
Synthesis, In Vivo Anticonvulsant Activity Evaluation and In Silico Studies of Some Quinazolin-4(3H)-One Derivatives

 ,
,  , , ,
, , , 
 , , , ,
, , , ,  and
and
Abstract
: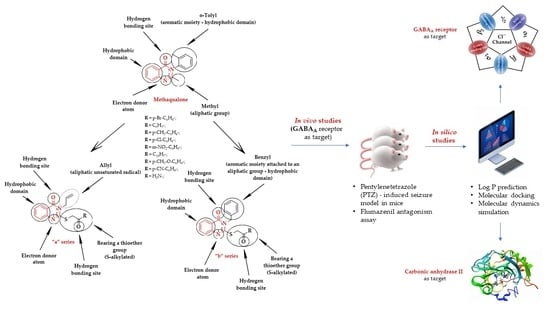
1. Introduction
2. Results and Discussion
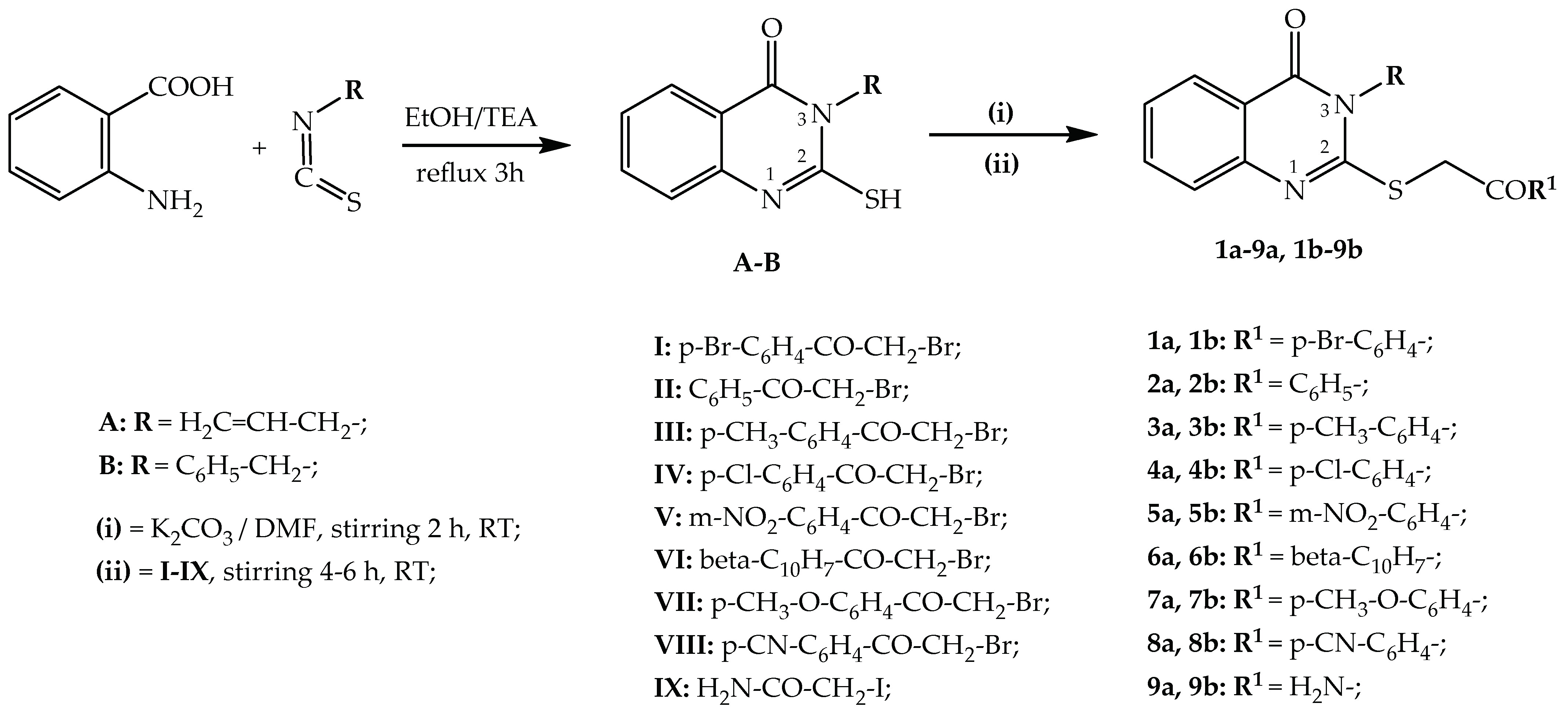
2.1. Chemical Synthesis
2.2. In Vivo Anticonvulsant Activity Evaluation
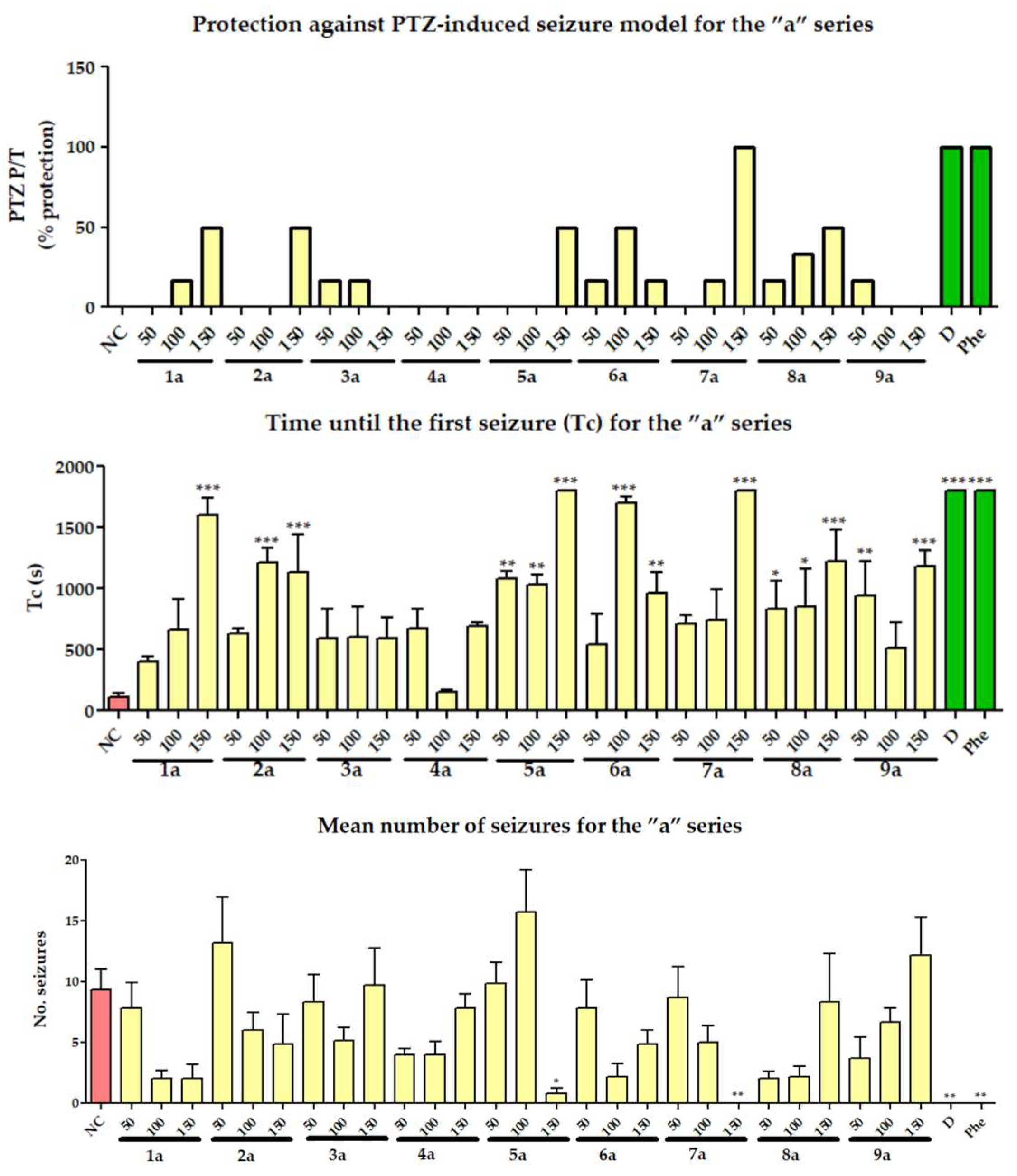
2.2.1. Pentylenetetrazole (PTZ)-Induced Seizures
2.2.2. Flumazenil Antagonism Assay
2.3. In Silico Studies
2.3.1. LogP Prediction
2.3.2. Molecular Docking
E-Zn2+-H2O ⇌ E-Zn2++-OH− + H+,
2.3.3. Molecular Dynamics Simulation
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis of Intermediate Compounds A and B
3.1.2. Synthesis of Compounds 1a–9a
3.1.3. Synthesis of Compounds 1b–9b
3.2. In Vivo Anticonvulsant Activity Evaluation
3.2.1. Animals and Ethics
3.2.2. Pentylenetetrazole (PTZ)-Induced Seizures
3.2.3. The Flumazenil Antagonism Assay
- Group A: 5 mg/kg flumazenil, 5 min later + 2 mg/kg diazepam (previously used dose), 30 min later + 70 mg/kg pentylenetetrazole (previously used dose);
- Group B: 7 mg/kg flumazenil, 5 min later + 2 mg/kg diazepam, 30 min later + 70 mg/kg pentylenetetrazole;
- Group C: 10 mg/kg flumazenil, 5 min later + 2 mg/kg diazepam, 30 min later + 70 mg/kg pentylenetetrazole.
- Group 1 (7a): 5 mg/kg flumazenil, 5 min later + 150 mg/kg compound 7a, 30 min later + 70 mg/kg pentylenetetrazole;
- Group 2 (8b): 5 mg/kg flumazenil, 5 min later + 150 mg/kg compound 8b, 30 min later + 70 mg/kg pentylenetetrazole.
3.3. In Silico Studies
3.3.1. LogP Prediction
3.3.2. Molecular Docking
3.3.3. Molecular Dynamics Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roche, V.F.; Zito, S.W.; Lemke, T.L. Sedative-hypnotics. In Foye’s Principles of Medicinal Chemistry; Williams, D.A., Ed.; LWW: Philadelphia, PA, USA, 2012; pp. 489–490. [Google Scholar]
