
Investigating Ligand Sphere Perturbations on MnIII–Alkylperoxo Complexes

 , , and
, , and
Abstract
:
1. Introduction
2. Results
2.1. Formation and Characterization of MnII(OTf)(6Medpaq5NO2) (1)
2.2. Formation and Characterization of [MnIII(OH)(6Medpaq5NO2)](OTf) (2)
2.3. Formation of [MnIII(OOtBu)(6Medpaq5NO2)]+ (3a) and [MnIII(OOCm)(6Medpaq5NO2)]+ (3b)
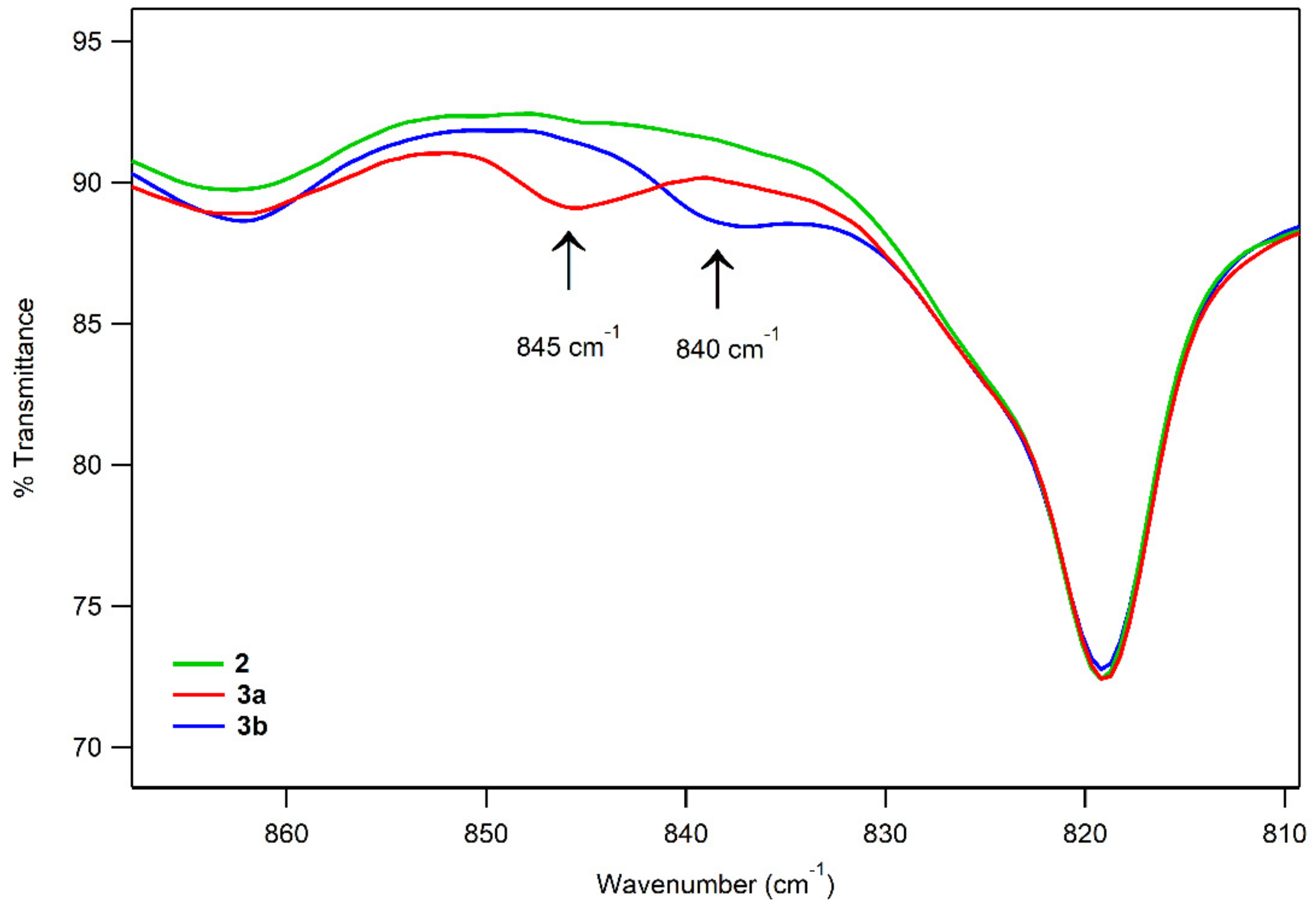
2.4. Spectroscopic Characterization of 3a and 3b
2.5. DFT Structures of 3a and 3b
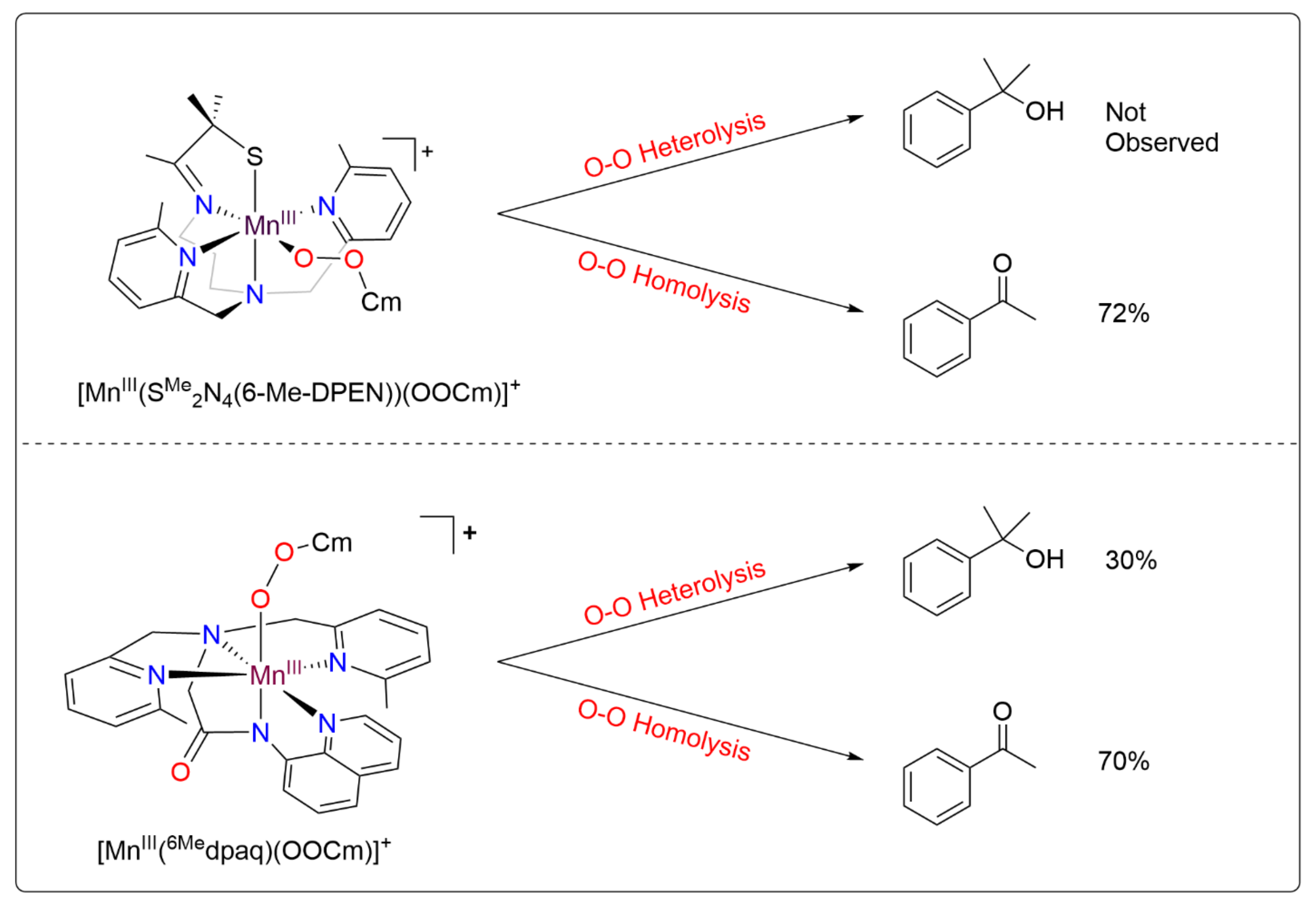
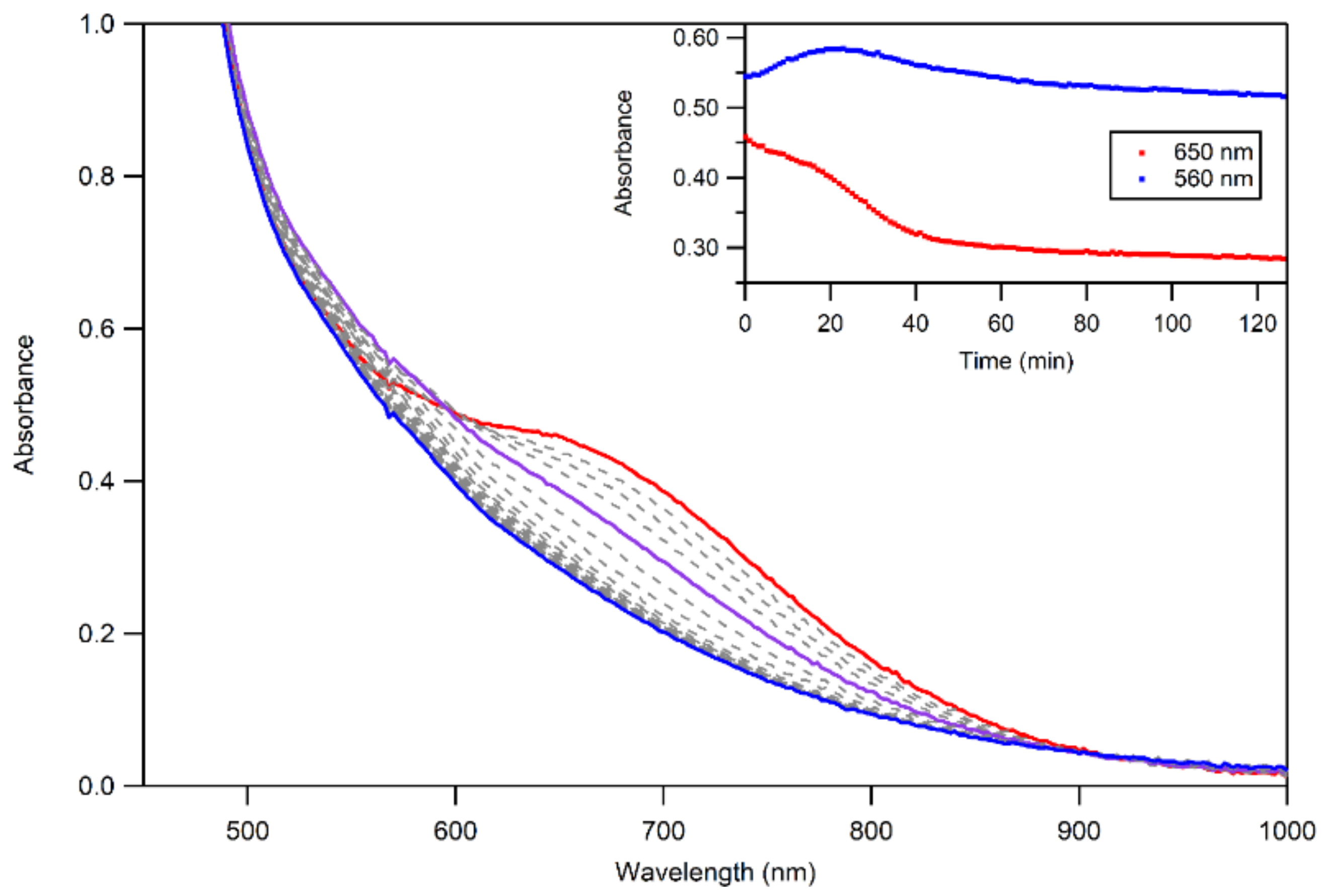
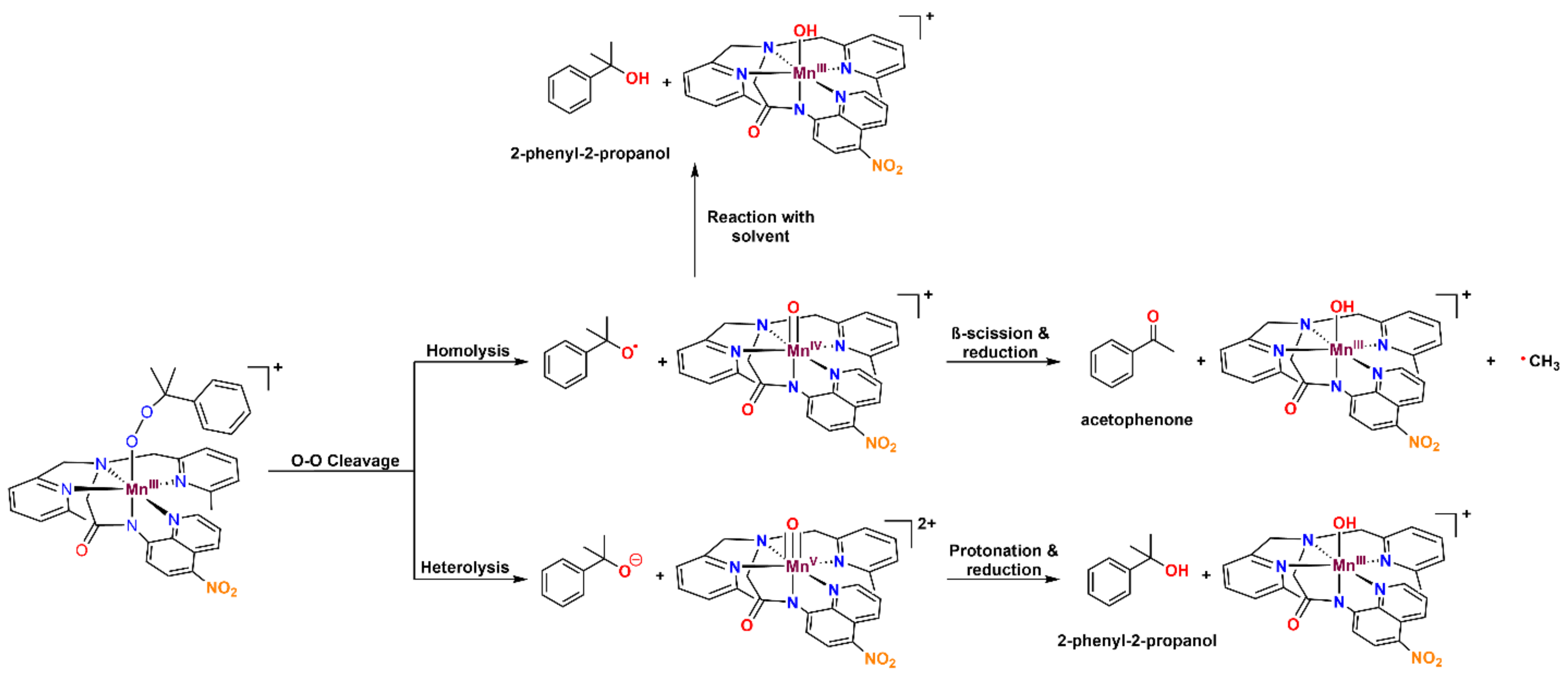
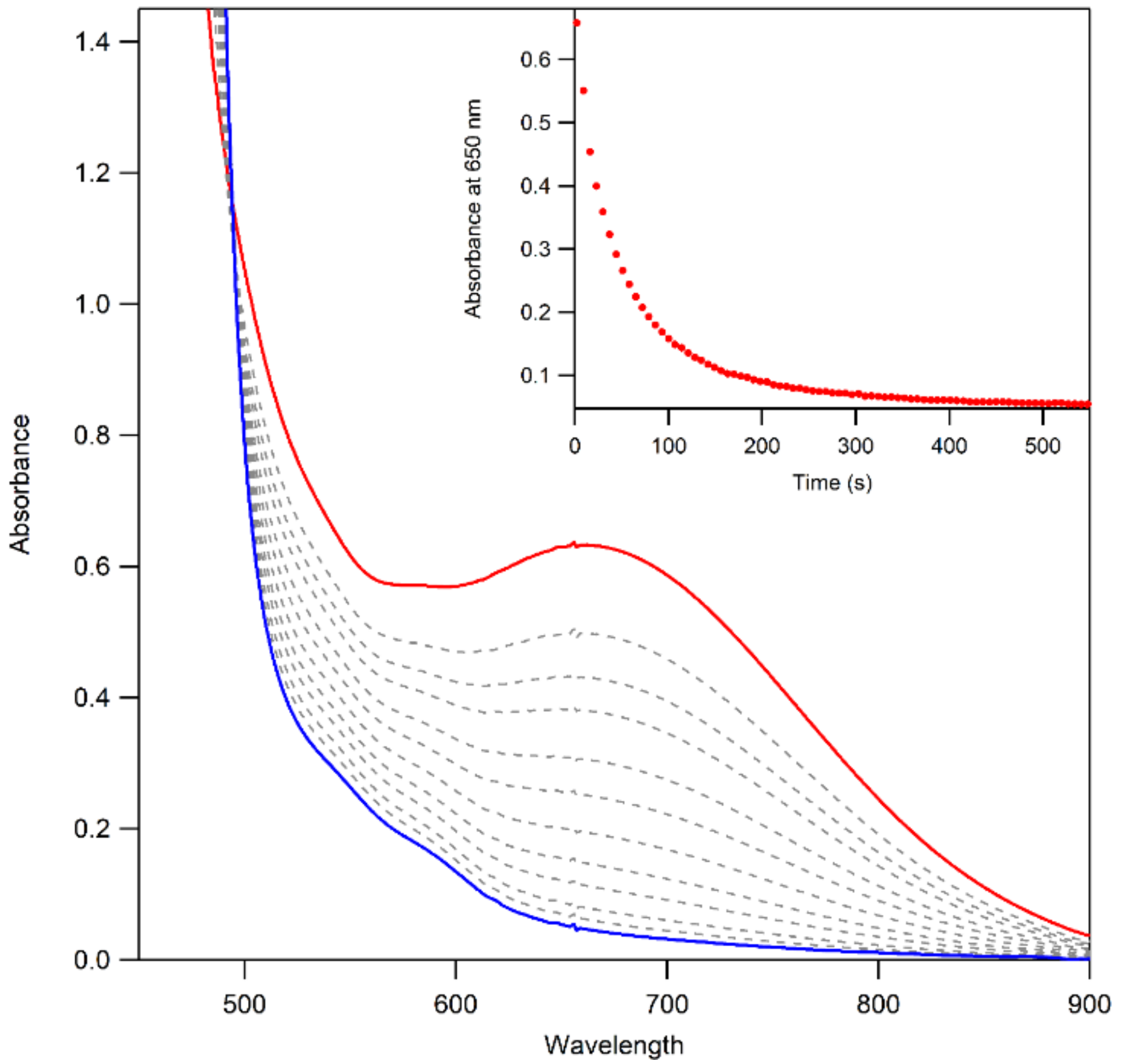
2.6. Thermal Decay Pathways of 3a and 3b
2.7. Oxidation of TEMPOH by the MnII–Hydroxo Complex 2
2.8. Oxidation of Triphenylphosphine by MnIII–Alkylperoxo Complexes
3. Discussion
4. Materials and Methods
4.1. General Methods and Instrumentation
4.2. Synthesis of (H6Medpaq5NO2)
4.3. Synthesis of MnII(OTf)(6Medpaq5NO2) (1), [MnIII(OH)(6Medpaq5NO2)]+ (2), and [MnIII(OOR)(6Medpaq5NO2)]+ (3a: R: tBu and 3b: R: Cm)
4.4. Thermal Decay Reactions of 3a and 3b and Reactivity with PPh3
4.5. Computational Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brash, A.R. Lipoxygenases: Occurrence, Functions, Catalysis, and Acquisition of Substrate. J. Biol. Chem. 1999, 274, 23679–23682. [Google Scholar] [PubMed]
- Oliw, E.H.; Jernerén, F.; Hoffmann, I.; Sahlin, M.; Garscha, U. Manganese lipoxygenase oxidizes bis-allylic hydroperoxides and octadecenoic acids by different mechanisms. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2011, 1811, 138–147. [Google Scholar]
- Su, C.; Oliw, E.H. Manganese Lipoxygenase: Purification and characterization. J. Biol. Chem. 1998, 273, 13072–13079. [Google Scholar] [PubMed]
- Su, C.; Sahlin, M.; Oliw, E.H. Kinetics of Manganese Lipoxygenase with a Catalytic Mononuclear Redox Center. J. Biol. Chem. 2000, 275, 18830–18835. [Google Scholar] [PubMed]
- Schramm, V.L.; Wedler, F.C. (Eds.) Manganese in Metabolism and Enzyme Function; Academic Press: Orlando, FL, USA, 1986. [Google Scholar]
