1. Introduction
In lymphocytes, Ca
2+ signals regulate various cellular functions, including differentiation, gene transcription and effector functions, such as vesicle exocytosis of lytic granules consisting of perforin and granzymes in cytotoxic T cells (CTLs) [
1,
2]. In T lymphocytes, T cell receptor (TCR) stimulation leads to activation of PLC-γ (phospholipase C gamma), which breaks down phosphatidylinositol-3,4-biphosphate (PIP
2) into two hydrolytic products, inositol-1,4,5-triphosphate (IP
3) and diacylglycerol (DAG). The endoplasmic reticulum (ER) Ca
2+ store depletion caused by the binding of IP
3 to IP
3 receptors on the membranes of ER triggers the opening of store-operated Ca
2+ release-activated Ca
2+ (CRAC) channels via stromal interaction molecule (STIM) 1/2-ORA1 interaction and elevates the intracellular Ca
2+ concentration [
2,
3]. Along with IP
3, TCR stimulation leads to generation of other second messengers, cyclic adenosine diphosphate ribose (cADPR) and nicotinic acid adenine dinucleotide phosphate (NAADP), which contribute to the rise in the intracellular Ca
2+ concentration [
3].
NAADP microinjection into intact Jurkat T cells was reported to stimulate Ca
2+ signaling in a dose-dependent manner. The role of NAADP in Ca
2+ release in Jurkat T lymphocytes was further elaborated by its self-inactivation property, where microinjection of a high concentration of NAADP (10 µM) along with cADPR/IP
3 completely inhibited the cADPR/IP
3-mediated Ca
2+ flux. NAADP (10 µM) microinjection prior to stimulation also completely blocked the anti-human CD3 monoclonal antibody OKT3-induced TCR/CD3-mediated Ca
2+ signaling [
4]. Gasser et al. measured the cellular NAADP following stimulation of Jurkat T cells with OKT3 using an enzyme assay and showed that the cellular NAADP rise followed biphasic kinetics and the NAADP rise was inhibited by tyrosine kinase inhibition [
5]. NAADP-mediated Ca
2+ signaling has been shown to be regulated by different ion channels/receptors, such as the ryanodine receptor (RyR), two-pore channels (TPC), transient receptor potential (TRP) channels, etc., and that varies from cell to cell preparation [
6]. Using photoaffinity labeling, the existence of different NAADP-binding proteins in different cell preparations, including sea urchin egg, T-lymphocytes and mammalian cells (SKBR3, HEK293, mouse pancreas), has been established [
7,
8,
9]. Two NAADP-binding proteins that appear to control Ca
2+ release via two-pore channels or RyR have been identified [
10,
11,
12,
13]. There are controversies regarding NAADP-mediated release of intracellular Ca
2+ either from endo/lysosomes/acidic stores or ER stores or both [
6,
14]. NAADP causes Ca
2+ release from reserve granules (lysosome-associated organelles) in sea urchin eggs [
15] and from endoplasmic reticulum via ryanodine receptors in Jurkat T lymphocytes [
16,
17,
18]. The reduction of NAADP-induced Ca
2+ spikes in the presence of either 8-NH
2-cADPR (cADPR antagonist) or heparin (IP
3 antagonist) in pancreatic acinar cells delineated the concept of “Ca
2+-induced Ca
2+ release”, where NAADP-mediated locally released Ca
2+ triggered global Ca
2+ signal by acting via the IP
3 and ryanodine receptors [
19]. Specific proteins, such as the progesterone receptor membrane component 1 (PGRMC1), can facilitate endosome/ER interaction [
20].
Ned-19, which was discovered via a virtual screening strategy, antagonized NAADP-mediated Ca
2+ flux in intact sea urchin eggs and NAADP-mediated Ca
2+ spiking in mouse pancreatic beta cells [
21]. It is thought to be an antagonist of the NAADP signaling pathway, although its direct binding partners have not been demonstrated. Using Ned-19 antagonism, Ali et al. have shown that lysosome-like acidic stores and ER stores serve as the principal and contributory sources, respectively, of NAADP-mediated Ca
2+ release in naïve and effector mouse T cells [
22]. In this study, since the profiles of Ca
2+ flux in memory cells are quite different from those in naïve and effector T cells, we asked whether NAADP-mediated Ca
2+ flux exists in memory CD4
+ T cells. If it exists, what are the sources of NAADP-mediated Ca
2+ release. Furthermore, we explored what downstream effector functions of memory CD4
+ T cells would be controlled by NAADP-mediated Ca
2+ signaling.
3. Discussion
NAADP-mediated Ca
2+ signaling and its effect on downstream effector functions in naïve, effector and natural regulatory T cells has already been reported by Ali et al. [
22]. However, this is the first study of NAADP-mediated Ca
2+ signaling and its impact on the downstream effector functions of primary memory CD4
+ T cells. We have utilized the NAADP pathway antagonist Ned-19 to investigate various downstream effector functions of Ca
2+ signaling mediated by NAADP in memory CD4
+ T cells.
Ned-19 was discovered via virtual screening as an efficient chemical probe for NAADP. Evidence supports the idea that cell-permeant Ned-19 specifically targets NAADP-mediated Ca
2+ signaling. In sea urchin egg homogenates, 100 μM Ned-19 was shown to have inhibited NAADP-mediated Ca
2+ release, but it did not have any effect on Ca
2+ release caused by either of two messengers, inositol 1,4,5-triphosphate and cyclic ADP-ribose [
21]. The competition of Ned-19 with [
32P] NAADP toward binding to the sea urchin egg binding protein and slow dissociation of bound Ned-19 from the sea urchin NAADP receptor established Ned-19 to be a noncompetitive antagonist of NAADP. The incubation of intact sea urchin eggs with 100 μM Ned-19 in artificial sea water, followed by NAADP injection, did not cause any Ca
2+ flux. When mouse pancreatic beta cells were incubated with 100 μM Ned-19, followed by application of NAADP (100 nM) using a patch pipette, it inhibited Ca
2+-dependent current traces, mediated by NAADP [
21]. The preincubation of rat uterine smooth muscle cells with 5 μM Ned-19 for 15 min, followed by 400 nM NAADP-AM addition, entirely inhibited the NAADP-mediated intracellular Ca
2+ surge [
30]. Ali et al. have shown that 1 h preincubation of naïve CD4
+ T cells with 100 μM Ned-19 inhibited the NAADP-mediated Ca
2+ flux [
22]. Much of our approach was pharmacological, using Ned-19 as a probe for NAADP-mediated Ca
2+ release.
