


17-β-Estradiol—β-Cyclodextrin Complex as Solid: Synthesis, Structural and Physicochemical Characterization

 ,
,  ,
,  ,
,  , ,
, , 
Abstract
:1. Introduction
2. Results and Discussion
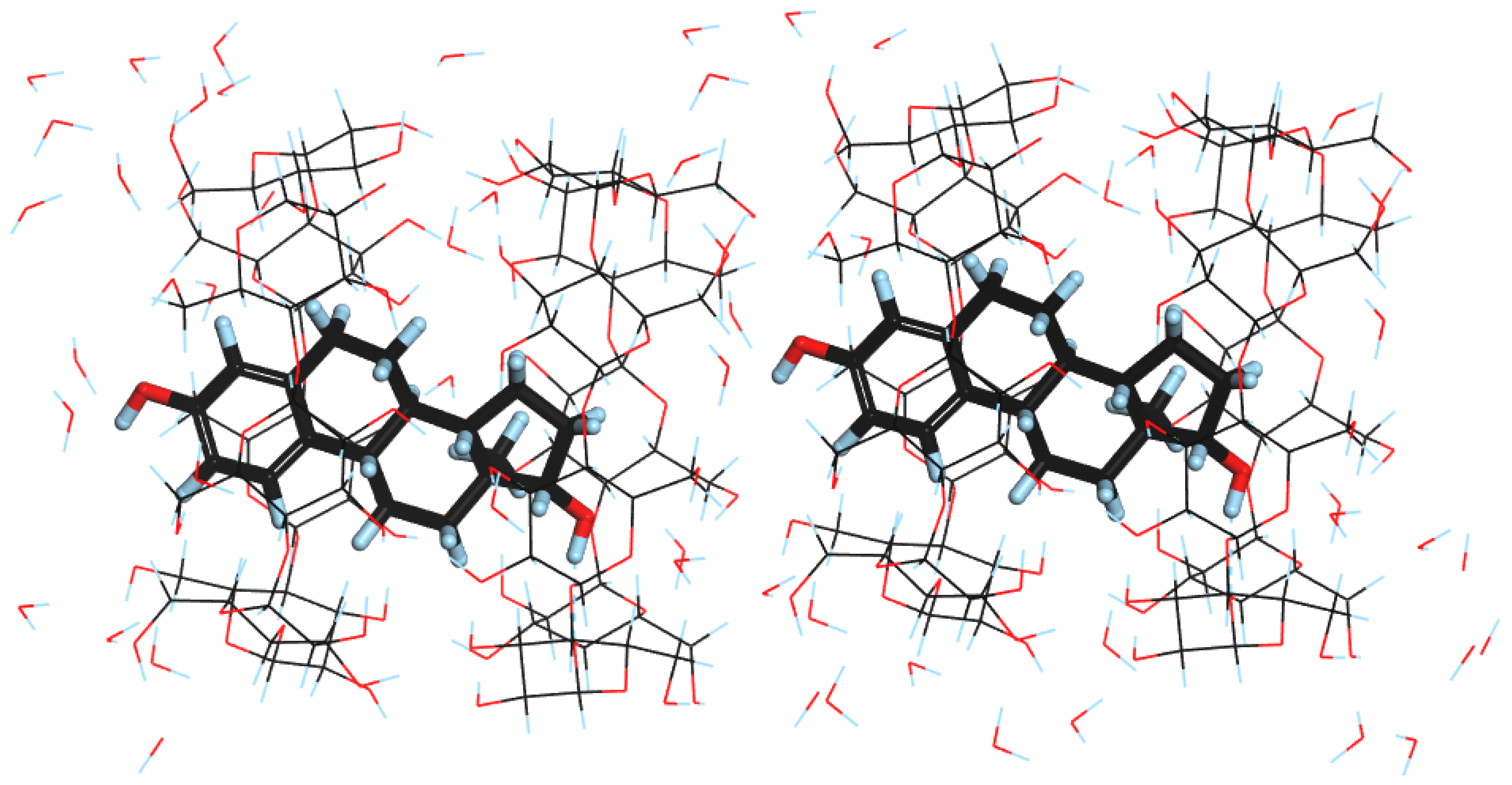
2.1. SCXRD Results
2.2. Periodic DFT Calculation Results
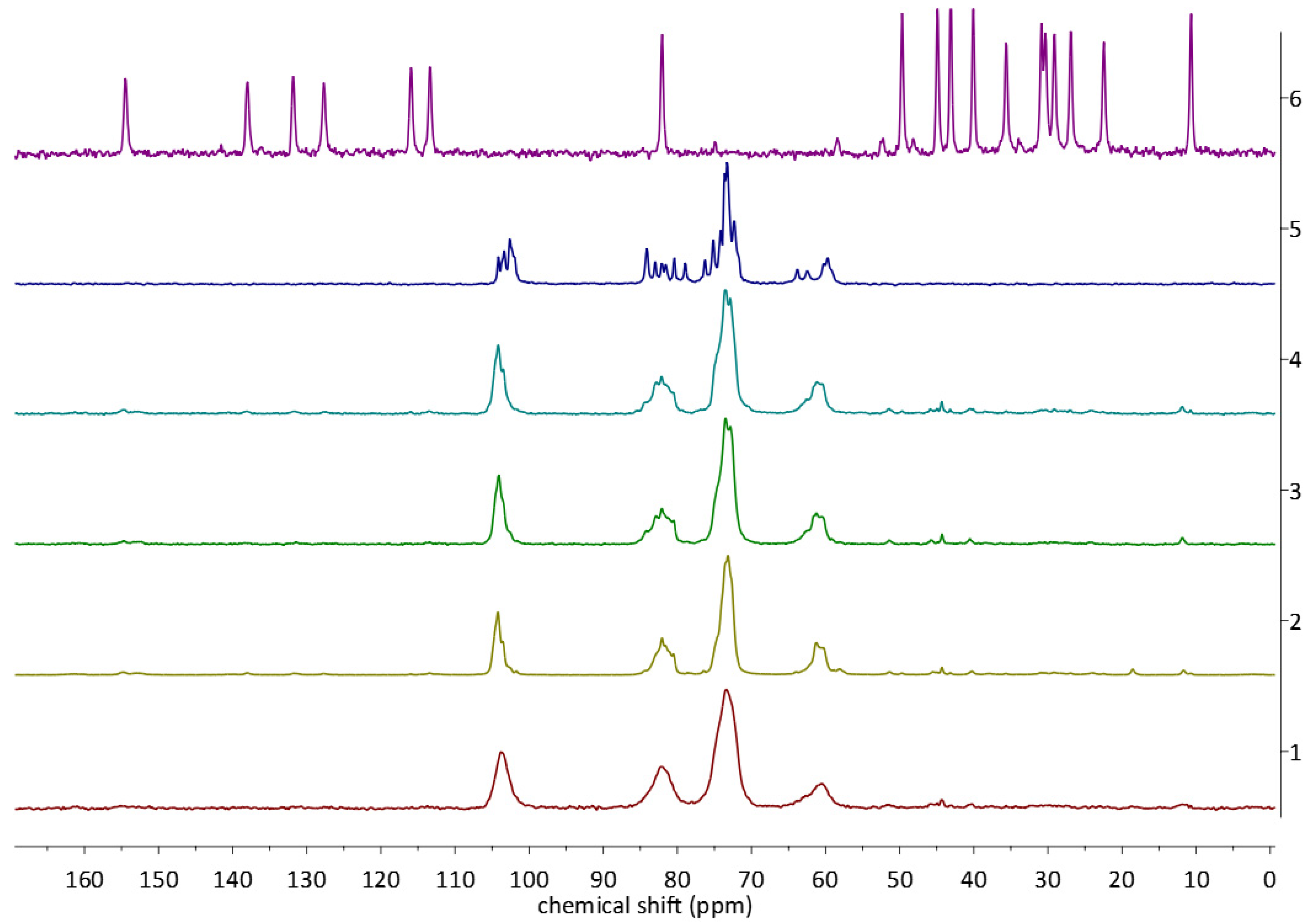
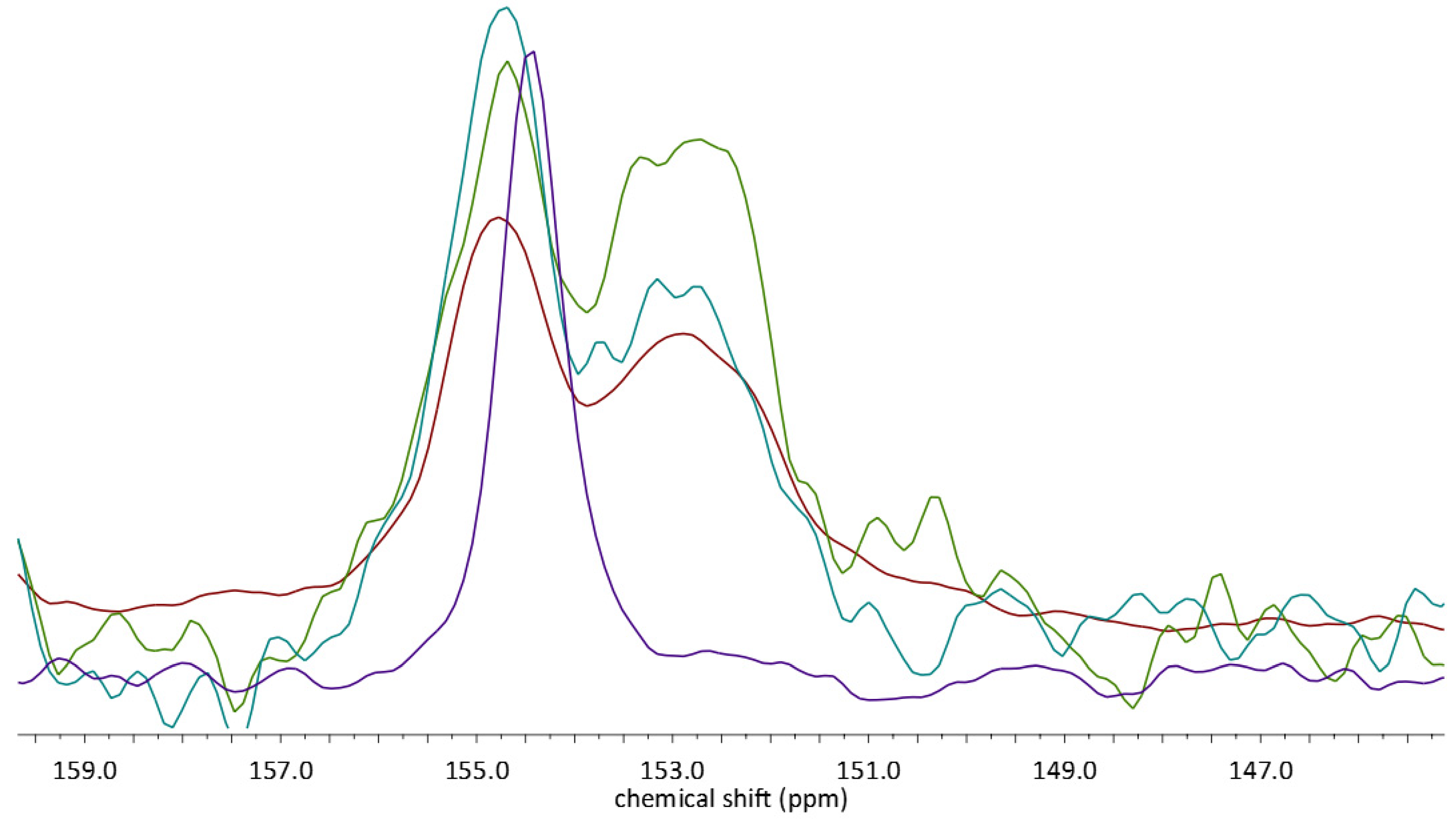
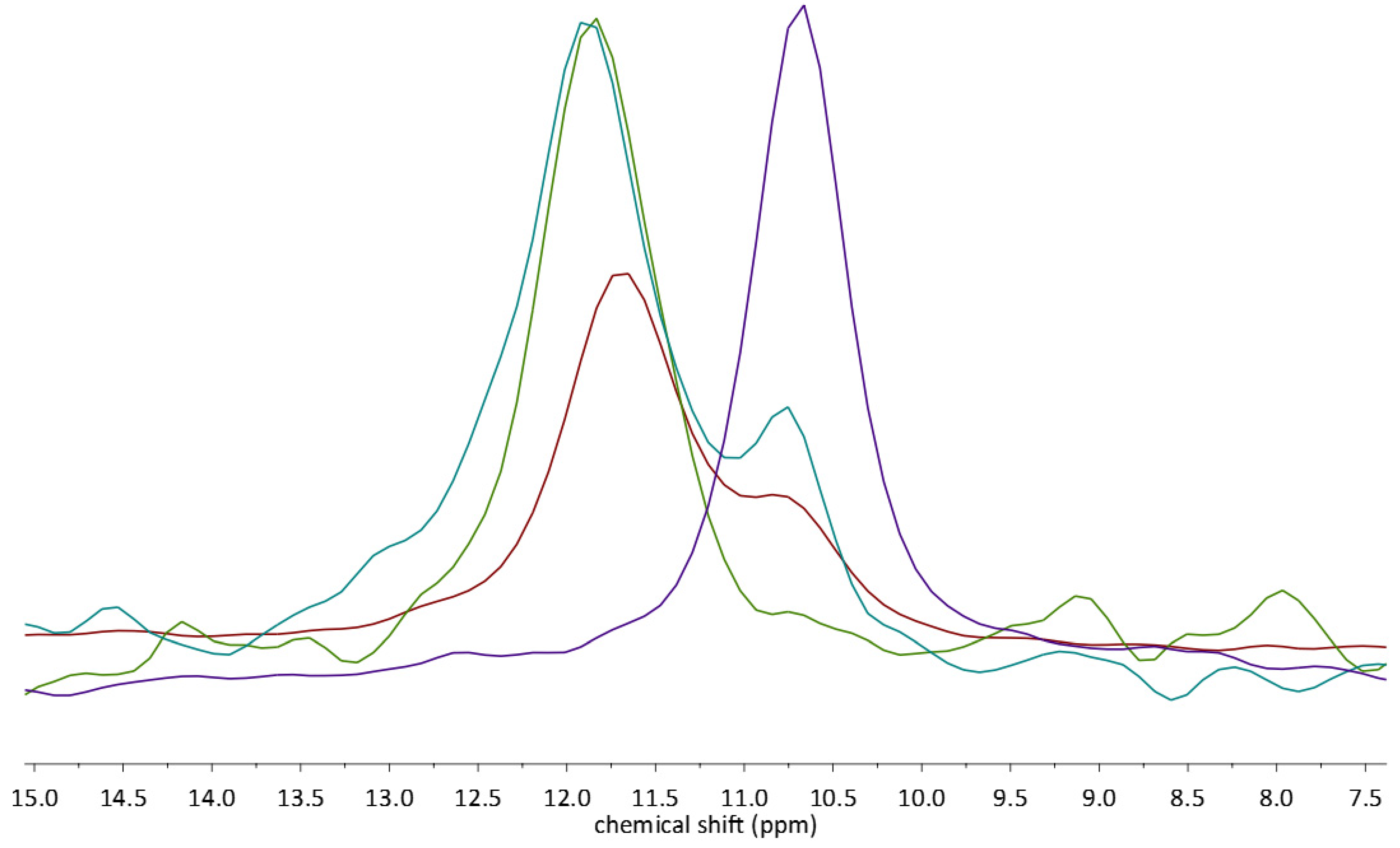
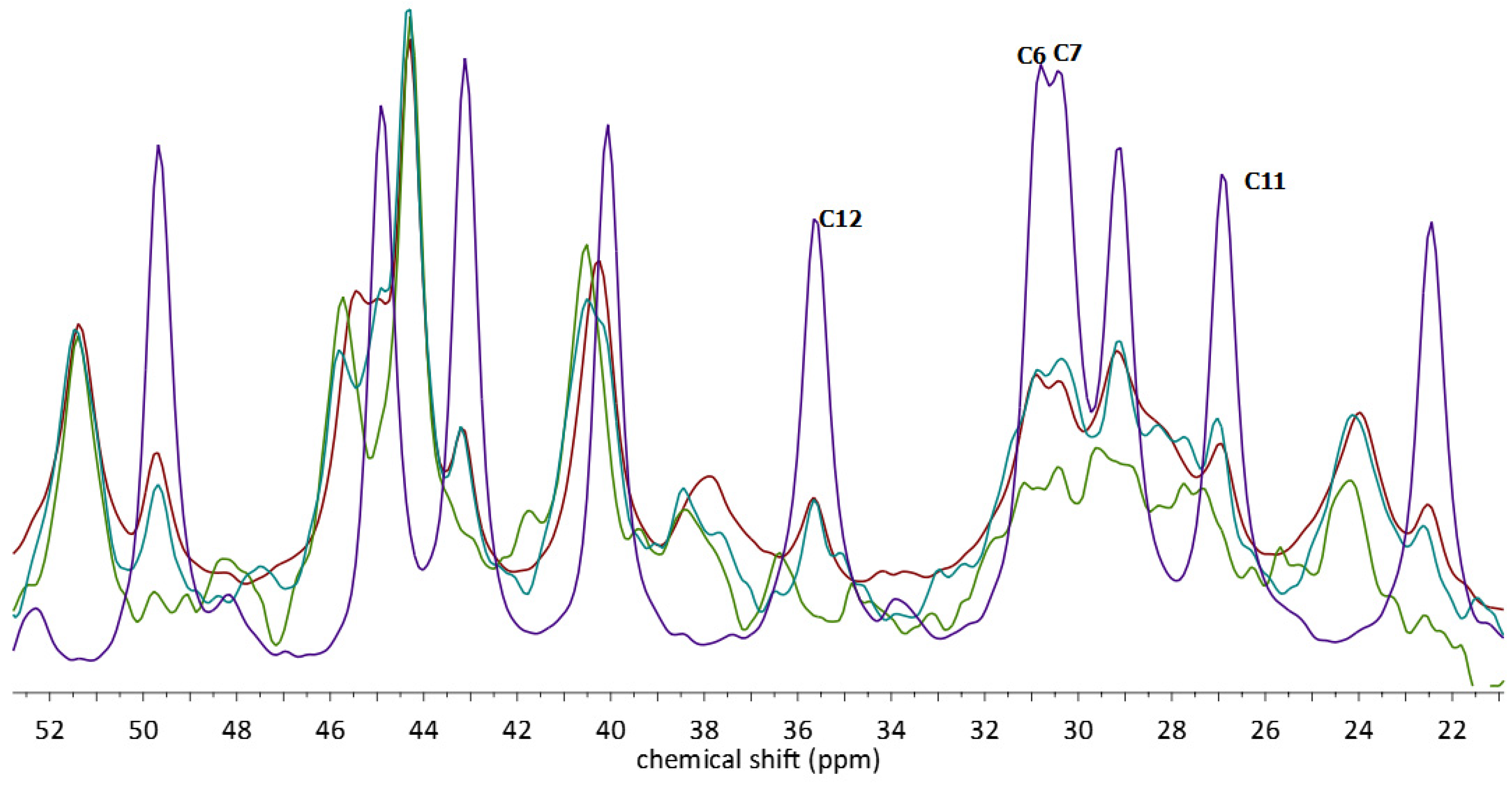
2.3. 13C CP MAS Solid State NMR Analysis
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Methods of Complex Preparation
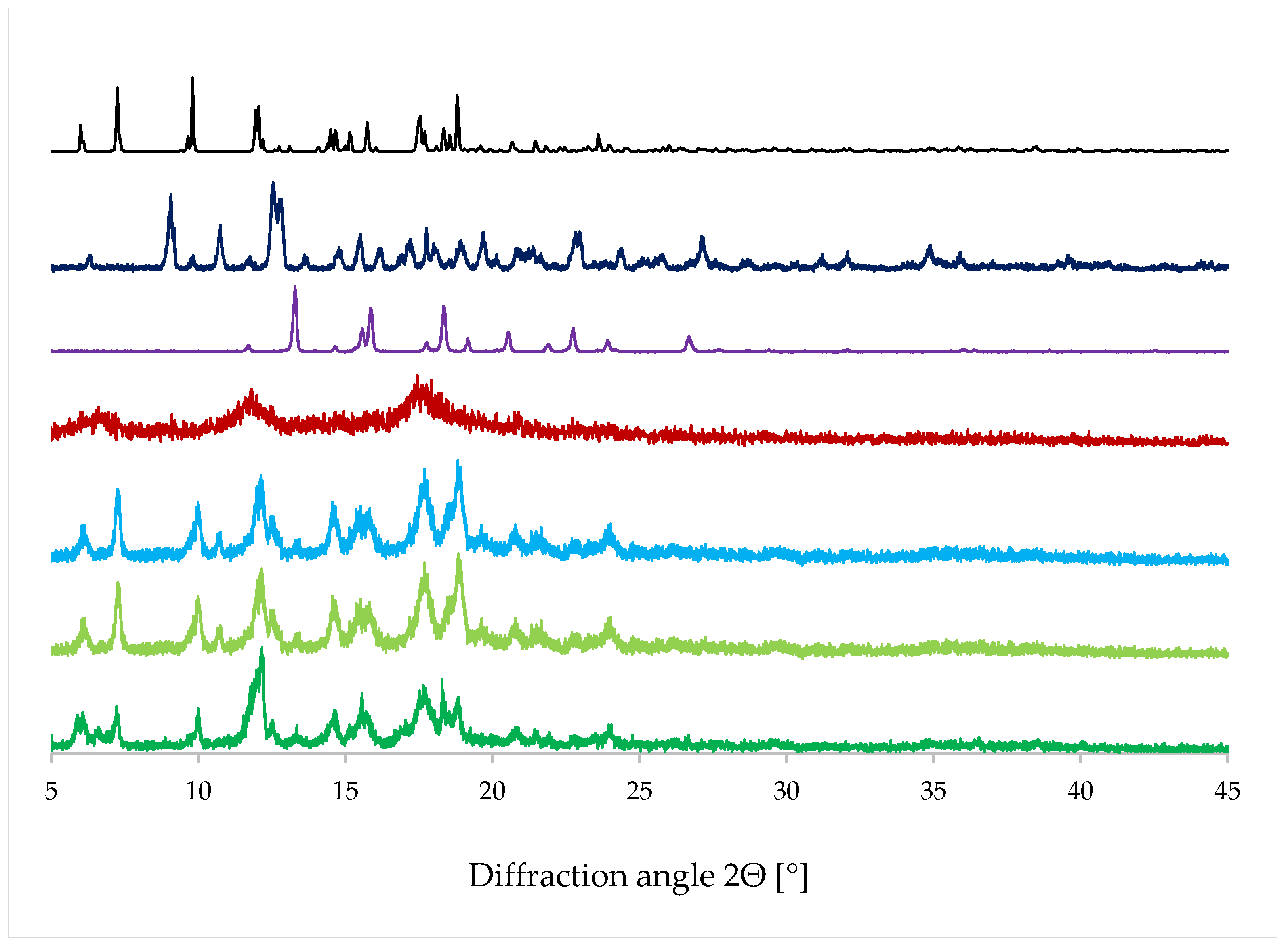
4.3. Powder X-ray Diffraction (PXRD)
4.4. Single-Crystal X-ray Diffraction (SCXRD)
4.5. Fourier-Transform Infrared Spectroscopy (FT-IR)
4.6. Cryo-Scanning Electron Microscopy (Cryo-SEM)
4.7. Differential Scanning Calorimetry and Thermogravimetry Analysis (DSC-TGA)
4.8. 13C CP MAS NMR
4.9. Periodic DFT Calculations
4.9.1. Geometry Optimization
4.9.2. NMR Parameter Calculations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Thomas, M.P.; Potter, B.V. The structural biology of oestrogen metabolism. J. Steroid Biochem. Mol. Biol. 2013, 137, 27–49. [Google Scholar] [CrossRef] [PubMed]
- Grandi, G.; Napolitano, A.; Cagnacci, A. Metabolic impact of combined hormonal contraceptives containing estradiol. Expert Opin. Drug Metab. Toxicol. 2016, 12, 779–787. [Google Scholar] [CrossRef]
- Santoro, N.; Epperson, C.N.; Mathews, S.B. Menopausal Symptoms and Their Management. Endocrinol. Metab. Clin. N. Am. 2015, 44, 497–515. [Google Scholar] [CrossRef] [PubMed]