- Cotruvo, J.A., Jr.; Stubbe, J. An Active Dimanganese(III)–Tyrosyl Radical Cofactor in Escherichia coli Class Ib Ribonucleotide Reductase. Biochemistry 2010, 49, 1297–1309. [Google Scholar] [PubMed]
- Dismukes, G.C. Manganese Enzymes with Binuclear Active Sites. Chem. Rev. 1996, 96, 2909–2926. [Google Scholar] [PubMed]
- Fraústo da Silva, J.J.R.; Williams, R.J.P. The Biological Chemistry of the Elements, 2nd ed.; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Requena, L.; Bornemann, S. Barley (Hordeum vulgare) oxalate oxidase is a manganese-containing enzyme. Biochem. J. 1999, 343, 185–190. [Google Scholar] [PubMed]
- Vinyard, D.J.; Ananyev, G.M.; Charles Dismukes, G. Photosystem II: The Reaction Center of Oxygenic Photosynthesis. Annu. Rev. Biochem. 2013, 82, 577–606. [Google Scholar] [PubMed]
- Wedler, F.C.; Denman, R.B.; Roby, W.G. Glutamine synthetase from ovine brain is a manganese(II) enzyme. Biochemistry 1982, 21, 6389–6396. [Google Scholar]
- Whiting, A.K.; Boldt, Y.R.; Hendrich, M.P.; Wackett, L.P.; Que, L. Manganese(II)-Dependent Extradiol-Cleaving Catechol Dioxygenase from Arthrobacter globiformis CM-2. Biochemistry 1996, 35, 160–170. [Google Scholar]
- Williams, R.J.P. Free manganese(II) and iron(II) cations can act as intracellular cell controls. FEBS Lett. 1982, 140, 3–10. [Google Scholar] [PubMed]
- Grove, L.E.; Brunold, T.C. Second-sphere tuning of the metal ion reduction potentials in iron and manganese superoxide dismutases. Comment Inorg. Chem. 2008, 29, 134–168. [Google Scholar]
- Miller, A.-F. Superoxide dismutases: Active sites that save, but a protein that kills. Curr. Opin. Chem. Biol. 2004, 8, 162–168. [Google Scholar] [PubMed]
- Sheng, Y.; Abreu, I.A.; Cabelli, D.E.; Maroney, M.J.; Miller, A.-F.; Teixeira, M.; Valentine, J.S. Superoxide Dismutases and Superoxide Reductases. Chem. Rev. 2014, 114, 3854–3918. [Google Scholar] [PubMed]
- Wu, A.J.; Penner-Hahn, J.E.; Pecoraro, V.L. Structural, Spectroscopic, and Reactivity Models for the Manganese Catalases. Chem. Rev. 2004, 104, 903–938. [Google Scholar] [PubMed]