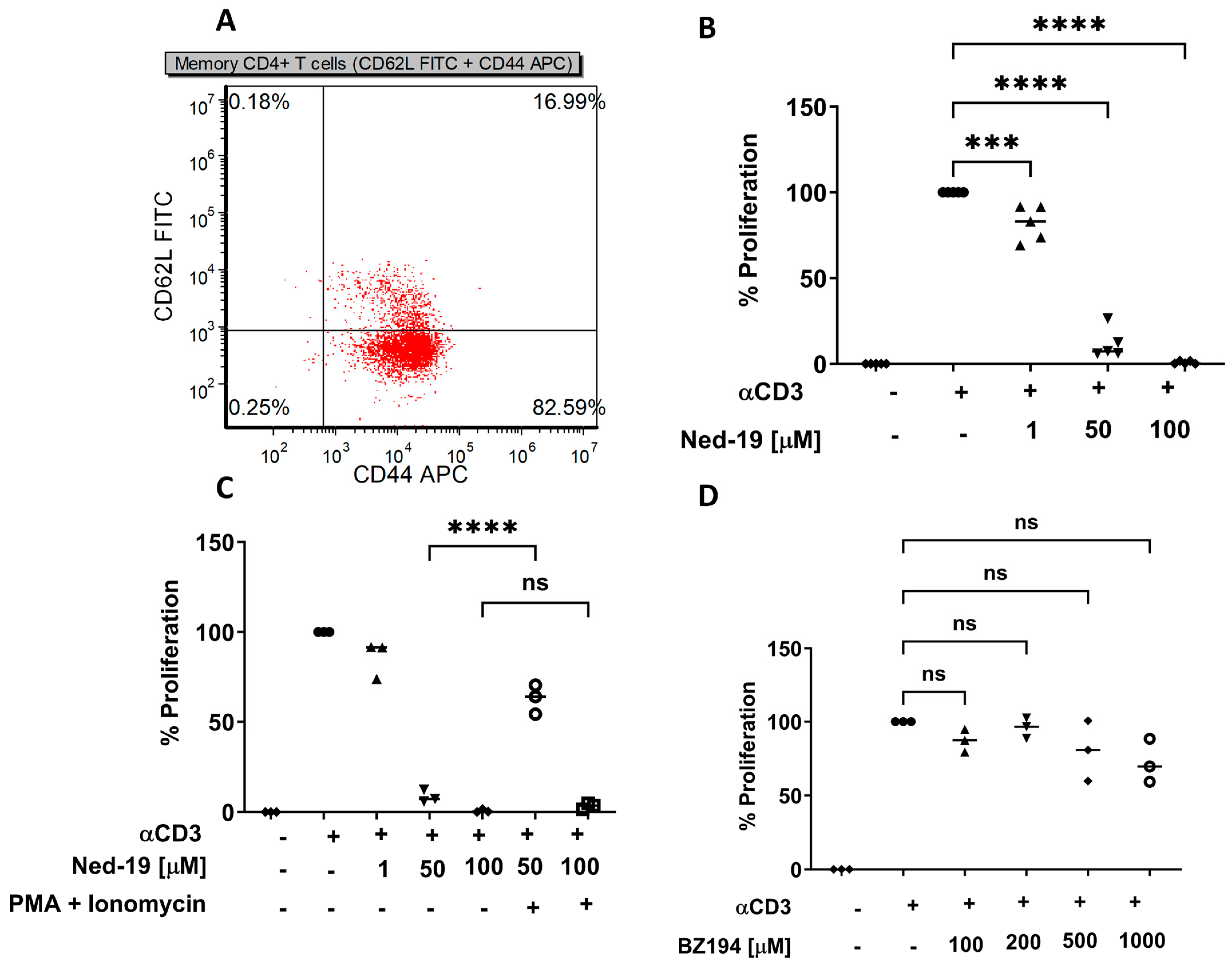
By using Ned-19, we have demonstrated that for memory CD4+ T cells (1) Ned-19 inhibited proliferation in a dose-dependent manner; (2) Ned-19 suppressed cytokine production (partial for IL-2, complete for IFN-γ and IL-10); (3) Ned-19 reduced IL-2 production and thereby intracellular accumulation; (4) Ned-19, between 50 μM and 200 μM, stimulated the TCR/CD3-mediated Ca2+ flux and 250 μM–300 μM inhibited the TCR/CD3-mediated Ca2+ flux in a concentration-dependent manner; (5) Ned-19 partially reduced NFAT and NF-κB nuclear translocation; and (6) Ned-19 suppressed NAADP-AM-mediated Ca2+ flux.
Naïve CD4
+ T cells proliferate in response to stimulation by anti-CD3 plus anti-CD28, whereas effector and memory CD4
+ T cells proliferate in response to anti-CD3 alone. Farber et al. have shown that mouse memory CD4
+ T cells (isolated based on the CD45RB levels, where CD45RB
lo cells are memory and CD45RB
hi cells are naïve) upon TCR/CD3 crosslinking generated a lesser number of tyrosine-phosphorylated protein species as compared to naïve cells and TCR signaling left ZAP-70 tyrosine kinase in an unphosphorylated and phosphorylated state in memory and naïve CD4
+ T cells, respectively. So, there are differences in the downstream signaling events between naïve and memory CD4
+ T cells [
31]. Presumably, this is why memory CD4
+ T cells required 10 μg/mL anti-CD3 stimulation to obtain optimum thymidine incorporation, whereas naïve CD4
+ T cells required 2 μg/mL anti-CD3 plus 2.5 μg/mL anti-CD28 stimulation in the same assay. Ned-19-mediated inhibition of memory CD4
+ T cell proliferation raised a question about the possible cytotoxic effect of Ned-19. PMA and ionomycin were able to rescue the proliferation of cells treated with to memory CD4
+ T cells incubated for one hour with 50 μM Ned-19, but not those treated with 100 μM Ned-19. The synthetic NAADP antagonist, BZ194, was reported to impair proliferation of antigen-experienced CD4
+ effector T cells. The preincubation times with BZ194 for the optimum inhibitory effect were 8 h and 16 h in Ca
2+ release and NFAT1 translocation experiments, respectively [
23]. With 5 h of preincubation, we did not observe any effect of BZ194 on memory CD4
+ T cell proliferation, and the same result was reported by Ali et al. with naïve T cells [
22]. Our finding is very much consistent with the impaired sensitivity of naïve and long-lived memory T cells to BZ194, as reported by Cordiglieri et al. [
32].