- Daulbayev, C.; Kaidar, B.; Sultanov, F.; Bakbolat, B.; Smagulova, G.; Mansurov, Z. The recent progress in pitch derived carbon fibers applications. A Review. S. Afr. J. Chem. Eng. 2021, 38, 9–20. [Google Scholar] [CrossRef]
- Guo, A.; Gong, X.; He, J.; Guo, Y.; Ning, L.; Chen, X.; Xu, J.; Guo, Y.; Wang, H. Study on Co-crystals of Estradiol. Her. Med. 2021, 40, 1716–1723. [Google Scholar]
- Wang, J.R.; Wang, X.; Yang, Y.; Chen, X.; Mei, X. Solid-state characterization of 17β-estradiol co-crystals presenting improved dissolution and bioavailability. CrystEngComm 2016, 18, 3498–3505. [Google Scholar] [CrossRef]
- Ning, L.; Gong, X.; Li, P.; Chen, X.; Wang, H.; Xu, J. Measurement and correlation of the solubility of estradiol and estradiol-urea co-crystal in fourteen pure solvents at temperatures from 273.15 K to 318.15 K. J. Mol. Liq. 2020, 304, 112599. [Google Scholar] [CrossRef]
- Kovacs, T.; Nagy, P.; Panyi, G.; Szente, L.; Varga, Z.; Zakany, F. Cyclodextrins: Only Pharmaceutical Excipients or Full-Fledged Drug Candidates? Pharmaceutics 2022, 14, 2559. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Available online: https://www.ema.europa.eu/en (accessed on 21 February 2023).
- U.S. Food & Drug Administration (FDA). Available online: https://www.fda.gov (accessed on 21 February 2023).
- Pharmaceutical and Medical Devices Agencty. Available online: https://www.pmda.go.jp/english/index.html (accessed on 21 February 2023).
- Sun, J.; Hong, H.; Zhu, N.; Han, L.; Suo, Q. Effect of preparation methods on tosufloxacin tosylate/hydroxypropyl-β-cyclodextrin inclusion complex. Braz. J. Pharm. Sci. 2022, 58, e18650. [Google Scholar] [CrossRef]