- Call, A.; Capocasa, G.; Palone, A.; Vicens, L.; Aparicio, E.; Choukairi Afailal, N.; Siakavaras, N.; López Saló, M.E.; Bietti, M.; Costas, M. Highly Enantioselective Catalytic Lactonization at Nonactivated Primary and Secondary γ-C–H Bonds. J. Am. Chem. Soc. 2023, 145, 18094–18103. [Google Scholar] [PubMed]
- Aneeja, T.; Neetha, M.; Afsina, C.M.A.; Anilkumar, G. Recent advances and perspectives in manganese-catalyzed C–H activation. Catal. Sci. Technol. 2021, 11, 444–458. [Google Scholar]
- Chen, J.; Jiang, Z.; Fukuzumi, S.; Nam, W.; Wang, B. Artificial nonheme iron and manganese oxygenases for enantioselective olefin epoxidation and alkane hydroxylation reactions. Coord. Chem. Rev. 2020, 421, 213443. [Google Scholar]
- Kasper, J.B.; Vicens, L.; de Roo, C.M.; Hage, R.; Costas, M.; Browne, W.R. Reversible Deactivation of Manganese Catalysts in Alkene Oxidation and H2O2 Disproportionation. ACS Catal. 2023, 13, 6403–6415. [Google Scholar]
- Ottenbacher, R.V.; Talsi, E.P.; Bryliakov, K.P. Chiral Manganese Aminopyridine Complexes: The Versatile Catalysts of Chemo- and Stereoselective Oxidations with H2O2. Chem. Rec. 2018, 18, 78–90. [Google Scholar]
- Ottenbacher, R.V.; Talsi, E.P.; Rybalova, T.V.; Bryliakov, K.P. Enantioselective Benzylic Hydroxylation of Arylalkanes with H2O2 in Fluorinated Alcohols in the Presence of Chiral Mn Aminopyridine Complexes. ChemCatChem 2018, 10, 5323–5330. [Google Scholar]
- Philip, R.M.; Radhika, S.; Abdulla, C.M.A.; Anilkumar, G. Recent Trends and Prospects in Homogeneous Manganese-Catalysed Epoxidation. Adv. Synth. Catal. 2021, 363, 1272–1289. [Google Scholar]
- Sun, W.; Sun, Q. Bioinspired Manganese and Iron Complexes for Enantioselective Oxidation Reactions: Ligand Design, Catalytic Activity, and Beyond. Acc. Chem. Res. 2019, 52, 2370–2381. [Google Scholar] [PubMed]
- Talsi, E.P.; Bryliakov, K.P. Chemo- and stereoselective CH oxidations and epoxidations/cis-dihydroxylations with H2O2, catalyzed by non-heme iron and manganese complexes. Coord. Chem. Rev. 2012, 256, 1418–1434. [Google Scholar]