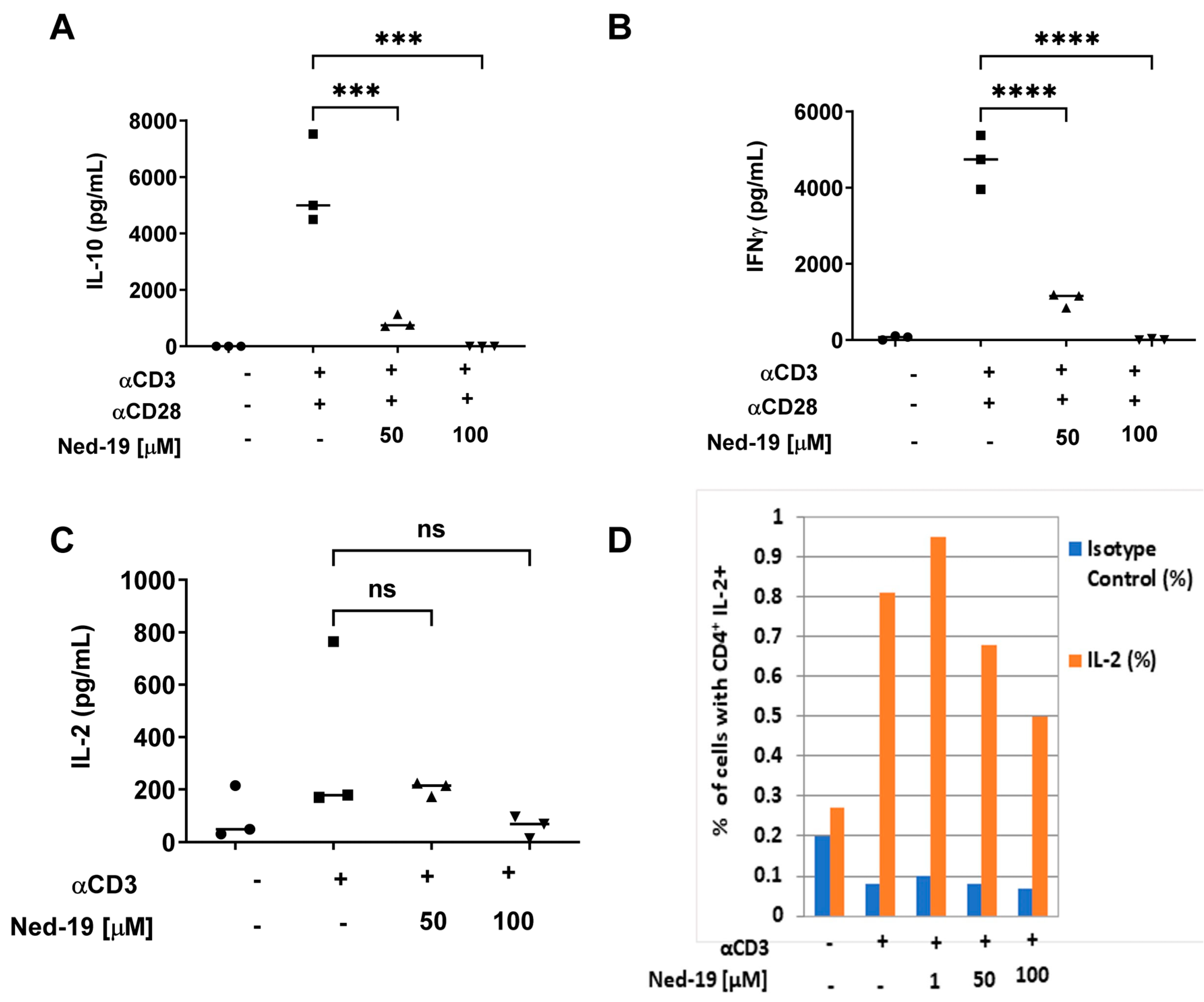
It has already been established that TCR activation leads to Ca
2+ influx, which activates the calcineurin-NFAT pathway, wherein the activated phosphatase calcineurin dephosphorylates NFAT, and as a consequence, NFAT proteins migrate into the nucleus [
2]. It has been shown that the IL-2 promoter has NFAT-binding sites and therefore IL-2 expression is regulated by NFAT [
33]. Sweetser et al. have demonstrated two strong NFAT-binding sites in the IFN-γ promoter and so NFAT promotes IFN-γ expression [
34]. We showed that Ned-19 partially suppressed IL-2 secretion and significantly inhibited IFN-γ and IL-10 secretion by stimulated memory CD4
+ T cells. The slight reduction (statistically not significant) in IL-2 secretion by Ned-19-treated stimulated memory CD4
+ T cells as compared to the control was substantiated by intracellular IL-2 staining of these cells in the presence or absence of Ned-19. Ned-19 also partially suppressed intracellular IL-2 production and that too in a dose-dependent manner.
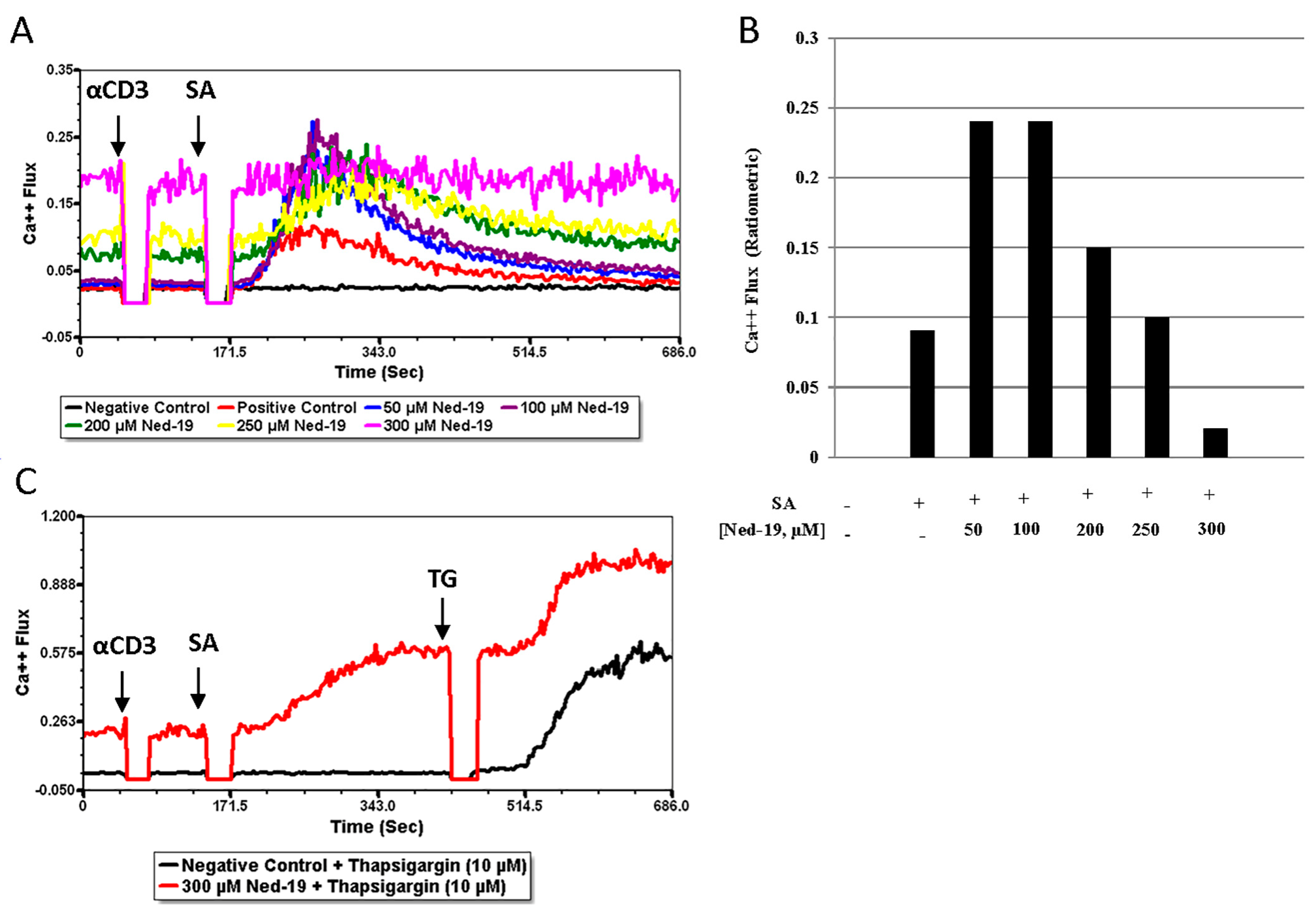
Sigova et al. have comparatively analyzed naïve and memory T cells (defined as ionomycin-resistant T cells) in terms of the response to Ca
2+-mobilizing agents, such as Con A, thimerosal, thapsigargin and ionomycin. They observed a dearth of intracellular Ca
2+ stores in memory T cells and insensitivity of memory T cells to Ca
2+-mobilizing agents [
35]. The above-mentioned findings and differences between naïve and memory CD4
+ T cells in downstream events of TCR signaling have prompted us to optimize the conventional receptor-mediated Ca
2+ flux assay for memory CD4
+ T cells. Memory CD4
+ T cells required a much higher concentration of biotinylated anti-CD3 (25 μg/mL) as compared to naïve CD4