- Cid-Samamed, A.; Rakmai, J.; Mejuto, J.C.; Simal-Gandara, J.; Astray, G. Cyclodextrins inclusion complex: Preparation methods, analytical techniques and food industry applications. Food Chem. 2022, 384, 132467. [Google Scholar] [CrossRef]
- Deckmann Nicoletti, C.; de Sá Haddad Queiroz, M.; de Souza Lima, C.G.; de Carvalho da Silva, F.; Futuro, D.O.; Ferreira, V.F. An improved method for the preparation of β-lapachone:2-hydroxypropyl-β-cyclodextrin inclusion complexes. J. Drug Deliv. Sci. Technol. 2020, 58, 101777. [Google Scholar] [CrossRef]
- Wdowiak, K.; Rosiak, N.; Tykarska, E.; Żarowski, M.; Płazińska, A.; Płaziński, W.; Cielecka-Piontek, J. Amorphous Inclusion Complexes: Molecular Interactions of Hesperidin and Hesperetin with HP-Β-CD and Their Biological Effects. Int. J. Mol. Sci. 2022, 23, 4000. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, A.H.; Szeleszczuk, Ł. A Review of Applications of Solid-State Nuclear Magnetic Resonance (ssNMR) for the Analysis of Cyclodextrin-Including Systems. Int. J. Mol. Sci. 2023, 24, 3648. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, A.H.; Szeleszczuk, Ł. Current Status of Quantum Chemical Studies of Cyclodextrin Host–Guest Complexes. Molecules 2022, 27, 3874. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, A.H.; Szeleszczuk, Ł.; Gubica, T. Application of Molecular Dynamics Simulations in the Analysis of Cyclodextrin Complexes. Int. J. Mol. Sci. 2021, 22, 9422. [Google Scholar] [CrossRef]
- Mazurek, A.H.; Szeleszczuk, Ł.; Pisklak, D.M. Periodic DFT Calculations—Review of Applications in the Pharmaceutical Sciences. Pharmaceutics 2020, 12, 415. [Google Scholar] [CrossRef]