- Vicens, L.; Olivo, G.; Costas, M. Rational Design of Bioinspired Catalysts for Selective Oxidations. ACS Catal. 2020, 10, 8611–8631. [Google Scholar]
- Bullock, R.M.; Chen, J.G.; Gagliardi, L.; Chirik, P.J.; Farha, O.K.; Hendon, C.H.; Jones, C.W.; Keith, J.A.; Klosin, J.; Minteer, S.D.; et al. Using nature’s blueprint to expand catalysis with Earth-abundant metals. Science 2020, 369, eabc3183. [Google Scholar]
- Nandy, A.; Adamji, H.; Kastner, D.W.; Vennelakanti, V.; Nazemi, A.; Liu, M.; Kulik, H.J. Using Computational Chemistry to Reveal Nature’s Blueprints for Single-Site Catalysis of C–H Activation. ACS Catal. 2022, 12, 9281–9306. [Google Scholar]
- Chen, J.; Song, W.; Yao, J.; Wu, Z.; Lee, Y.-M.; Wang, Y.; Nam, W.; Wang, B. Hydrogen Bonding-Assisted and Nonheme Manganese-Catalyzed Remote Hydroxylation of C–H Bonds in Nitrogen-Containing Molecules. J. Am. Chem. Soc. 2023, 145, 5456–5466. [Google Scholar]
- Leto, D.F.; Jackson, T.A. Peroxomanganese complexes as an aid to understanding redox-active manganese enzymes. J. Biol. Inorg. Chem. 2014, 19, 1–15. [Google Scholar]
- Lin, Y.-H.; Kutin, Y.; van Gastel, M.; Bill, E.; Schnegg, A.; Ye, S.; Lee, W.-Z. A Manganese(IV)-Hydroperoxo Intermediate Generated by Protonation of the Corresponding Manganese(III)-Superoxo Complex. J. Am. Chem. Soc. 2020, 142, 10255–10260. [Google Scholar]
- Sankaralingam, M.; Lee, Y.-M.; Jeon, S.H.; Seo, M.S.; Cho, K.-B.; Nam, W. A mononuclear manganese(iii)–hydroperoxo complex: Synthesis by activating dioxygen and reactivity in electrophilic and nucleophilic reactions. Chem. Comm. 2018, 54, 1209–1212. [Google Scholar] [PubMed]
- So, H.; Park, Y.J.; Cho, K.-B.; Lee, Y.-M.; Seo, M.S.; Cho, J.; Sarangi, R.; Nam, W. Spectroscopic Characterization and Reactivity Studies of a Mononuclear Nonheme Mn(III)–Hydroperoxo Complex. J. Am. Chem. Soc. 2014, 136, 12229–12232. [Google Scholar] [PubMed]