+ T cells (5 μg/mL). Thereafter, the study of receptor-mediated Ca
2+ flux in memory CD4
+ T cells in the presence of Ned-19 concluded that 50, 100 and 200 μM concentrations of Ned-19 stimulated receptor-mediated Ca
2+ flux and 250 and 300 μM of Ned-19 exhibited concentration-dependent inhibition of receptor-mediated Ca
2+ flux. The thapsigargin-mediated Ca
2+ flux of cells pre-incubated with 300 μM Ned-19 confirmed the viability of the cells. The reduction in the Ca
2+ flux in the absence of extracellular Ca
2+ showed that Ca
2+ influx from the extracellular medium, presumably via store-operated Ca
2+ channels, contributed to the receptor-mediated Ca
2+ flux. The complete inhibition of the receptor-mediated Ca
2+ flux in the case of thapsigargin-preincubated cells substantiated the contribution of the ER Ca
2+ store to the receptor-mediated Ca
2+ flux. The partial inhibition of the receptor-mediated Ca
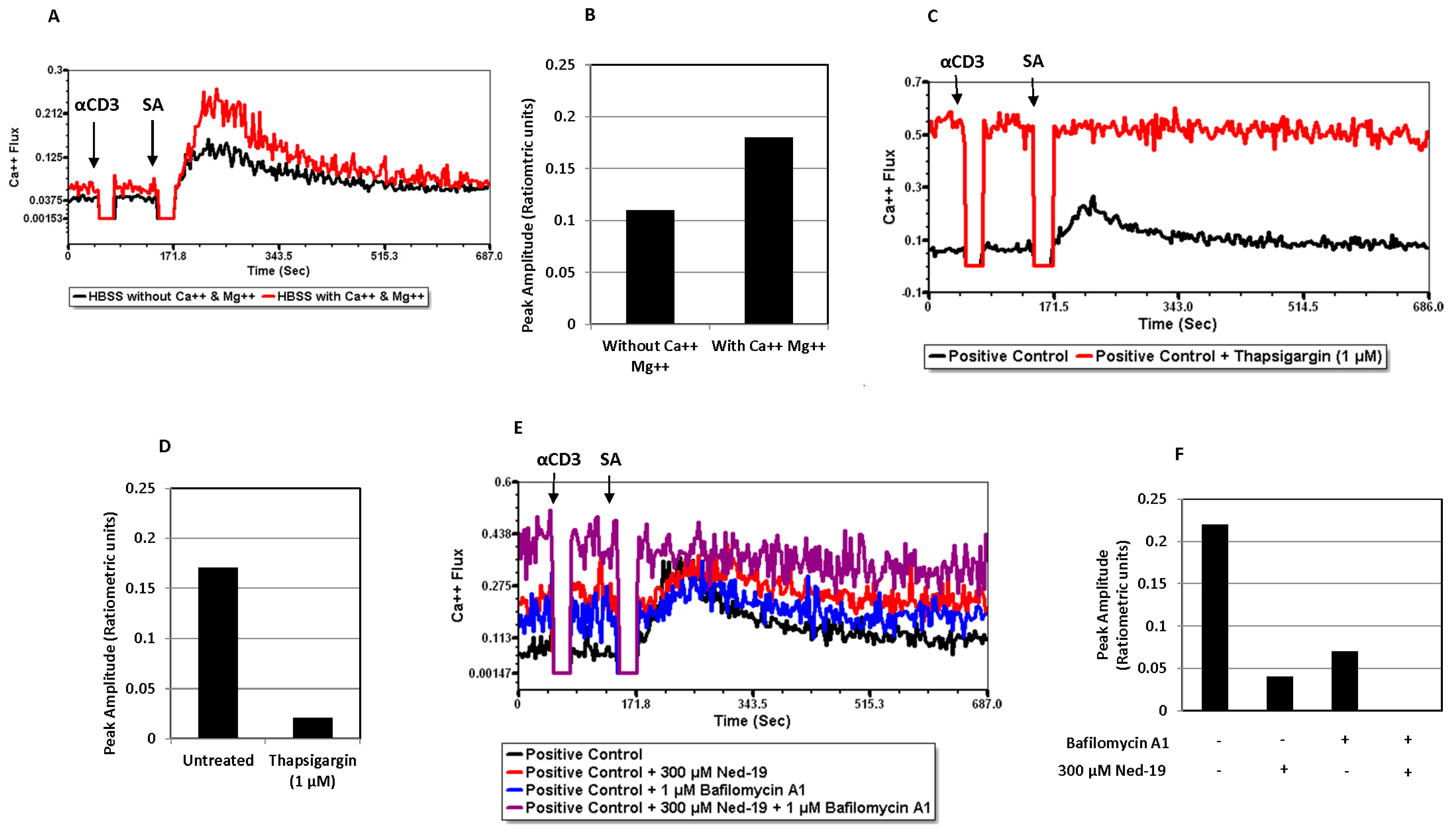
2+ flux in the case of bafilomycin A1-preincubated cells revealed the partial contribution of lysosome/acidic Ca
2+ stores in receptor-mediated Ca
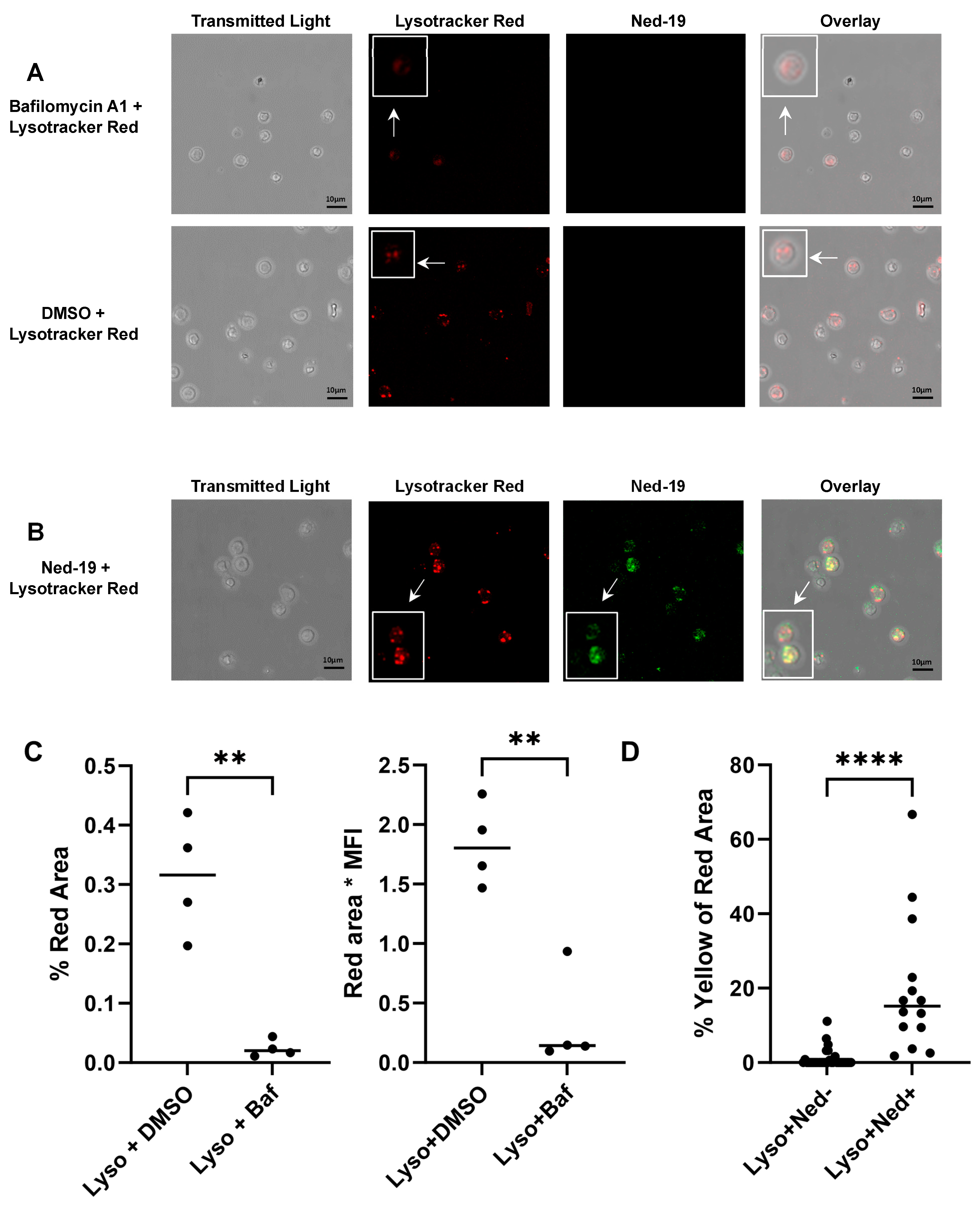
2+ flux. The action of bafilomycin A1 was confirmed by showing the disruption of the pH gradient of the lysosomes or acidic vesicles, as demonstrated by loss of Lysotracker Red staining of bafilomycin A1-preincubated memory CD4
+ T cells. In addition to this, pre-treatment of memory CD4
+ T cells with Ned-19 and Lysotracker Red, followed by confocal microscopy, showed weak co-localization in lysosomes or acidic vesicles, indicating the possible location of the Ned-19 target. This also showed that Ned-19 did not disrupt the pH gradient required for Lysotracker Red staining. This result was very much in alignment with a study where NAADP receptors in mouse pancreatic beta cells were fluorescently labeled using Ned-19 [