- Gallez, A.; Palazzo, C.; Blacher, S.; Tskitishvili, E.; Noël, A.; Foidart, J.M.; Evrard, B.; Pequeux, C.; Piel, G. Liposomes and drug-in-cyclodextrin-in-liposomes formulations encapsulating 17β-estradiol: An innovative drug delivery system that prevents the activation of the membrane-initiated steroid signaling (MISS) of estrogen receptor α. Int. J. Pharm. 2020, 573, 118861. [Google Scholar] [CrossRef]
- Schwarz, D.H.; Engelke, A.; Wenz, G. Solubilizing steroidal drugs by β-cyclodextrin derivatives. Int. J. Pharm. 2017, 531, 559–567. [Google Scholar] [CrossRef]
- Cai, W.; Yao, X.; Shao, X.; Pan, Z. Bimodal Complexations of Steroids with Cyclodextrins by a Flexible Docking Algorithm. J. Incl. Phenom. Macrocycl. Chem. 2005, 51, 41–51. [Google Scholar] [CrossRef]
- Silva, M.C.G.D.; Silva, J.F.D.; Santos, T.P.; Silva, N.P.C.D.; Santos, A.R.D.; Andrade, A.L.C.; Souza, E.H.L.D.S.; Sales Cadena, M.R.; Sá, F.B.; Silva Junior, V.A.D.; et al. The complexation of steroid hormones into cyclodextrin alters the toxic effects on the biological parameters of zebrafish (Danio rerio). Chemosphere 2019, 214, 330–340. [Google Scholar] [CrossRef]
- Haimhoffer, Á.; Vas, A.; Árvai, G.; Fenyvesi, É.; Jicsinszky, L.; Budai, I.; Bényei, A.; Regdon, G., Jr.; Rusznyák, Á.; Vasvári, G.; et al. Investigation of the Drug Carrier Properties of Insoluble Cyclodextrin Polymer Microspheres. Biomolecules 2022, 12, 931. [Google Scholar] [CrossRef]
- Lin, Z.H.; Wang, X.X.; Kou, S.B.; Shi, J.H. Exploring the inclusion interaction of estradiol with β-CD and HP-β-CD with the help of molecular dynamics simulation as well as multi-spectroscopic approaches. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 269, 120764. [Google Scholar] [CrossRef] [PubMed]