- Roche, V.; Zito, S.W.; Lemke, T.L.; Williams, D.A. Drugs to treat seizure disorders. In Foye’s Principles of Medicinal Chemistry; LWW: Philadelphia, PA, USA, 2019; pp. 468–474. [Google Scholar]
- Thijs, R.D.; Surges, R.; O’Brien, T.J.; Sander, J.W. Epilepsy in adults. Lancet 2019, 393, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Milligan, T.A. Epilepsy: A Clinical Overview. Am. J. Med. 2021, 134, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.S.; Cross, J.H.; French, J.A.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L.; Moshé, S.L.; Peltola, J.; Roulet Perez, E.; et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 522–530. [Google Scholar] [CrossRef]
- Ugale, V.G.; Bari, S.B. Quinazolines: New horizons in anticonvulsant therapy. Eur. J. Med. Chem. 2014, 80, 447–501. [Google Scholar] [CrossRef] [PubMed]
- Hammer, H.; Bader, B.M.; Ehnert, C.; Bundgaard, C.; Bunch, L.; Hoestgaard-Jensen, K.; Schroeder, O.H.-U.; Bastlund, J.F.; Gramowski-Voß, A.; Jensen, A.A. A Multifaceted GABA A Receptor Modulator: Functional Properties and Mechanism of Action of the Sedative-Hypnotic and Recreational Drug Methaqualone (Quaalude). Mol. Pharmacol. 2015, 88, 401–420. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.W. GABAA receptor: Positive and negative allosteric modulators. Neuropharmacology 2018, 136, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Ghit, A.; Assal, D.; Al-Shami, A.S.; Hussein, D.E.E. GABAA receptors: Structure, function, pharmacology, and related disorders. J. Genet. Eng. Biotechnol. 2021, 19, 123. [Google Scholar] [CrossRef]
- Treiman, D.M. GABAergic Mechanisms in Epilepsy. Epilepsia 2001, 42, 8–12. [Google Scholar] [CrossRef]
- Safavynia, S.A.; Keating, G.; Speigel, I.; Fidler, J.A.; Kreuzer, M.; Rye, D.B.; Jenkins, A.; García, P.S. Effects of γ-Aminobutyric Acid Type A Receptor Modulation by Flumazenil on Emergence from General Anesthesia. Anesthesiology 2016, 125, 147–158. [Google Scholar] [CrossRef]
