
Stabilizing Highly Active Ru Sites by Electron Reservoir in Acidic Oxygen Evolution



Abstract
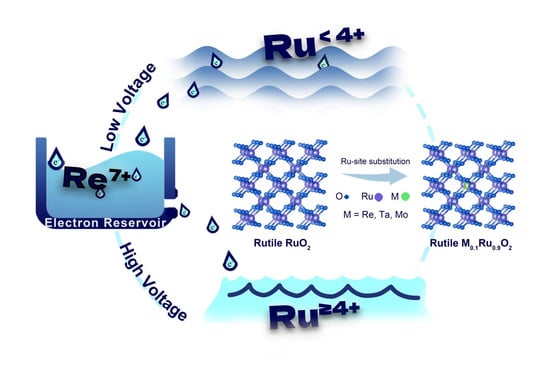
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Characterization of the M0.1Ru0.9O2 Catalysts
2.2. Acidic OER Performance
2.3. Investigation of Relationship between OER Stability and Electron Structure
3. Materials and Methods
3.1. Chemicals
3.2. Preparation of M0.1Ru0.9O2 Catalysts
3.3. Physical Characterization
3.4. Electrochemical Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gasteiger, H.A.; Marković, N.M. Just a Dream—Or Future Reality? Science 2009, 324, 48–49. [Google Scholar] [CrossRef]
- Carmo, M.; Fritz, D.L.; Mergel, J.; Stolten, D. A Comprehensive Review on PEM Water Electrolysis. Int. J. Hydrogen Energy 2013, 38, 4901–4934. [Google Scholar] [CrossRef]
- Montoya, J.H.; Seitz, L.C.; Chakthranont, P.; Vojvodic, A.; Jaramillo, T.F.; Nørskov, J.K. Materials for Solar Fuels and Chemicals. Nat. Mater. 2016, 16, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Liang, X.; Wang, L.; Sun, K.; Wang, Y.; Xie, Z.; Wu, Q.; Bai, X.; Hamdy, M.S.; Chen, H.; et al. Status and Perspectives of Key Materials for PEM Electrolyzer. Nano Res. Energy 2022, 1, 9120032. [Google Scholar] [CrossRef]
- Wang, Y.; Pang, Y.; Xu, H.; Martinez, A.; Chen, K.S. PEM Fuel Cell and Electrolysis Cell Technologies and Hydrogen Infrastructure Development—A Review. Energy Environ. Sci. 2022, 15, 2288–2328. [Google Scholar] [CrossRef]
- Chu, S.; Majumdar, A. Opportunities and Challenges for a Sustainable Energy Future. Nature 2012, 488, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.A. Sustainable Hydrogen Production. Science 2004, 305, 972–974. [Google Scholar] [CrossRef] [PubMed]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Nørskov, J.K.; Jaramillo, T.F. Combining Theory and Experiment in Electrocatalysis: Insights into Materials Design. Science 2017, 355, 146. [Google Scholar] [CrossRef] [PubMed]
- Millet, P.; Ngameni, R.; Grigoriev, S.A.; Mbemba, N.; Brisset, F.; Ranjbari, A.; Etiévant, C. PEM Water Electrolyzers: From Electrocatalysis To Stack Development. Int. J. Hydrogen Energy 2010, 35, 5043–5052. [Google Scholar] [CrossRef]
- Wang, Q.; Cheng, Y.; Tao, H.B.; Liu, Y.; Ma, X.; Li, D.S.; Yang, H.B.; Liu, B. Long-Term Stability Challenges and Opportunities in Acidic Oxygen Evolution Electrocatalysis. Angew. Chem. Int. Ed. 2023, 62, 202216645. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, Q.; Wang, A.; Duan, J.; Zhou, W.; Sang, Y.; Liu, H. Iron Oxide Embedded Titania Nanowires—An Active and Stable Electrocatalyst for Oxygen Evolution in Acidic Media. Nano Energy 2018, 45, 118–126. [Google Scholar] [CrossRef]
- Gao, J.; Tao, H.; Liu, B. Progress of Nonprecious-Metal-Based Electrocatalysts for Oxygen Evolution in Acidic Media. Adv. Mater. 2021, 33, 2003786. [Google Scholar] [CrossRef] [PubMed]