- Sadlej-Sosnowska, N. Fluorometric determination of association constants of three estrogens with cyclodextrins. J. Fluoresc. 1997, 7, 195–200. [Google Scholar] [CrossRef]
- van Uden, W.; Woerdenbag, H.J.; Pras, N. Cyclodextrins as a useful tool for bioconversions in plant cell biotechnology. Plant Cell Tiss Organ Cult. 1994, 38, 103–113. [Google Scholar] [CrossRef]
- Vicatos, A.I.; Hoossen, Z.; Caira, M.R. Inclusion complexes of the steroid hormones 17β-estradiol and progesterone with β- and γ-cyclodextrin hosts: Syntheses, X-ray structures, thermal analyses and API solubility enhancements. Beilstein J. Org. Chem. 2022, 18, 1749–1762. [Google Scholar] [CrossRef]
- Mentzafos, D.; Mavridis, I.M.; Le Bas, G.; Tsoucaris, G. Structure of the 4-It Tert-Butylbenzyl Alcohol–β-Cyclodextrin Complex. Common Features in the Geometry of β-Cyclodextrin Dimeric Complexes. Acta Crystallogr. Sect. B 1991, 47, 746–757. [Google Scholar] [CrossRef]
- Commodari, F.; Sclavos, G.; Ibrahimi, S.; Khiat, A.; Boulanger, Y. Comparison of 17beta-estradiol structures from X-ray diffraction and solution NMR. Magn. Reson. Chem. 2005, 43, 444–450. [Google Scholar] [CrossRef]
- Rekik, N.; Issaoui, N.; Ghalla, H.; Oujia, B.; Wójcik, M.J. Infrared spectral density of H-bonds within the strong anharmonic coupling theory: Indirect relaxation effect. J. Mol. Struct. 2007, 844–845, 21–31. [Google Scholar] [CrossRef]
- Brela, M.Z.; Klimas, O.; Surmiak, E.; Boczar, M.; Nakajima, T.; Wójcik, M.J. Comparison of the Hydrogen Bond Interaction Dynamics in the Guanine and Cytosine Crystals: Ab Initio Molecular Dynamics and Spectroscopic Study. J. Phys. Chem. A 2019, 123, 10757–10763. [Google Scholar] [CrossRef]