- Pitsikas, N.; Tarantilis, P.A. The GABAA-Benzodiazepine Receptor Antagonist Flumazenil Abolishes the Anxiolytic Effects of the Active Constituents of Crocus sativus L. Crocins in Rats. Molecules 2020, 25, 5647. [Google Scholar] [CrossRef]
- Shaye, H.; Stauch, B.; Gati, C.; Cherezov, V. Molecular mechanisms of metabotropic GABA B receptor function. Sci. Adv. 2021, 7, eabg3362. [Google Scholar] [CrossRef] [PubMed]
- Asiedu, M.N.; Mejia, G.L.; Hübner, C.A.; Kaila, K.; Price, T.J. Inhibition of Carbonic Anhydrase Augments GABAA Receptor-Mediated Analgesia via a Spinal Mechanism of Action. J. Pain 2014, 15, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Ruusuvuori, E.; Kaila, K. Carbonic Anhydrases and Brain pH in the Control of Neuronal Excitability. In Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications. Subcellular Biochemistry; Frost, S.C., McKenna, R., Eds.; Springer: Dordrecht, The Netherlands, 2014; pp. 271–290. [Google Scholar]
- Leniger, T.; Thöne, J.; Wiemann, M. Topiramate modulates pH of hippocampal CA3 neurons by combined effects on carbonic anhydrase and Cl−/HCO 3−exchange. Br. J. Pharmacol. 2004, 142, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Wahan, S.K.; Sharma, B.; Chawla, P.A. Medicinal perspective of quinazolinone derivatives: Recent developments and structure–activity relationship studies. J. Heterocycl. Chem. 2022, 59, 239–257. [Google Scholar] [CrossRef]
- Auti, P.S.; George, G.; Paul, A.T. Recent advances in the pharmacological diversification of quinazoline/quinazolinone hybrids. RSC Adv. 2020, 10, 41353–41392. [Google Scholar] [CrossRef]
- Patel, H.M.; Noolvi, M.N.; Shirkhedkar, A.A.; Kulkarni, A.D.; Pardeshi, C.V.; Surana, S.J. Anti-convulsant potential of quinazolinones. RSC Adv. 2016, 6, 44435–44455. [Google Scholar] [CrossRef]
- El-Helby, A.A.; Abdel Wahab, M.H. Design and synthesis of some new derivatives of 3H-quinazolin-4-one with promising anticonvulsant activity. Acta Pharm. 2003, 53, 127–138. [Google Scholar] [PubMed]