- Schuler, T.; Kimura, T.; Schmidt, T.J.; Büchi, F.N. Towards a Generic Understanding of Oxygen Evolution Reaction Kinetics in Polymer Electrolyte Water Electrolysis. Energy Environ. Sci. 2020, 13, 2153–2166. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Z.; Fang, J.; Li, M.; Sendeku, M.G.; Wang, X.; Wu, H.; Li, Y.; Ge, J.; Zhuang, Z.; et al. Eliminating Over-oxidation of Ruthenium Oxides by Niobium for Highly Stable Electrocatalytic Oxygen Evolution in Acidic Media. Joule 2023, 7, 558–573. [Google Scholar] [CrossRef]
- Li, N.; Cai, L.; Wang, C.; Lin, Y.; Huang, J.; Sheng, H.; Pan, H.; Zhang, W.; Ji, Q.; Duan, H.; et al. Identification of the Active-Layer Structures for Acidic Oxygen Evolution from 9R-BaIrO3 Electrocatalyst with Enhanced Iridium Mass Activity. J. Am. Chem. Soc. 2021, 143, 18001–18009. [Google Scholar] [CrossRef]
- Yin, J.; Jin, J.; Lu, M.; Huang, B.; Zhang, H.; Peng, Y.; Xi, P.; Yan, C.-H. Iridium Single Atoms Coupling with Oxygen Vacancies Boosts Oxygen Evolution Reaction in Acid Media. J. Am. Chem. Soc. 2020, 142, 18378–18386. [Google Scholar] [CrossRef]
- Su, J.W.; Ge, R.X.; Jiang, K.M.; Dong, Y.; Hao, F.; Tian, Z.Q.; Chen, G.X.; Chen, L. Assembling Ultrasmall Copper-Doped Ruthenium Oxide Nanocrystals into Hollow Porous Polyhedra: Highly Robust Electrocatalysts for Oxygen Evolution in Acidic Media. Adv. Mater. 2018, 30, 1801351. [Google Scholar] [CrossRef]
- Yu, J.; He, Q.; Yang, G.; Zhou, W.; Shao, Z.; Ni, M. Recent Advances and Prospective in Ruthenium-Based Materials for Electrochemical Water Splitting. ACS Catal. 2019, 9, 9973–10011. [Google Scholar] [CrossRef]
- Spöri, C.; Kwan, J.T.H.; Bonakdarpour, A.; Wilkinson, D.P.; Strasser, P. The Stability Challenges of Oxygen Evolving Catalysts: Towards a Common Fundamental Understanding and Mitigation of Catalyst Degradation. Angew. Chem. Int. Ed. 2017, 56, 5994–6021. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.R.; Kolb, M.J.; Giordano, L.; Pedersen, A.F.; Katayama, Y.; Hwang, J.; Mehta, A.; You, H.; Lunger, J.R.; Zhou, H.; et al. Operando Identification of Site-dependent Water Oxidation Activity on Ruthenium Dioxide Single-crystal Surfaces. Nat. Catal. 2020, 3, 516–525. [Google Scholar] [CrossRef]
- Roy, C.; Rao, R.R.; Stoerzinger, K.A.; Hwang, J.; Rossmeisl, J.; Chorkendorff, I.; Shao-Horn, Y.; Stephens, I.E.L. Trends in Activity and Dissolution on RuO2 under Oxygen Evolution Conditions: Particles versus Well-defined Extended Surfaces. ACS Energy Lett. 2018, 3, 2045–2051. [Google Scholar] [CrossRef]
- Deka, N.; Jones, T.E.; Falling, L.J.; Sandoval-Diaz, L.-E.; Lunkenbein, T.; Velasco-Velez, J.-J.; Chan, T.-S.; Chuang, C.-H.; Knop-Gericke, A.; Mom, R.V. On the Operando Structure of Ruthenium Oxides during the Oxygen Evolution Reaction in Acidic Media. ACS Catal. 2023, 13, 7488–7498. [Google Scholar] [CrossRef]
- Shi, Z.; Li, J.; Wang, Y.; Liu, S.; Zhu, J.; Yang, J.; Wang, X.; Ni, J.; Jiang, Z.; Zhang, L.; et al. Customized Reaction Route for Ruthenium Oxide towards Stabilized Water Oxidation in High-Performance PEM Electrolyzers. Nat. Commun. 2023, 14, 843. [Google Scholar] [CrossRef]