- Rekik, N.; Issaoui, N.; Ghalla, H.; Oujia, B.; Wójcik, M.J. IR spectral density of H-bonds. Both intrinsic anharmonicity of the fast mode and the H-bond bridge. Part I: Anharmonic coupling parameter and temperature effects. J. Mol. Struct. Theochem. 2007, 821, 9–21. [Google Scholar] [CrossRef]
- Wójcik, J.M. Theoretical Modeling of Vibrational Spectra and Proton Tunneling in Hydrogen-Bonded Systems. Adv. Chem. Phys. 2016, 160. [Google Scholar] [CrossRef]
- Pereva, S.; Nikolova, V.; Angelova, S.; Spassov, T.; Dudev, T. Water inside β-cyclodextrin cavity: Amount, stability and mechanism of binding. Beilstein J. Org. Chem. 2019, 15, 1592–1600. [Google Scholar] [CrossRef]
- Bruker. APEX 3, SAINT, SADABS; Bruker AXS Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Sheldrick, G.M. It SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with It SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. It ShelXle: A Qt Graphical User Interface for It SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef]
- Schüttelkopf, A.W.; van Aalten, D.M.F. PRODRG: A Tool for High-Throughput Crystallography of Protein–Ligand Complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef]
- Thorn, A.; Dittrich, B.; Sheldrick, G.M. Enhanced Rigid-Bond Restraints. Acta Crystallogr. Sect. A Found. Crystallogr. 2012, 68, 448–451. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. It OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. It Mercury CSD 2.0—New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- The Pymol Molecular Graphics System, Version 1.8; Schrödinger, Inc.: New York, NY, USA, 2015.