- De Simone, G.; Bua, S.; Supuran, C.T.; Alterio, V. Benzyl alcohol inhibits carbonic anhydrases by anchoring to the zinc coordinated water molecule. Biochem. Biophys. Res. Commun. 2021, 548, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Matias, M.; Silvestre, S.; Falcao, A.; Alves, G. Recent Highlights on Molecular Hybrids Potentially Useful in Central Nervous System Disorders. Mini-Rev. Med. Chem. 2017, 17, 486–517. [Google Scholar] [CrossRef]
- Nikolic, M.V.; Mijajlovic, M.Z.; Tomovic, D.L.; Bukonjic, A.M.; Jevtic, V.V.; Ratkovic, Z.R.; Trifunovic, S.R.; Radic, G.P. Synthesis and Characterization of Zinc(II)-Complexes with S-Alkyl Derivatives of Thiosalicylic Acid. Serbian J. Exp. Clin. Res. 2018, 19, 113–117. [Google Scholar] [CrossRef]
- Sigel, E.; Ernst, M. The Benzodiazepine Binding Sites of GABA A Receptors. Trends Pharmacol. Sci. 2018, 39, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Richter, L.; De Graaf, C.; Sieghart, W.; Varagic, Z.; Mörzinger, M.; De Esch, I.J.P.; Ecker, G.F.; Ernst, M. Diazepam-bound GABAA receptor model identify new benzodiazepine binding-site ligands. Nat. Chem. Biol. 2012, 8, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Borghese, C.M.; Herman, M.; Snell, L.D.; Lawrence, K.J.; Lee, H.-Y.; Backos, D.S.; Vanderlinden, L.A.; Harris, R.A.; Roberto, M.; Hoffman, P.L.; et al. Novel Molecule Exhibiting Selective Affinity for GABAA Receptor Subtypes. Sci. Rep. 2017, 7, 6230. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.-K.; El-Adl, K.; Al-Karmalawy, A.A. Design, synthesis, molecular docking and anticonvulsant evaluation of novel 6-iodo-2-phenyl-3-substituted-quinazolin-4(3H)-ones. Bull. Fac. Pharm. Cairo Univ. 2015, 53, 101–116. [Google Scholar] [CrossRef]
- Pele, R.; Marc, G.; Stana, A.; Ionuț, I.; Nastasă, C.; Tiperciuc, B.; Oniga, I.; Pîrnău, A.; Vlase, L.; Oniga, O. Synthesis of New Phenolic Derivatives of Quinazolin-4(3H)-One as Potential Antioxidant Agents—In Vitro Evaluation and Quantum Studies. Molecules 2022, 27, 2599. [Google Scholar] [CrossRef] [PubMed]