21]. Trufanov et al. have illustrated a great extent of cis-NED 19 and Lysotracker co-localization in rat aorta smooth muscle cells [
28].
NAADP, being negatively charged, does not readily enter cells unless it is taken up by a transport mechanism, such as connexin-43 hemichannels [
22,
36]. Parkesh et al. have discussed NAADP-AM, an acetoxy methyl derivative of NAADP, which could readily go inside the cell and then be converted into NAADP by esterases [
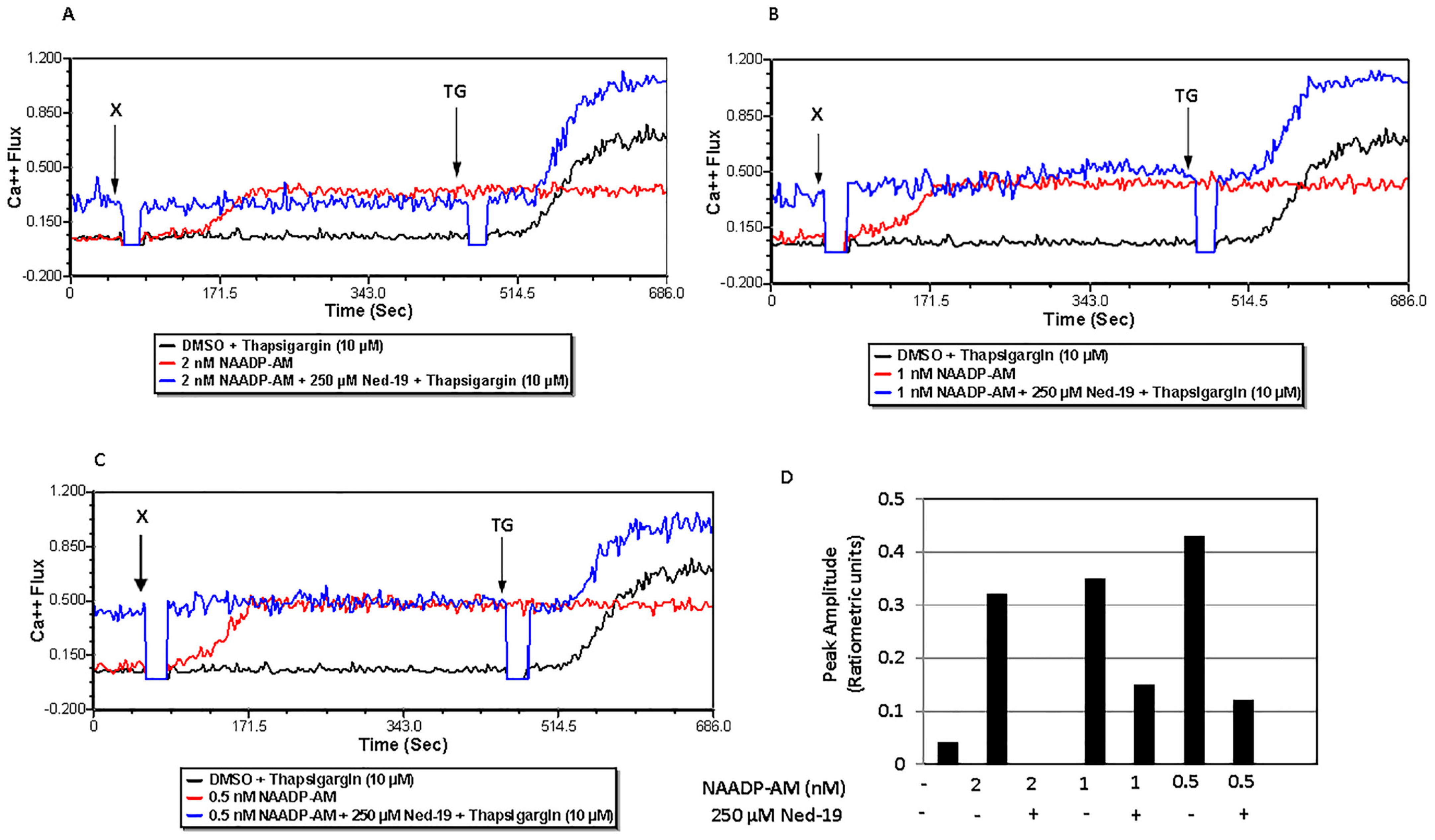
29]. In our study, only 2, 1 and 0.5 nM NAADP-AM caused Ca
2+ flux. Preincubation of memory CD4
+ T cells with 250 μM Ned-19 suppressed the Ca
2+ flux caused by all three concentrations of NAADP-AM. However, the activity of different preparations of NAADP-AM was variable, reflecting the instability of the reagent. The viability of cell samples that were incubated for 1 h with Ned-19, followed by the addition of NAADP-AM during the Ca
2+ flux, was confirmed by the addition of thapsigargin at a later time point, which caused a Ca
2+ flux. Hence, the presence of Ned-19 did suppress the NAADP-mediated Ca
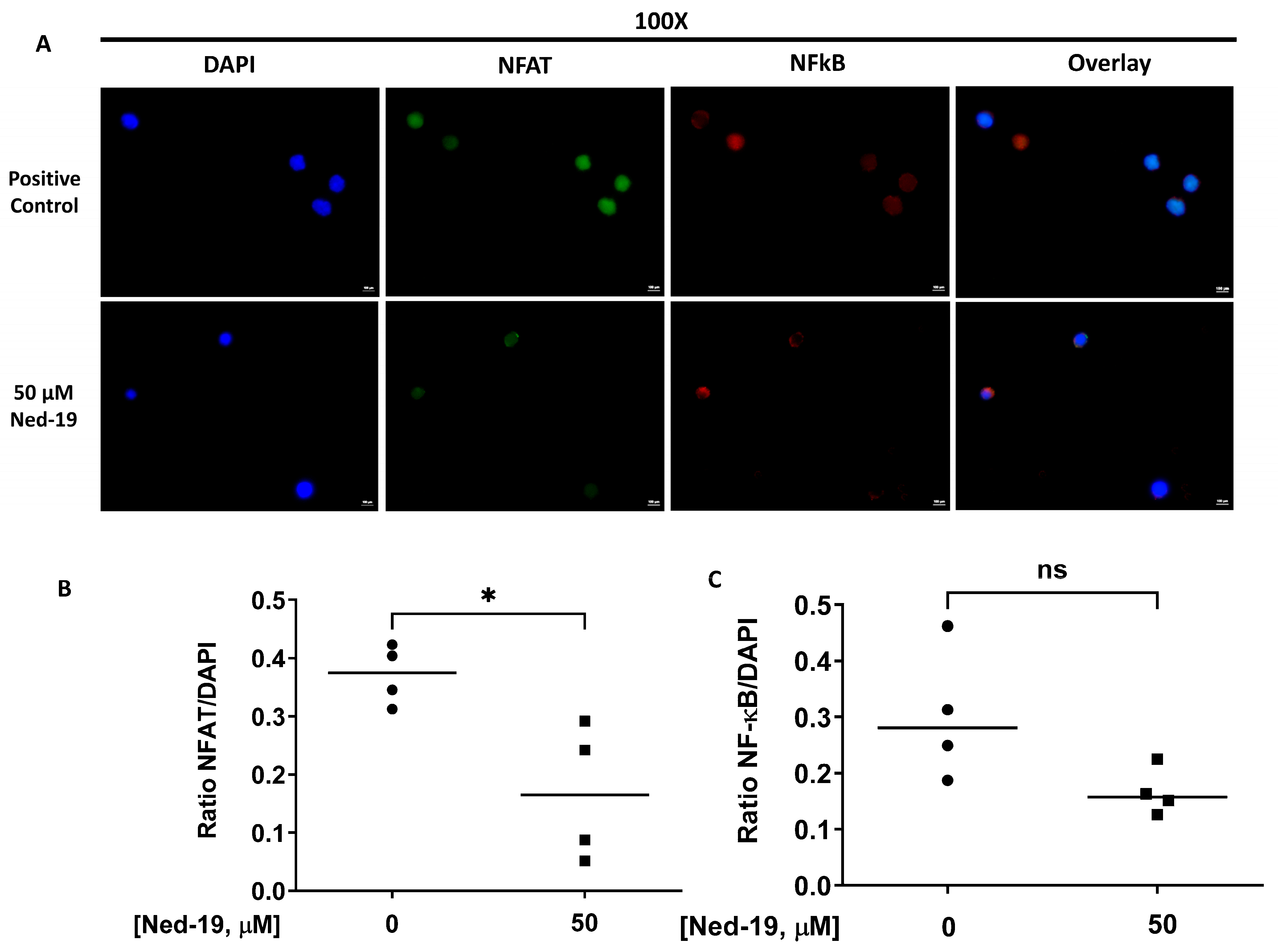
2+ flux when NAADP was shown to be present. In reference to the Ned-19-mediated partial inhibition of IL-2 secretion and significant inhibition of IFN-γ and IL-10 secretion by anti-CD3-stimulated memory CD4
+ T cells, we showed that Ned-19 mediated the partial inhibition of NFAT and NF-κB (not significant) nuclear translocation and therefore could mediate the inhibition of cytokine production by stimulated memory CD4
+ T cells. If we compare the effect of NAADP-mediated Ca