- Tian, Y.-C.; Jiang, Y.; Lin, Y.-H.; Zhang, P.; Wang, C.-C.; Ye, S.; Lee, W.-Z. Hydrogen Atom Transfer Thermodynamics of Homologous Co(III)- and Mn(III)-Superoxo Complexes: The Effect of the Metal Spin State. JACS Au 2022, 2, 1899–1909. [Google Scholar] [PubMed]
- Chavez, F.A.; Rowland, J.M.; Olmstead, M.M.; Mascharak, P.K. Syntheses, Structures, and Reactivities of Cobalt(III)–Alkylperoxo Complexes and Their Role in Stoichiometric and Catalytic Oxidation of Hydrocarbons. J. Am. Chem. Soc. 1998, 120, 9015–9027. [Google Scholar]
- Chen, Y.; Shi, H.; Lee, C.-S.; Yiu, S.-M.; Man, W.-L.; Lau, T.-C. Room Temperature Aerobic Peroxidation of Organic Substrates Catalyzed by Cobalt(III) Alkylperoxo Complexes. J. Am. Chem. Soc. 2021, 143, 14445–14450. [Google Scholar] [PubMed]
- Costas, M.; Mehn, M.P.; Jensen, M.P.; Que, L. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev. 2004, 104, 939–986. [Google Scholar]
- Lehnert, N.; Ho, R.Y.N.; Que, L., Jr.; Solomon, E.I. Electronic Structure of High-Spin Iron(III)–Alkylperoxo Complexes and Its Relation to Low-Spin Analogues: Reaction Coordinate of O–O Bond Homolysis. J. Am. Chem. Soc. 2001, 123, 12802–12816. [Google Scholar]
- Lehnert, N.; Ho, R.Y.N.; Que, L.; Solomon, E.I. Spectroscopic Properties and Electronic Structure of Low-Spin Fe(III)–Alkylperoxo Complexes: Homolytic Cleavage of the O–O Bond. J. Am. Chem. Soc. 2001, 123, 8271–8290. [Google Scholar] [PubMed]
- Widger, L.R.; Jiang, Y.; McQuilken, A.C.; Yang, T.; Siegler, M.A.; Matsumura, H.; Moënne-Loccoz, P.; Kumar, D.; de Visser, S.P.; Goldberg, D.P. Thioether-ligated iron(ii) and iron(iii)-hydroperoxo/alkylperoxo complexes with an H-bond donor in the second coordination sphere. Dalton Trans. 2014, 43, 7522–7532. [Google Scholar]
- Du, J.; Miao, C.; Xia, C.; Lee, Y.-M.; Nam, W.; Sun, W. Mechanistic Insights into the Enantioselective Epoxidation of Olefins by Bioinspired Manganese Complexes: Role of Carboxylic Acid and Nature of Active Oxidant. ACS Catal. 2018, 8, 4528–4538. [Google Scholar]