- Pele, R.; Marc, G.; Ionuț, I.; Nastasă, C.; Fizeșan, I.; Pîrnău, A.; Vlase, L.; Palage, M.; Oniga, S.; Oniga, O. Antioxidant and Cytotoxic Activity of New Polyphenolic Derivatives of Quinazolin-4(3H)-one: Synthesis and In Vitro Activities Evaluation. Pharmaceutics 2022, 15, 136. [Google Scholar] [CrossRef] [PubMed]
- Abulkhair, H.S.; El-Gamal, K.M.; El-Adl, K.; Fadl, M.F. Molecular Docking, Synthesis and Biological Evaluation of Some Novel 2-Substituted-3-allyl-4(3H)-quinazolinone Derivatives as Anticonvulsant Agents. Med. Chem. 2016, 6, 593–603. [Google Scholar] [CrossRef]
- Krasovskii, A.N.; Bulgakov, A.K.; Chumakova, L.Y.; Krasovskii, I.A.; Dyachenko, A.M.; Bokun, A.A.; Kravchenko, N.A.; Demchenko, A.M. Synthesis and Antibacterial Activity of S-Substituted 2-Thioquinazolin-4(3H)-ones. Pharm. Chem. J. 1999, 33, 15–16. [Google Scholar] [CrossRef]
- Gatadi, S.; Lakshmi, T.V.; Nanduri, S. 4(3H)-Quinazolinone derivatives: Promising antibacterial drug leads. Eur. J. Med. Chem. 2019, 170, 157–172. [Google Scholar] [CrossRef]
- Laddha, S.S.; Bhatnagar, S.P. Novel fused quinazolinones: Further studies on the anticonvulsant activity of 1,2,9,11-tetrasubstituted- 7H -thieno[2′,3′:4,5]pyrimido[6,1-b]-quinazolin-7-one and 1,3,10,12-tetrasubstituted- 8H -pyrido[2´,3´:4,5]pyrimido[6,1-b]quinazolin-8-one. Future Med. Chem. 2010, 2, 565–573. [Google Scholar] [CrossRef]
- Łączkowski, K.Z.; Sałat, K.; Misiura, K.; Podkowa, A.; Malikowska, N. Synthesis and anticonvulsant activities of novel 2-(cyclopentylmethylene)hydrazinyl-1,3-thiazoles in mouse models of seizures. J. Enzym. Inhib. Med. Chem. 2016, 31, 1576–1582. [Google Scholar] [CrossRef] [PubMed]
- Lader, M. Benzodiazepine harm: How can it be reduced? Br. J. Clin. Pharmacol. 2014, 77, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Baias, A.; Cristina, R.T.; Chiurciu, V. Guidelines for euthanasia of laboratory animals used in biomedical research. Vet. Drug 2012, 6, 57–62. [Google Scholar]