2+ signaling in naïve and memory CD4
+ T cells based on this study and the study conducted by Ali et al. [
22], we will conclude that Ned-19 suppresses proliferation, cytokine production, NFAT and NF-κB translocation to the nucleus in both the cells. Ned-19 inhibits Ca
2+ flux in a dose-dependent manner, with complete inhibition at 100 μM in naïve CD4
+ cells, whereas in memory CD4
+ cells, it stimulates Ca
2+ flux between 50 and 200 μM and inhibits Ca
2+ flux between 250 and 300 μM.
In conclusion, NAADP-mediated Ca2+ signaling generated via TCR activation has significant effects in terms of proliferation, cytokine (IL-2, IFN-γ and IL-10) secretion, and other downstream effector functions of memory CD4+ T cells. The memory CD4+ T cells utilized here were generated against antigens present in the environment. In the future, the study of NAADP-mediated Ca2+ signaling in memory CD4+ T cells generated against a specific antigen would be clinically beneficial and therefore enable us to look for better approaches to manipulate Ca2+ flux in memory cells.
4. Materials and Methods
4.1. Experimental Animals
Fifteen- to forty-week-old female C57BL/6J mice, obtained from Jackson Laboratory (Bar Harbor, ME, USA), were used for the experiments. The mice were kept under daily supervision in a specific-pathogen-free environment in the animal facility of the University of Toledo Health Science Campus. All the experiments with mice complied with the U.S. NIH guidelines for the Care and Use of Laboratory Animals. The protocol used for the project was approved by the Institutional Animal Care and Use Committee (IACUC). Approval Code: 107556 Approval Dates: 16 October 2014, 14 November 2017, 13 November 2020, 12 November 2023.