- Wang, H.; Zhai, T.; Wu, Y.; Zhou, T.; Zhou, B.; Shang, C.; Guo, Z. High-Valence Oxides for High Performance Oxygen Evolution Electrocatalysis. Adv. Sci. 2023, 10, 2301706. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Y.; Yang, B.; Li, Z.; Qin, X.; Zhang, Q.; Lei, L.; Qiu, M.; Wu, G.; Hou, Y. Highly Active Ruthenium Sites Stabilized by Modulating Electron-feeding for Sustainable Acidic Oxygen-evolution Electrocatalysis. Energy Environ. Sci. 2022, 15, 2356–2365. [Google Scholar] [CrossRef]
- Hou, L.; Li, Z.; Jang, H.; Wang, Y.; Cui, X.; Gu, X.; Kim, M.G.; Feng, L.; Liu, S.; Liu, X. Electronic and Lattice Engineering of Ruthenium Oxide towards Highly Active and Stable Water Splitting. Adv. Energy Mater. 2023, 13, 2300177. [Google Scholar] [CrossRef]
- Wen, Y.; Liu, C.; Huang, R.; Zhang, H.; Li, X.; García de Arquer, F.P.; Liu, Z.; Li, Y.; Zhang, B. Introducing Brønsted Acid Sites to Accelerate the Bridging-oxygen-assisted Deprotonation in Acidic Water Oxidation. Nat. Commun. 2022, 13, 4871. [Google Scholar] [CrossRef]
- He, J.; Li, W.; Xu, P.; Sun, J. Tuning Electron Correlations of RuO2 by Co-doping of Mo and Ce for Boosting Electrocatalytic Water Oxidation in Acidic Media. Appl. Catal. B-Environ. 2021, 298, 120528. [Google Scholar] [CrossRef]
- Wang, X.; Jang, H.; Liu, S.; Li, Z.; Zhao, X.; Chen, Y.; Kim, M.G.; Qin, Q.; Liu, X. Enhancing the Catalytic Kinetics and Stability of Ru Sites for Acidic Water Oxidation by Forming Brønsted Acid Sites in Tungsten Oxide Matrix. Adv. Energy Mater. 2023, 13, 2301673. [Google Scholar] [CrossRef]
- Lee, G.R.; Kim, J.; Hong, D.; Kim, Y.J.; Jang, H.; Han, H.J.; Hwang, C.-K.; Kim, D.; Kim, J.Y.; Jung, Y.S. Efficient and Sustainable Water Electrolysis Achieved by Excess Electron Reservoir Enabling Charge Replenishment to Catalysts. Nat. Commun. 2023, 14, 5402. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Hung, S.F.; Lin, Z.Y.; Zhang, Y.; Zhang, C.; Hao, Y.; Liu, S.; Kuo, C.H.; Chen, H.Y.; Peng, J.; et al. Valence Oscillation of Ru Active Sites for Efficient and Robust Acidic Water Oxidation. Adv. Mater. 2023, 35, 2305939. [Google Scholar] [CrossRef] [PubMed]
- Danilovic, N.; Subbaraman, R.; Chang, K.-C.; Chang, S.H.; Kang, Y.J.; Snyder, J.; Paulikas, A.P.; Strmcnik, D.; Kim, Y.-T.; Myers, D.; et al. Activity–Stability Trends for the Oxygen Evolution Reaction on Monometallic Oxides in Acidic Environments. J. Phys. Chem. Lett. 2014, 5, 2474–2478. [Google Scholar] [CrossRef] [PubMed]
- Dickens, C.F.; Nørskov, J.K. A Theoretical Investigation into the Role of Surface Defects for Oxygen Evolution on RuO2. J. Phys. Chem. C 2017, 121, 18516–18524. [Google Scholar] [CrossRef]
- Li, K.; Xue, D. Estimation of Electronegativity Values of Elements in Different Valence States. J. Phys. Chem. A 2006, 110, 11332–11337. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Yu, T.; Deng, S.; Zhou, X.-Y.; Lin, D.; Zhang, Q.; Jin, Z.; Zhang, D.; He, Y.-B.; Qiu, H.-J.; et al. RuO2 Electronic Structure and Lattice Strain Dual Engineering for Enhanced Acidic Oxygen Evolution Reaction Performance. Nat. Commun. 2022, 13, 3784. [Google Scholar] [CrossRef]