- Miao, C.; Wang, B.; Wang, Y.; Xia, C.; Lee, Y.-M.; Nam, W.; Sun, W. Proton-Promoted and Anion-Enhanced Epoxidation of Olefins by Hydrogen Peroxide in the Presence of Nonheme Manganese Catalysts. J. Am. Chem. Soc. 2016, 138, 936–943. [Google Scholar]
- Coggins, M.K.; Martin-Diaconescu, V.; DeBeer, S.; Kovacs, J.A. Correlation Between Structural, Spectroscopic, and Reactivity Properties Within a Series of Structurally Analogous Metastable Manganese(III)–Alkylperoxo Complexes. J. Am. Chem. Soc. 2013, 135, 4260–4272. [Google Scholar]
- Coggins, M.K.; Kovacs, J.A. Structural and Spectroscopic Characterization of Metastable Thiolate-Ligated Manganese(III)–Alkylperoxo Species. J. Am. Chem. Soc. 2011, 133, 12470–12473. [Google Scholar]
- Downing, A.N.; Coggins, M.K.; Poon, P.C.Y.; Kovacs, J.A. Influence of Thiolate versus Alkoxide Ligands on the Stability of Crystallographically Characterized Mn(III)-Alkylperoxo Complexes. J. Am. Chem. Soc. 2021, 143, 6104–6113. [Google Scholar]
- Parham, J.D.; Wijeratne, G.B.; Rice, D.B.; Jackson, T.A. Spectroscopic and Structural Characterization of Mn(III)-Alkylperoxo Complexes Supported by Pentadentate Amide-Containing Ligands. Inorg. Chem. 2018, 57, 2489–2502. [Google Scholar] [PubMed]
- Opalade, A.A.; Parham, J.D.; Day, V.W.; Jackson, T.A. Characterization and chemical reactivity of room-temperature-stable MnIII–alkylperoxo complexes. Chem. Sci. 2021, 12, 12564–12575. [Google Scholar]
- Brunclik, S.A.; Opalade, A.A.; Jackson, T.A. Electronic structure contributions to O–O bond cleavage reactions for MnIII-alkylperoxo complexes. Dalton Trans. 2023, 52, 13878–13894. [Google Scholar]
- Dawson, J.H.; Holm, R.H.; Trudell, J.R.; Barth, G.; Linder, R.E.; Bunnenberg, E.; Djerassi, C.; Tang, S.C. Magnetic circular dichroism studies. 43. Oxidized cytochrome P-450. Magnetic circular dichroism evidence for thiolate ligation in the substrate-bound form. Implications for the catalytic mechanism. J. Am. Chem. Soc. 1976, 98, 3707–3709. [Google Scholar] [PubMed]
- Wijeratne, G.B.; Corzine, B.; Day, V.W.; Jackson, T.A. Saturation Kinetics in Phenolic O–H Bond Oxidation by a Mononuclear Mn(III)–OH Complex Derived from Dioxygen. Inorg. Chem. 2014, 53, 7622–7634. [Google Scholar]