4.2. Medium
The T cell medium consisted of RPMI 1640 with L-glutamine (Corning Cellgro, Corning, CA, USA), 10% heat-inactivated fetal bovine serum/HI-FBS (Atlanta Biologicals, Minneapolis, MN, USA), 3 × 10−5 M 2-mercaptoethanol (Fisher Scientific, Hampton, NH, USA), 2 × 10−3 M L-glutamine, pH 7.4, 10 mM HEPES buffer (Sigma Aldrich, St. Louis, MO, USA), 100 U/mL penicillin, 100 µg/mL streptomycin, and 1% media additions (0.06 g folic acid (Gibco), 0.36 g L-asparagine (Gibco, Carlsbad, CA, USA), 1.16 g L-arginine (Hazleton Biologics, Inc., Lenexa, KS, USA), 2.16 g L-glutamine (MP Biomedicals, LLC, Irvine, CA, USA), and 1.10 g sodium pyruvate (MP Biomedicals, LLC) in 100 mL PBS). The memory CD4+ T cell medium (used during isolation) consisted of PBS with 2% HI-FBS and 1 mM EDTA. The FACS medium consisted of 0.05% bovine serum albumin (BSA, Fisher Scientific) and 0.1% sodium azide (Fluka, Charlotte, NC, USA) in PBS. The medium for the Ca2+ flux experiments using flow cytometry consisted of 2% HI-FBS in Hank’s balanced salt solution (HBSS) with CaCl2, MgCl2 (Gibco by Life Technologies, Carlsbad, CA, USA).
4.3. Memory CD4+ T Cell Isolation
The mouse spleen was disrupted using a sterile loose-fitting glass homogenizer in a medium composed of 2% HI-FBS in HBSS without calcium, magnesium or phenol red, followed by filtration through a 70 µm nylon mesh sterile cell strainer (Fisher Scientific). Then, the cells were resuspended in memory CD4+ T cell medium after centrifugation, followed by aspirating off the supernatant. The number of nucleated cells in that cell suspension was determined after lysing red blood cells in an aliquot of the cells. This was chosen because RBC Lysing Buffer could not be used with the isolation kit. Finally, after repeated centrifugation, the spleen cell pellet was resuspended in 1 mL memory CD4+ T cell medium. Thereafter, 108 nucleated spleen cells were processed using EasySepTM Mouse Memory CD4+ T Cell Isolation Kit (Stemcell Technologies, Vancouver, BC, Canada). FACS analysis showed that there were 82.6% CD62LlowCD44high cells (effector memory CD4+ T cells) and 17.0% CD62LhighCD44high (central memory CD4+ T cells). All the memory cell experiments were performed using similarly isolated cells.
4.4. Memory CD4+ T Cell Proliferation Assays
A flat bottom, tissue culture-treated polystyrene, sterile 96-well plate with a lid (CytoOne, USA Scientific, Ocala, FL, USA) was coated overnight with eBioscienceTM anti-mouse CD3 in PBS (10 µg/mL) or PBS. After isolation, memory CD4+ T cells in T cell medium (5 × 105 cells/mL) were incubated for 1 h with DMSO or increasing concentrations of Ned-19 in DMSO in the incubator at 37 °C and 5% CO2. When using BZ194, the incubation period of the memory CD4+ T cells with DMSO or BZ194 was 5 h. The washed plate was seeded with cells (105 cells/well) as per the proliferation plan. In some wells, phorbol 12-myristate 13-acetate (PMA) (100 ng/mL, Sigma-Aldrich) and ionomycin (500 ng/mL, Calbiochem, Burlington, MA, USA) were added to cells already incubated for an hour just before they were seeded into the plate. The plate was then incubated at 37 °C and 5% CO2 for 48 h. Thereafter, [3H] thymidine (specific activity: 7.0 Ci/mmol and concentration: 1.0 mCi/mL) from Moravek, Inc. (Brea, CA, USA) in T cell medium was added to each well (1 µCi/well) and the plate was incubated at 37 °C and 5% CO2 for another 24 h. The cells were harvested onto a filter plate (UniFilter-96 GF/B, PerkinElmer, Waltham, MA, USA). The plate was dried overnight at room temperature. On the next day, the bottom of the plate was sealed and then 40 µL of liquid scintillation cocktail (MP Biomedicals, Irvine, CA, USA) was added to each well. The top of the plate was sealed with a transparent sticker and the plate was read in a Perkin Elmer Top Count NXT.
4.5. ELISA Assays (IL-2, IFN-γ, and IL-10)
A flat bottom 96-well plate was coated overnight with anti-mouse CD3 (eBioscience, ThermoFisher, Carlsbad, CA, USA) in PBS (10 µg/mL for IL-2/2 µg/mL for IFN-γ and IL-10 ELISA) or PBS at 4 °C. Isolated memory CD4+ T cells in T cell medium (5 × 105 cells/mL) were incubated for 1 h with DMSO, 50 µM, and 100 µM Ned-19 in DMSO at 37 °C and 5% CO2. Meanwhile, the plate was washed twice with PBS. Thereafter, 1 µg/mL soluble anti-mouse CD28 (eBioscience) was added to those cell suspensions that would be seeded to wells coated with anti-mouse CD3 (for IL-10 and IFN-γ ELISA). Thereafter, the plate was seeded with cells (105 cells/well) and the plate was incubated at 37 °C and 5% CO2 for 48 h (96 h for IL-10 and IFN-γ ELISA). The supernatant was collected and stored in the freezer at −80 °C until the ELISA was performed. The ELISA was performed using IL-2 Mini ABTS ELISA Development Kit/Murine IL-10 Mini TMB ELISA Development Kit/Murine IFN-γ Standard TMB ELISA Development Kit (all from Peprotech, Carlsbad, CA, USA). TMB one-component HRP Microwell Substrate (Surmodics, Eden Prairie, MN, USA) was used as substrate. The plate was read at 620 nm (for IL-2 ELISA)/450 nm with wavelength correction set at 620 nm (for IL-10 and IFN-γ ELISA) using a CLARIOstar microplate reader from BMG Labtech (Ortenberg, Germany).
4.6. Flow Cytometry
Immunophenotyping was performed to determine the purity of the isolated cells. A BD Accuri C6 flow cytometer was used for the analysis and the following anti-mouse antibodies from eBioscience were used for immunophenotyping: CD4APC (final concentration 1 µg/mL), CD8a FITC (2.5 µg/mL), CD4 FITC (2.5 µg/mL), CD8a APC (1 µg/mL), CD62L FITC (2.5 µg/mL), and CD44 APC (1 µg/mL). The cell pellet was suspended in Fc block for 10 min, followed by diluted primary antibodies and incubation in ice for 1 h. The cells in FACS medium were washed once through a cushion of HI-FBS and FACS medium was added into the pellets for flow cytometry. All the flow experiments analyzed at least 10,000 cells per group.