- Wang, Y.; Lei, X.; Zhang, B.; Bai, B.; Das, P.; Azam, T.; Xiao, J.; Wu, Z.-S. Breaking the Ru-O-Ru Symmetry of a RuO2 Catalyst for Sustainable Acidic Water Oxidation. Angew. Chem. Int. Ed. 2024, 136, 202316903. [Google Scholar] [CrossRef]
- Xue, Y.; Zhao, J.; Huang, L.; Lu, Y.-R.; Malek, A.; Gao, G.; Zhuang, Z.; Wang, D.; Yavuz, C.T.; Lu, X. Stabilizing Ruthenium Dioxide with Cation-anchored Sulfate for Durable Oxygen Evolution in Proton-exchange Membrane Water Electrolyzers. Nat. Commun. 2023, 14, 8093. [Google Scholar] [CrossRef]
- Hao, S.; Liu, M.; Pan, J.; Liu, X.; Tan, X.; Xu, N.; He, Y.; Lei, L.; Zhang, X. Dopants Fixation of Ruthenium for Boosting Acidic Oxygen Evolution Stability and Activity. Nat. Commun. 2020, 11, 5368. [Google Scholar] [CrossRef]
- Liu, J.; Banis, M.N.; Li, X.; Lushington, A.; Cai, M.; Li, R.; Sham, T.-K.; Sun, X. Atomic Layer Deposition of Lithium Tantalate Solid-State Electrolytes. J. Phys. Chem. C 2013, 117, 20260–20267. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, Y.; Peng, F.; Meng, F.; Zha, J.; Ma, L.; Du, Y.; Peng, N.; Ma, L.; Zhang, Q.; et al. Intercalation-Activated Layered MoO3 Nanobelts as Biodegradable Nanozymes for Tumor-Specific Photo-Enhanced Catalytic Therapy. Angew. Chem. Int. Ed. 2022, 61, 202115939. [Google Scholar] [CrossRef]
- Liu, X.; Xi, S.; Kim, H.; Kumar, A.; Lee, J.; Wang, J.; Tran, N.Q.; Yang, T.; Shao, X.; Liang, M.; et al. Restructuring Highly Electron-Deficient Metal-Metal Oxides for Boosting Stability in Acidic Oxygen Evolution Reaction. Nat. Commun. 2021, 12, 5676. [Google Scholar] [CrossRef]
- Oh, Y.; Theerthagiri, J.; Min, A.; Moon, C.J.; Yu, Y.; Choi, M.Y. Pulsed Laser Interference Patterning of Transition-Metal Carbides for Stable Alkaline Water Electrolysis Kinetics. Carbon Energy 2024, e448. [Google Scholar] [CrossRef]
- Oh, Y.; Theerthagiri, J.; Aruna Kumari, M.L.; Min, A.; Moon, C.J.; Choi, M.Y. Electrokinetic-mechanism of Water and Furfural Oxidation on Pulsed Laser-interlaced Cu2O and CoO on Nickel Foam. J. Energy Chem. 2024, 91, 145–154. [Google Scholar] [CrossRef]
- Jin, H.; Liu, X.; An, P.; Tang, C.; Yu, H.; Zhang, Q.; Peng, H.-J.; Gu, L.; Zheng, Y.; Song, T.; et al. Dynamic Rhenium Dopant Boosts Ruthenium Oxide for Durable Oxygen Evolution. Nat. Commun. 2023, 14, 354. [Google Scholar] [CrossRef]
- Zhao, Z.L.; Wang, Q.; Huang, X.; Feng, Q.; Gu, S.; Zhang, Z.; Xu, H.; Zeng, L.; Gu, M.; Li, H. Boosting the Oxygen Evolution Reaction Using Defect-rich Ultra-thin Ruthenium Oxide Nanosheets in Acidic Media. Energy Environ. Sci. 2020, 13, 5143–5151. [Google Scholar] [CrossRef]
- Wu, Y.; Yao, R.; Zhao, Q.; Li, J.; Liu, G. La-RuO2 Nanocrystals with Efficient Electrocatalytic Activity for Overall Water Splitting in Acidic Media: Synergistic Effect of La Doping and Oxygen Vacancy. Chem. Eng. J. 2022, 439, 135699. [Google Scholar] [CrossRef]