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist.-Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- BIOVIA. Materials Studio. Available online: http://accelrys.com/products/collaborative-science/biovia-materials-studio (accessed on 23 February 2023).
- Koelling, D.D.; Harmon, B.N. Technique for relativistic spin-polarized calculations. J. Phys. C Solid State Phys. 1977, 10, 3107–3114. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Tkatchenko, A.; Scheffler, M. Accurate Molecular van der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef]
- Packwood, D.; Kermode, J.; Mones, L.; Bernstein, N.; Woolley, J.; Gould, N.; Ortner, C.; Csányi, G. A universal preconditioner for simulating condensed phase materials. J. Chem. Phys. 2016, 144, 164109. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations—A reply. Phys. Rev. B 1977, 16, 1748–1749. [Google Scholar]
- Pickard, C.J.; Mauri, F. All-electron magnetic response with pseudopotentials: NMR chemical shifts. Phys. Rev. B 2001, 63, 63–77. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Data | EST/ß-CD |
|---|---|
| CCDC No. | 2250781 |
| Complex formula in the asymmetric unit | (C42H70O35)·0.5(C18H24O2)·10.5H2O |
| Formula weight | 1439.16 |
| Crystal system, space group | Monoclinic, C2 |
| Temperature (K) | 100 |
| a, b, c (Å) | 19.1245 (15), 24.4180 (18), 15.6004 (11) |
| α, β, γ (°) | 109.500 (5) |
| V (Å3) | 6867.2 (9) |
| Z | 4 |
| Radiation type | Cu Ka |
| μ (mm−1) | 1.09 |
| Crystal size (mm3) | 0.4 × 0.27 × 0.13 |
| Data collection | |
| Tmin, Tmax | 0.593, 0.754 |
| No. of measured, independent, and observed [I > 2σ(I)] reflections | 105,516, 11,945, 10,450 |
| Rint | 0.074 |
| (sin θ/λ)max (Å−1) | 0.595 |
| Refinement | |
| R1 [F2 > 2σ(F2)], wR2(F2), GooF | 0.095, 0.269, 1.04 |
| No. of reflections | 11,945 |
| No. of parameters | 950 |
| No. of restraints | 202 |
| Δρmax, Δρmin (e Å−3) | 0.78, −0.44 |
| Exp. | ADAD | DAAD | |
|---|---|---|---|
| a [Å] | 15.515011 | 15.742737 | 15.484059 |
| b [Å] | 15.515011 | 15.314397 | 15.484028 |
| c [Å] | 31.188400 | 31.718429 | 31.699391 |
| α [°] | 101.86701 | 100.93157 | 101.31461 |
| β [°] | 101.86701 | 99.83545 | 101.31466 |
| γ [°] | 103.84663 | 104.42976 | 101.44811 |
| Relative energy [kcal/mol] | 0 | −5.152 |
| Atom Number | δ EST Exp. | δ EST GIPAW | δ EST Exp.—δ EST GIPAW | δ EST + ß-CD Exp. | δ ADAD GIPAW (1) | δ ADAD GIPAW (2) | (EST + ß-CD Exp.)—ADAD GIPAW (1) | (EST + βCD Exp.)—ADAD GIPAW (2) | δ DAAD GIPAW (1) | δ DAAD GIPAW (2) | (EST + ß-CD Exp.)—DAAD GIPAW (1) | (EST + ß-CD Exp.)—DAAD GIPAW (2) | δ EST Exp.—(EST/ß-CD Exp.) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 127.65 | 129.59 | −1.94 | 127.78 | 127.77 | 127.76 | 0.01 | 0.02 | 124.65 | 124.71 | 3.13 | 3.07 | −0.13 |
| 2 | 113.33 | 111.18 | 2.15 | 113.43 | 109.86 | 109.82 | 3.57 | 3.61 | 109.61 | 109.65 | 3.82 | 3.78 | −0.1 |
| 3 | 154.42 | 156.06 | −1.64 | 154.8/152.9 | 157.21 | 157.23 | −2.41 | −2.43 | 155.72 | 155.71 | −2.92 | −2.91 | 1.62 |
| 4 | 115.89 | 118.35 | −2.46 | 115.97 | 113.2 | 113.2 | 2.77 | 2.77 | 113.24 | 113.27 | 2.73 | 2.7 | −0.08 |
| 5 | 137.94 | 139.1 | −1.16 | 137.98 | 140.12 | 139.98 | −2.14 | −2.00 | 139.55 | 139.65 | −1.57 | −1.67 | −0.04 |
| 6 | 30.83 | 30.75 | 0.08 | 30.92 | 28.79 | 28.81 | 2.13 | 2.11 | 28.58 | 28.56 | 2.34 | 2.36 | −0.09 |
| 7 | 30.36 | 29.98 | 0.38 | 30.36 | 25.09 | 25.09 | 5.27 | 5.27 | 26.49 | 26.49 | 3.87 | 3.87 | 0.00 |
| 8 | 40.05 | 38.54 | 1.51 | 40.31 | 38 | 37.94 | 2.31 | 2.37 | 37.13 | 37.12 | 3.18 | 3.19 | −0.26 |
| 9 | 44.89 | 43.52 | 1.37 | 45.5 | 45.24 | 45.2 | 0.26 | 0.30 | 43.73 | 43.74 | 1.77 | 1.76 | −0.61 |