For the intracellular IL-2 staining, memory CD4+ T cells from 2 spleens (1.75 × 106 cells/mL) were incubated for 1 h with DMSO or increasing concentrations of Ned-19 at 37 °C and 5% CO2. The cells were seeded in an anti-mouse CD3 (eBioscience) pre-coated (10 µg/mL) flat-bottom 96-well plate and the plate was incubated at 37 °C and 5% CO2 for 6.5 h, followed by 1000× brefeldin A solution (eBioscience) in T cell medium (final concentration in culture: 1:1000) and further incubation of the plate for an additional 7.5 h at 37 °C and 5% CO2. The cells were stained first with primary antibody for CD4 glycoprotein, and then fixed and permeabilized using Intracellular Fixation and Permeabilization Buffer Set (eBioscience) following the protocol, followed by staining for intracellular IL-2. The acquisition of the samples was performed in a BD Biosciences FACS Calibur flow cytometer and the data were processed using FCS Express7 Flow Cytometry software. The anti-mouse antibodies from eBioscience used for intracellular IL-2 staining analysis were CD4 APC, CD4 Alexa Fluor 488, IL-2 APC, and rat IgG2b kappa isotype control APC.
4.7. Calcium Flux
Calcium flux was determined via flow cytometry. After isolation, memory CD4+ T cells (about 105 cells/tube) were taken into 5 mL polystyrene tubes with cap (Evergreen Scientific, Bothell, WA, USA) as per the design of the experiment and washed once with FACS medium for Ca2+ flux. Then, the cells were loaded with the acetoxymethyl (AM) ester derivative of Fluo-4 or Fluo-4, AM (1.6 µM) and the acetoxymethyl (AM) ester derivative of Fura Red or Fura Red, AM (3.06 µM) cell permeant fluorescent dyes (Invitrogen by Thermo Fisher Scientific) and incubated at 37 °C and 5% CO2 for 30 min. The cells were incubated at 37 °C and 5% CO2 with trans-Ned19 (Tocris, BioTechne, Minneapolis, MN, USA) for one hour, thapsigargin (Calbiochem, Burlington, MA, USA) for 30 min, and bafilomycin A1 (LC Laboratories, Woburn, MA, USA) for 30 min. Just before the calcium flux, the cells were washed once with FACS medium for Ca2+ flux at room temperature. Calcium flux, defined as the ratio of FL1 to FL3, was conducted using a BD Biosciences FACS Calibur flow cytometer. The population of live cells was gated via forward and side scatter and fluorescence at FL1 (530 nm) and FL3 (670 nm) was monitored. For the TCR-mediated Ca2+ flux, the cells were stimulated via addition of 25 µg/mL (final concentration after addition) biotinylated anti-mouse CD3 monoclonal antibody or Anti-Mo CD3e, eBioscienceTM Biotin, functional grade (Invitrogen by Thermo Fisher Scientific) at 50 s followed by 50 µg/mL (final concentration after addition) of streptavidin (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) addition at 2 min 30 s. For the NAADP-mediated calcium flux of memory CD4+ T cells, NAADP-AM (AAT Bioquest, Pleasanton, CA, USA) was added at 1 min. Acquisition was briefly interrupted for each reagent addition. The data were analyzed using FCS Express 7 Flow Cytometry software.
4.8. Imaging of the Lysosomes of Memory CD4+ T Cells
Memory CD4+ T cells were incubated with 100 µM Ned-19 for 30 min at 37 °C and 5% CO2. Then, 60 nM Lysotracker Red DND-99 (Molecular Probes, Fisher Scientific, Hampton, NH, USA), 1 µM bafilomycin A, or DMSO were added and the incubation continued for another 30 min at 37 °C and 5% CO2. The cells were plated on glass-bottom microwell dishes (P50G-1.5-30-F, MatTek Corporation, Ashland, MA, USA) pre-coated with poly-L-lysine (100 µg/mL). The images were captured with a Leica TCS SP5 laser scanning confocal microscope equipped with 5 conventional lasers plus multi-photon excitation, which produces laser excitation lines: 458, 488, 514, 561, 633, and a tunable Ti-Sapphire MP laser 710–990 nm. While capturing the Lysotracker Red DND-99-stained images, the 577/590 nm excitation/emission setting was used. The multi-photon laser excitation at 760 nm and the collected emissions at 415–488 nm were utilized during capturing of the Ned-19-stained image. Quantitation was performed using ImageJ 1.54g software.
4.9. NFAT and NF-κB Translocation Analysis
Memory CD4+ T cells were stimulated by anti-mouse CD3 (10 µg/mL) from eBioscience in the presence of DMSO or 50 µM Ned-19 for 48 h at 37 °C and 5% CO2. Then, the cells were transferred onto microscope cover glass (Fisher Scientific), which was coated overnight with poly-L-lysine in water (50 µg/mL) in a flat-bottom with a lid 6-well cell culture plate (Corning Incorporated, Corning, NY, USA). The cell suspension (3.15 × 105 cells/mL for DMSO treated and 2.9 × 105/mL for 50 µM Ned-19 treated sample) was incubated in the wells at 37 °C and 5% CO2 overnight. On the next day, the cells were fixed using 2% paraformaldehyde at room temperature for 15 min. After 3 washes with PBS, the cells were permeabilized using 0.3% Triton X 100 for 25 min. After 3 washes with PBS, 1% BSA in PBS was added and incubated for 30 min to 1 h to block any nonspecific binding. Thereafter, mouse monoclonal antibody IgG1 NFATc1 (7A6) sc-7294 and rabbit polyclonal antibody NF-κB p65 (C-20): sc-372 from Santa Cruz Biotechnology (Dallas, TX, USA) were added at 3 µg/mL (final concentration) for immunohistochemical staining of NFAT and NF-κB in permeabilized cells. The cells were kept overnight at 4 °C. On the next day, after washing 3 times with PBS, secondary antibodies Texas Red labeled donkey anti-rabbit IgG (1:1000; Jackson ImmunoResearch) and Alexa Fluor 488 affinipure donkey anti-mouse IgG (1:1000; Jackson ImmunoResearch) were added and incubated for 1 h at room temperature. The cover glasses were mounted onto slides with ProLong Gold Antifade Mountant with DAPI (Molecular Probes). The immunofluorescence images were captured using a Nikon Eclipse Ti microscope with 100× oil objective. The quantification of the images was performed with ImageJ 1.54g software and the quantification of NFAT and NF-κB translocation to the nucleus was performed by taking the ratio of the integrated intensity (the product of the selected area in square pixels and mean gray value) of NFAT or NF-κB to DAPI. The average of 4 cells was determined in the analysis, as very few cells stuck to the cover glasses.
4.10. Statistical Analysis
The statistical analysis was performed using GraphPad Prism 10.0.2 software. The normal distribution analysis was conducted with the Kolmogorov–Smirnov or Shapiro–Wilk test. The proliferation and IL-10 and IFNγ ELISAs were analyzed using a one-way analysis of variance (ANOVA) with Tukey’s Multiple Comparison Tests, where **** p < 0.0001, *** p < 0.001, ** p < 0.01, * p < 0.1 and ns = not significant. IL-2 production was analyzed using the Kruskal–Wallis test. Lysosome staining and transcription factor translocation were analyzed using an unpaired t test, * p < 0.05. Ned-19 staining was analyzed using the Mann–Whitney test.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