- Liu, C.X.; Jiang, Y.B.; Wang, T.; Li, Q.S.; Liu, Y.Z. Nano Si-doped Ruthenium Oxide Particles from Caged Precursors for High-performance Acidic Oxygen Evolution. Adv. Sci. 2023, 10, 2207429. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C.; Jiang, H.; Chen, Z.Y.; Chen, Q.; Ma, M.Y.; Zhen, L.; Song, B.; Xu, C.Y. Bifunctional WC-supported RuO2 Nanoparticles for Robust Water Splitting in Acidic Media. Angew. Chem. Int. Ed. 2022, 61, 202202519. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-Y.; Chen, F.-Y.; Li, B.; Yu, S.-W.; Finfrock, Y.Z.; Meira, D.M.; Yan, Q.-Q.; Zhu, P.; Chen, M.-X.; Song, T.-W.; et al. Non-iridium-based Electrocatalyst for Durable Acidic Oxygen Evolution Reaction in Proton Exchange Membrane Water Electrolysis. Nat. Mater. 2022, 22, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Lin, C.L.; Yu, G.Q.; Du, P.; Xie, X.Y.; He, X.; Zheng, Z.C.; Sun, N.; Tang, H.L.; Li, X.B.; et al. Ru/Se-RuO2 Composites via Controlled Selenization Strategy for Enhanced Acidic Oxygen Evolution. Adv. Funct. Mater. 2023, 33, 2211102. [Google Scholar] [CrossRef]
- Feng, T.L.; Yu, G.T.; Tao, S.Y.; Zhu, S.J.; Ku, R.Q.; Zhang, R.; Zeng, Q.S.; Yang, M.X.; Chen, Y.X.; Chen, W.H.; et al. A Highly Efficient Overall Water Splitting Ruthenium-cobalt Alloy Electrocatalyst across a Wide pH Range Electronic Coupling with Carbon Dots. J. Mater. Chem. A 2020, 8, 9638–9645. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, R.; Ding, Y.; Zhang, B.; Li, H.; Bai, B.; Li, M.; Cui, Y.; Xiao, J.; Wu, Z.-S. Unraveling Oxygen Vacancy Site Mechanism of Rh-doped RuO2 Catalyst for Long-lasting Acidic Water Oxidation. Nat. Commun. 2023, 14, 1412. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wan, X.; Qin, X.; Chen, C.; Qian, X.; Guo, Y.; Xu, Q.; Cai, W.-B.; Yang, H.; Jiang, K. Electronic Structure Modulation of RuO2 by TiO2 Enriched with Oxygen Vacancies to Boost Acidic O2 Evolution. ACS Catal. 2022, 12, 9437–9445. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Qiu, Z.; Zhang, J.; Song, H.; Cui, Z.; Du, L. Stabilizing Highly Active Ru Sites by Electron Reservoir in Acidic Oxygen Evolution. Molecules 2024, 29, 785. https://doi.org/10.3390/molecules29040785
Wu J, Qiu Z, Zhang J, Song H, Cui Z, Du L. Stabilizing Highly Active Ru Sites by Electron Reservoir in Acidic Oxygen Evolution. Molecules. 2024; 29(4):785. https://doi.org/10.3390/molecules29040785
Chicago/Turabian StyleWu, Jiayan, Zhongjie Qiu, Jiaxi Zhang, Huiyu Song, Zhiming Cui, and Li Du. 2024. "Stabilizing Highly Active Ru Sites by Electron Reservoir in Acidic Oxygen Evolution" Molecules 29, no. 4: 785. https://doi.org/10.3390/molecules29040785
APA StyleWu, J., Qiu, Z., Zhang, J., Song, H., Cui, Z., & Du, L. (2024). Stabilizing Highly Active Ru Sites by Electron Reservoir in Acidic Oxygen Evolution. Molecules, 29(4), 785. https://doi.org/10.3390/molecules29040785