| 10 | 131.77 | 133.53 | −1.76 | 131.84 | 132.39 | 132.31 | −0.55 | −0.47 | 130.46 | 130.52 | 1.38 | 1.32 | −0.07 |
| 11 | 26.9 | 25.78 | 1.12 | 26.95 | 26.27 | 26.29 | 0.68 | 0.66 | 23.63 | 23.61 | 3.32 | 3.34 | −0.05 |
| 12 | 35.59 | 36.07 | −0.48 | 37.76 | 35.08 | 35.15 | 2.68 | 2.61 | 35.84 | 35.84 | 1.92 | 1.92 | −2.17 |
| 13 | 43.09 | 42.62 | 0.47 | 44.3 | 43.5 | 43.38 | 0.8 | 0.92 | 42.08 | 42.07 | 2.22 | 2.23 | −1.21 |
| 14 | 49.65 | 48.09 | 1.56 | 51.39 | 51.23 | 51.21 | 0.16 | 0.18 | 50.06 | 50.04 | 1.33 | 1.35 | −1.74 |
| 15 | 22.45 | 21.06 | 1.39 | 23.98 | 20.61 | 20.61 | 3.37 | 3.37 | 21.45 | 21.43 | 2.53 | 2.55 | −1.53 |
| 16 | 29.12 | 28.09 | 1.03 | 29.18 | 26.62 | 26.61 | 2.56 | 2.57 | 29.53 | 29.52 | −0.35 | −0.34 | −0.06 |
| 17 | 82.04 | 82.63 | −0.59 | 82.06 | 84.46 | 84.38 | −2.4 | −2.32 | 85.32 | 85.28 | −3.26 | −3.22 | −0.02 |
| 18 | 10.67 | 7.54 | 3.13 | 11.71 | 8.46 | 8.5 | 3.25 | 3.21 | 8.54 | 8.52 | 3.17 | 3.19 | −1.04 |
| STAND | DSC | (1) Onset temp. 29.26 °C | (3) Onset temp. 152.0 °C |
| Peak temp. 61.83 °C | Peak temp. 177.96 °C | ||
| Enthalpy 83.82 J/g | Enthalpy 10.74 J/g | ||
| (2) Onset temp. 101.1 °C | (4) Onset temp. 202.0 °C | ||
| Peak temp. 111.27 °C | Peak temp. 212.69 °C | ||
| Enthalpy 18.53 J/g | Enthalpy 6.434 J/g | ||
| TGA | Temp. range of dehydration: 27–200 °C | ||
| Associated mass loss: 6.911% | |||
| STANDSHORT | DSC | Onset temp. 26.38 °C | |
| Peak temp. 45.33 °C | |||
| Enthalpy 185.1 J/g | |||
| TGA | Temp. range of dehydration: 25–200 °C | ||
| Associated mass loss: 7.034% | |||
| MECH | DSC | Onset temp. 30.55 °C | |
| Peak temp. 69.66 °C | |||
| Enthalpy 209.8 J/g | |||
| TGA | Temp. range of dehydration: 20–200 °C | ||
| Associated mass loss: 9.408% | |||
| LYS | DSC | Onset temp. 41.0 °C | |
| Peak temp. 55.01 °C | |||
| Enthalpy 47.53 J/g | |||
| TGA | Temp. range of dehydration: 25–200 °C | ||
| Associated mass loss: 4.065% | |||
| EST | DSC | (1) Onset temp. 81.88 °C | |
| Peak temp. 104.81 °C | |||
| Enthalpy 19.70 J/g | |||
| (2) Onset temp. 175.91 °C | |||
| Peak temp. 178.20 °C | |||
| Enthalpy 91.06 J/g | |||
| TGA | Temp. range of dehydration: 20–200 °C | ||
| Associated mass loss: 6.20% | |||
| ß-CD | DSC | Onset temp. 56.31 °C | |
| Peak temp. 91.48 °C | |||
| Enthalpy 380.8 J/g | |||
| TGA | Temp. range of dehydration: 20–100 °C | ||
| Associated mass loss: 12.95% | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazurek, A.H.; Szeleszczuk, Ł.; Bethanis, K.; Christoforides, E.; Dudek, M.K.; Zielińska-Pisklak, M.; Pisklak, D.M. 17-β-Estradiol—β-Cyclodextrin Complex as Solid: Synthesis, Structural and Physicochemical Characterization. Molecules 2023, 28, 3747. https://doi.org/10.3390/molecules28093747
Mazurek AH, Szeleszczuk Ł, Bethanis K, Christoforides E, Dudek MK, Zielińska-Pisklak M, Pisklak DM. 17-β-Estradiol—β-Cyclodextrin Complex as Solid: Synthesis, Structural and Physicochemical Characterization. Molecules. 2023; 28(9):3747. https://doi.org/10.3390/molecules28093747
Chicago/Turabian StyleMazurek, Anna Helena, Łukasz Szeleszczuk, Kostas Bethanis, Elias Christoforides, Marta Katarzyna Dudek, Monika Zielińska-Pisklak, and Dariusz Maciej Pisklak. 2023. "17-β-Estradiol—β-Cyclodextrin Complex as Solid: Synthesis, Structural and Physicochemical Characterization" Molecules 28, no. 9: 3747. https://doi.org/10.3390/molecules28093747
APA StyleMazurek, A. H., Szeleszczuk, Ł., Bethanis, K., Christoforides, E., Dudek, M. K., Zielińska-Pisklak, M., & Pisklak, D. M. (2023). 17-β-Estradiol—β-Cyclodextrin Complex as Solid: Synthesis, Structural and Physicochemical Characterization. Molecules, 28(9), 3747. https://doi.org/10.3390/molecules28093747