions of Morita–Baylis–Hillman Acetates. Molecules 2020, 25, 5168. [Google Scholar] [CrossRef]
- Imanzadeh, G.H.; Zare, A.; Kalafi-Hezhad, A.; Hasaninejad, A.; Moosavi Zare, A.R.; Parhami, A. Microwave-assisted Michael addition of sulfonamides to α,β-unsaturated esters: A rapid entry to protected β-amino acid synthesis. J. Iran. Chem. Soc. 2007, 4, 467–475. [Google Scholar] [CrossRef]
- Zare, A.; Hasaninejad, A.; Khalafi-Nezhad, A.; Moosavi Zare, A.R.; Parhami, A. An efficient solventless method for the synthesis of N,N-dialkyl sulfonamide derivatives. J. Iran. Chem. Soc. 2008, 5, 617–622. [Google Scholar] [CrossRef]
- Wang, D.; Wei, Y.; Shi, M. Synthesis of highly functionalized aminoindolizines by titanium(IV) chloride mediated cycloisomerization and phosphine-catalyzed aza-Michael addition reactions. Asian J. Org. Chem. 2013, 2, 480–485. [Google Scholar] [CrossRef]
- Huang, F.-P.; Dong, Z.-W.; Zhang, H.-R.; Hui, X.-P. Tandem aza-Michael-aldol reactions: One-pot synthesis of functionalized piperidine derivatives. J. Heterocycl. Chem. 2013, 51, 532–538. [Google Scholar] [CrossRef]
- Douraki, S.M.; Massah, A.R. The zeolite ZSM-5-SO3H catalyzed aza-Michael addition of amines and sulfonamides to electron-deficient alkenes under solvent-free conditions. Indian J. Chem. Sect. B Org. Chem. Med. Chem. 2015, 54, 1346–1349. [Google Scholar] [CrossRef]
- Luo, H.; Yan, X.; Chen, L.; Li, Y.; Liu, N.; Yin, G. Enantioselective catalytic domino aza-Michael-Henry reactions: One-pot asymmetric synthesis of 3-nitro-1,2-dihydroquinolines via iminium activation. Eur. J. Org. Chem. 2016, 2016, 1702–1707. [Google Scholar] [CrossRef]
- Zhai, X.-D.; Yang, Z.-D.; Luo, Z.; Xu, H.-T. Asymmetric catalyzed intramolecular aza-Michael reaction mediated by quinine-derived primary amines. Chin. Chem. Lett. 2017, 28, 1793–1797. [Google Scholar] [CrossRef]
- Roy, T.K.; Parhi, B.; Ghorai, P. Chinchonamine squaramide catalyzed asymmetric aza-Michael reaction: Dihydroisoquinolines and tetrahydropyridines. Angew. Chem. Int. Ed. 2018, 57, 9397–9401. [Google Scholar] [CrossRef] [PubMed]
- Bunce, R.A.; Smith, C.L.; Knight, C.L. N-(Nitrophenyl) benzamide and benzenesulfonamide derivatives by nucleophilic aromatic substitution. Org. Prep. Proced. Int. 2004, 36, 482–486. [Google Scholar] [CrossRef]
- Bordwell, F.G.; Algrim, D. Nitrogen acids. 1. Carboxamides and sulfonamides. J. Org. Chem. 1976, 41, 2507–2508. [Google Scholar] [CrossRef]
- Bordwell, F.G.; Fried, H.E.; Hughes, D.L.; Lynch, T.Y.; Satish, A.V.; Whang, Y.E. Acidities of carboxamides, hydroxamic acids, carbohydrazides, benzenesulfonamides, and benzenesulfonohydrazides in DMSO solution. J. Org. Chem. 1990, 55, 3330–3336. [Google Scholar] [CrossRef]
- Trost, B.M. The atom economy–A search for synthetic efficiency. Science 1991, 254, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M. Atom economy—A challenge for organic synthesis: Homogeneous catalysis leads the way. Angew. Chem. Int. Ed. 1995, 90, 259–281. [Google Scholar] [CrossRef]
- Trost, B.M. Atom economy: A challenge for enhanced synthetic efficiency. In Handbook of Green Chemistry; Wiley Online Library: Hoboken, NJ, USA, 2012; pp. 1–33. [Google Scholar] [CrossRef]
- Kim, J.N.; Lee, H.J.; Lee, K.Y.; Kim, H.S. Synthesis of 3-quinolinecarboxylic acid esters from the Baylis–Hillman adducts of 2-halobenzaldehyde N-tosylimines. Tetrahedron Lett. 2001, 42, 3737–3740. [Google Scholar] [CrossRef]