- Rice, D.B.; Munasinghe, A.; Grotemeyer, E.N.; Burr, A.D.; Day, V.W.; Jackson, T.A. Structure and Reactivity of (μ-Oxo)dimanganese(III,III) and Mononuclear Hydroxomanganese(III) Adducts Supported by Derivatives of an Amide-Containing Pentadentate Ligand. Inorg. Chem. 2019, 58, 622–636. [Google Scholar]
- Eaton, G.R.; Eaton, S.S.; Barr, D.P.; Weber, R.T. Quantitative EPR, 1st ed.; Springer: Vienna, Austria, 2010; p. 185. [Google Scholar]
- Opalade, A.A.; Grotemeyer, E.N.; Jackson, T.A. Mimicking Elementary Reactions of Manganese Lipoxygenase Using Mn-hydroxo and Mn-alkylperoxo Complexes. Molecules 2021, 26, 7151. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar]
- Neese, F. Software update: The ORCA program system—Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta—Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [PubMed]
- Neese, F.; Wennmohs, F.; Hansen, A. Efficient and accurate local approximations to coupled-electron pair approaches: An attempt to revive the pair natural orbital method. J. Chem. Phys. 2009, 130, 114108. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | 1 | [MnII(OH2)(L)](OTf) | [MnII(L)](OTf) |
|---|---|---|---|
| a L = 6Medpaq | b L = dpaq | ||
| Mn–O1 | 2.169(7) | 2.108(3) | 2.079(2) |
| Mn–N1 | 2.260(6) | 2.233(3) | 2.214(3) |
| Mn–N2 | 2.150(7) | 2.152(4) | 2.191(3) |
| Mn–N3 | 2.281(6) | 2.280(3) | 2.314(3) |
| Mn–N4 | 2.328(7) | 2.354(4) | 2.244(3) |
| Mn–N5 | 2.357(8) | 2.417(3) | 2.286(3) |
| Bond | [MnIII(OH)(L)](OTf) | [MnIIIMnIII(µ–O)(L)2](OTf)2 | ||
|---|---|---|---|---|
| L = 6Medpaq5NO2 | a L = 6Medpaq | b L = dpaq | c L = dpaq5NO2 | |
| Mn–O1 | 1.826(4) | 1.806(6) | 1.806(13) | 1.7918(4) |
| Mn–N1 | 2.043(4) | 2.041(7) | 2.072(14) | 2.054(2) |
| Mn–N2 | 1.959(5) | 1.962(6) | 1.975(14) | 1.973(2) |
| Mn–N3 | 2.114(4) | 2.130(6) | 2.173(14) | 2.199(2) |
| Mn–N4 | 2.397(4) | 2.322(6) | 2.260(14) | 2.186(2) |
| Mn–N5 | 2.342(5) | 2.381(7) | 2.216(15) | 2.288(3) |
| 2 | 3a | 3b | |
|---|---|---|---|
| H-quinoline | 67.0 | 67.3 | 68.3 |
| NA a | 58.4 | ||
| H-pyridine | 54.0 | 48.1 | 47.6 |
| H-pyridine | 45.6 | 46.5 | 47.0 |
| NA a | 12.7 | 9.5 | |
| NA a | 9.9 | 8.8 | |
| H-pyridine | −16.8 | ||
| H-quinoline | −19.4 | −21.5 | −21.8 |
| H-quinoline | −60.7 | −58.9 | −59.5 |
| Bond | 3a | 3b | [MnIII(OOCm)(L)](OTf) |
|---|---|---|---|
| L = 6Medpaq5NO2 | L = 6Medpaq5NO2 | a L = 6Medpaq | |
| Mn–O1 | 1.833 | 1.831 | 1.849(3) |
| Mn–N1 | 2.045 | 2.037 | 2.044(4) |
| Mn–N2 | 1.958 | 1.950 | 1.955(4) |
| Mn–N3 | 2.153 | 2.150 | 2.100(4) |
| Mn–N4 | 2.355 | 2.370 | 2.284(4) |
| Mn–N5 | 2.347 | 2.296 | 2.394(4) |
| O1–O2 | 1.454 | 1.451 | 1.466(4) |
| Mn–O1–O2 | 107.3 | 114.2 | 110.4(2) |
| Source | DFT | DFT | X-Ray |
| Complex | Solvent | 2-Phenyl-2-propanol | Acetophenone |
|---|---|---|---|
| 3b | CH3CN | 49.7 ± 3.5% | 35.2 ± 2.8% |
| a [MnIII(OOCm)(6Medpaq)]+ | CH3CN | 61.3 ± 0.1% | 25.7 ± 0.1% |
| 3b | CD3CN | 24.0 ± 1.6% | 71.5 ± 4.3% |
| a [MnIII(OOCm)(6Medpaq)]+ | CD3CN | 50 ± 0.3% | 40 ± 0.3% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brunclik, S.A.; Grotemeyer, E.N.; Aghaei, Z.; Mian, M.R.; Jackson, T.A. Investigating Ligand Sphere Perturbations on MnIII–Alkylperoxo Complexes. Molecules 2024, 29, 1849. https://doi.org/10.3390/molecules29081849
Brunclik SA, Grotemeyer EN, Aghaei Z, Mian MR, Jackson TA. Investigating Ligand Sphere Perturbations on MnIII–Alkylperoxo Complexes. Molecules. 2024; 29(8):1849. https://doi.org/10.3390/molecules29081849
Chicago/Turabian StyleBrunclik, Samuel A., Elizabeth N. Grotemeyer, Zahra Aghaei, Mohammad Rasel Mian, and Timothy A. Jackson. 2024. "Investigating Ligand Sphere Perturbations on MnIII–Alkylperoxo Complexes" Molecules 29, no. 8: 1849. https://doi.org/10.3390/molecules29081849
APA StyleBrunclik, S. A., Grotemeyer, E. N., Aghaei, Z., Mian, M. R., & Jackson, T. A. (2024). Investigating Ligand Sphere Perturbations on MnIII–Alkylperoxo Complexes. Molecules, 29(8), 1849. https://doi.org/10.3390/molecules29081849