- Niu, Q.; Mao, H.; Yuan, G.; Gao, J.; Liu, H.; Tu, Y.; Wang, X.; Lv, X. Copper-catalyzed domino SN2/Coupling reaction: A versatile and facile synthesis of cyclic compounds from Baylis-Hillman acetates. Adv. Synth. Catal. 2013, 355, 1185–1192. [Google Scholar] [CrossRef]
- Chen, L.; Wilder, P.T.; Drennen, B.; Tran, J.; Roth, B.M.; Chesko, K.; Shapiro, P.; Fletcher, S. Structure-based design of 3-carboxy-substituted 1,2,3,4-tetrahydroquinolines as inhibitors of myeloid cell leukemia-1 (Mcl-1). Org. Biomol. Chem. 2015, 14, 5505–5510. [Google Scholar] [CrossRef]
- Okolotowicz, K.J.; Dwyer, M.; Ryan, D.; Cheng, J.; Cashman, E.A.; Moore, S.; Mercola, M.; Cashman, J.R. Novel tertiary sulfonamides as potent anti-cancer agents. Bioorganic Med. Chem. 2018, 26, 4441–4451. [Google Scholar] [CrossRef]
- Peerzada, M.N.; Khan, P.; Ahmad, K.; Hassan, M.I.; Azam, A. Synthesis, characterization and biological evaluation of tertiary sulfonamide derivatives of pyridyl-indole based heteroaryl chalcone as potential carbonic anhydrase IX inhibitors and anticancer agents. Eur. J. Med. Chem. 2018, 155, 13–23. [Google Scholar] [CrossRef]
- Durgapal, S.D.; Soman, S.S. Evaluation of novel coumarin-proline sulfonamide hybrids as anticancer and antidiabetic agents. Synth. Commun. 2019, 49, 2869–2883. [Google Scholar] [CrossRef]
- Taslimi, P.; Sujayev, A.; Mamedova, S.; Kalın, P.; Gulçin, P.; Sadeghian, N.; Beydemir, S.; Kufrevioglu, O.I.; Alwasel, S.H.; Farzaliyev, V.; et al. Synthesis and bioactivity of several new hetaryl sulfonamides. J. Enzym. Inhib. Med. Chem. 2017, 32, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Pero, J.E.; Matthews, J.M.; Behm, D.J.; Brnardic, E.J.; Brooks, C.; Budzik, B.W.; Costell, M.H.; Donatelli, C.A.; Eisennagel, S.H.; Erhard, K.; et al. Design and optimization of sulfone pyrrolidine sulfonamide antagonists of transient receptor potential vanilloid-4 with in vivo activity in a pulmonary edema model. J. Med. Chem. 2018, 61, 11209–11220. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S. Sulfonamide synthesis under green conditions. Synth. Commun. 2021, 51, 1023–1044. [Google Scholar] [CrossRef]
- Procopiou, P.A.; Baugh, S.P.D.; Flack, S.S.; Inglis, G.G.A. An extremely powerful acylation reaction of alcohols with acid anhydrides catalyzed by trimethylsilyl trifluoromethanesulfonate. J. Org. Chem. 1998, 63, 2342–2347. [Google Scholar] [CrossRef]
- Gnanasekaran, K.K.; Yoon, J.; Bunce, R.A. Nucleophilic additions to polarized vinylarenes. Tetrahedron Lett. 2016, 57, 3190–3193. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, R.; Yousaf, M.; Naqvi, S.A.R.; Irfan, M.; Zahoor, A.F.; Hussain, A.I.; Chatha, S.A.S. Synthesis of ciprofloxacin-based compounds: A review. Synth. Commun. 2016, 46, 1849–1879. [Google Scholar] [CrossRef]
- Flynn, A.J.; Ford, A.; Maguire, A.R. Synthetic and mechanistic aspects of sulfonyl migrations. Org. Biomol. Chem. 2020, 18, 2549–2610. [Google Scholar] [CrossRef]
- Xin, X.; Wang, D.; Li, X.; Wan, B. Highly regioselective migration of the sulfonyl group: Easy access to functionalized pyrroles. Angew. Chem. Int. Ed. 2012, 51, 1693–1697. [Google Scholar] [CrossRef]
- Tummatorn, J.; Thongsornkleeb, C.; Ruchirawat, S.; Gettongsong, T. Synthesis of 2,4-unsubstituted quinoline-3-carboxylic acid ethyl esters from arylmethyl azides via a domino process. Org. Biomol. Chem. 2013, 11, 1463–1467. [Google Scholar] [CrossRef]
- Bruker. APEX3, version 2018.7-2; Bruker Inc.: Madison, WI, USA, 2018.
- Bruker. SAINT, version 8.38A; Bruker Inc.: Madison, WI, USA, 2016.
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXT-Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| X | Y | Z | Pdt | Yield (%) |
|---|---|---|---|---|
| CO2Et | 2-F-5-NO2 | CH | 13 | 98 a |
| CO2Et | 2-F-5-CN | CH | 14 | 98 a |
| CO2Et | 2-F-5-CF3 | CH | 15 | 92 |
| CO2Et | 2-F-5-Cl | CH | 16 | 93 |
| CO2Et | 2,4,5-tri-F | CH | 17 | 93 |
| CO2Et | 2-F | N | 18 | 96 a |
| CN | 2-F-5-NO2 | CH | 19 | 98 a |
| CN | 2-F-5-CN | CH | 20 | 98 a |
| CN | 2-F-5-CF3 | CH | 21 | 93 |
| CN | 2-F-5-Cl | CH | 22 | 95 |
| COCH3 | 2-F-5-NO2 | CH | 23 | 96 |
| COCH3 | 2-F-5-CN | CH | 24 | 98 |
| COCH3 | 2-F-5-CF3 | CH | 25 | 95 |
| COCH3 | 2,4,5-tri-F | CH | 26 | 97 |
| COCH3 | 2-F | N | 27 | 89 |

| Substrate | Pdt | X | Y | Z | Yield (%) |
|---|---|---|---|---|---|
| 13 (X = CO2Et; Y = 2-F-5-NO2; Z = CH) | 28-Ms | CO2Et | 6-NO2 | CH | 96 |
| 13 (X = CO2Et; Y = 2-F-5-NO2; Z = CH) | 28-Ts | CO2Et | 6-NO2 | CH | 91 |
| 14 (X = CO2Et; Y = 2-F-5-CN; Z = CH) | 29 | CO2Et | 6-CN | CH | 93 |
| 15 (X = CO2Et; Y = 2-F-5-CF3; Z = CH) | 30 | CO2Et | 6-CF3 | CH | 96 |
| 16 (X = CO2Et; Y = 2-F-5-Cl; Z = CH) | 31 | CO2Et | 6-Cl | CH | 91 |
| 17 (X = CO2Et; Y = 2,4,5-tri-F; Z = CH) | 32 | CO2Et | 6,7-di-F | CH | 93 |
| 18 (X = CO2Et; Y = 2-F; Z = N) | 33 | CO2Et | H | N | 92 |
| 19 (X = CN; Y = 2-F-5-NO2; Z = CH) | 34 | CN | 6-NO2 | CH | 96 |
| 20 (X = CN; Y = 2-F-5-CN; Z = CH) | 35 | CN | 6-CN | CH | 0 a |
| 21 (X = CN; Y = 2-F-5-CF3; Z = CH) | 36 | CN | 6-CF3 | CH | 0 a |
| 22 (X = CN; Y = 2-F-5-Cl; Z = CH) | 37 | CN | 6-Cl | CH | 0 a |
| 23 (X = COMe; Y = 2-F-5-NO2; Z = CH) | 38 | COMe | 6-NO2 | CH | 93 |
| 24 (X = COMe; Y = 2-F-5-CN; Z = CH) | 39 | COMe | CN | CH | 88 |
| 25 (X = COMe; Y = 2-F-5-CF3; Z = CH) | 40 | COMe | CF3 | CH | 93 |
| 26 (X = COMe; Y = 2,4,5-tri-F; Z = CH) | 41 | COMe | 6,7-di-F | CH | 84 |
| 27 (X = COMe; Y = 2-F; Z = CH) | 42 | COMe | H | N | 87 |

| Substrate | X | Y | Z | Pdt | Yield (%) |
|---|---|---|---|---|---|
| 28-Ms | CO2Et | 6-NO2 | CH | 43 | 83 |
| 28-Ts | CO2Et | 6-NO2 | CH | 43 | 85 |
| 29 | CO2Et | 6-CN | CH | 44 | 86 |
| 30 | CO2Et | 6-CF3 | CH | 45 | 87 |
| 31 | CO2Et | 6-Cl | CH | 46 | 96 |
| 32 | CO2Et | 6,7-di-F | CH | 47 | 0 a |
| 34 | CN | 6-NO2 | CH | 48 | 95 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ametsetor, E.; Fobi, K.; Bunce, R.A. Synthesis and Elimination Pathways of 1-Methanesulfonyl-1,2-dihydroquinoline Sulfonamides. Molecules 2023, 28, 3256. https://doi.org/10.3390/molecules28073256
Ametsetor E, Fobi K, Bunce RA. Synthesis and Elimination Pathways of 1-Methanesulfonyl-1,2-dihydroquinoline Sulfonamides. Molecules. 2023; 28(7):3256. https://doi.org/10.3390/molecules28073256
Chicago/Turabian StyleAmetsetor, Ebenezer, Kwabena Fobi, and Richard A. Bunce. 2023. "Synthesis and Elimination Pathways of 1-Methanesulfonyl-1,2-dihydroquinoline Sulfonamides" Molecules 28, no. 7: 3256. https://doi.org/10.3390/molecules28073256
APA StyleAmetsetor, E., Fobi, K., & Bunce, R. A. (2023). Synthesis and Elimination Pathways of 1-Methanesulfonyl-1,2-dihydroquinoline Sulfonamides. Molecules, 28(7), 3256. https://doi.org/10.3390/molecules28073256