- Gawad, N.M.A.; Georgey, H.H.; Youssef, R.M.; Sayed, N.A.E. Design, synthesis, and anticonvulsant activity of novel quinazolinone analogues. Med. Chem. Res. 2011, 20, 1280–1286. [Google Scholar] [CrossRef]
- Zayed, M.; Ihmaid, S.; Ahmed, H.; El-Adl, K.; Asiri, A.; Omar, A. Synthesis, Modelling, and Anticonvulsant Studies of New Quinazolines Showing Three Highly Active Compounds with Low Toxicity and High Affinity to the GABA-A Receptor. Molecules 2017, 22, 188. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Al-Shamary, D.S.; Al-Alshaikh, M.A.; Kheder, N.A.; Mabkhot, Y.N.; Badshah, S.L. Molecular docking and biological evaluation of some thioxoquinazolin-4(3H)-one derivatives as anticancer, antioxidant and anticonvulsant agents. Chem. Cent. J. 2017, 11, 48. [Google Scholar] [CrossRef]
- Al-Salem, H.S.A.; Hegazy, G.H.; El-Taher, K.E.H.; El-Messery, S.M.; Al-Obaid, A.M.; El-Subbagh, H.I. Synthesis, anticonvulsant activity and molecular modeling study of some new hydrazinecarbothioamide, benzenesulfonohydrazide, and phenacylacetohydrazide analogues of 4(3H)-quinazolinone. Bioorg. Med. Chem. Lett. 2015, 25, 1490–1499. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 3467–3474. [Google Scholar] [CrossRef]
- El-Azab, A.S.; Abdel-Aziz, A.A.-M.; Bua, S.; Nocentini, A.; El-Gendy, M.A.; Mohamed, M.A.; Shawer, T.Z.; AlSaif, N.A.; Supuran, C.T. Synthesis of benzensulfonamides linked to quinazoline scaffolds as novel carbonic anhydrase inhibitors. Bioorg. Chem. 2019, 87, 78–90. [Google Scholar] [CrossRef]
- Magheru, C.; Magheru, S.; Coltau, M.; Hoza, A.; Moldovan, C.; Sachelarie, L.; Gradinaru, I.; Hurjui, L.L.; Marc, F.; Farcas, D.M. Antiepileptic Drugs and Their Dual Mechanism of Action on Carbonic Anhydrase. J. Clin. Med. 2022, 11, 2614. [Google Scholar] [CrossRef] [PubMed]
- Shukralla, A.A.; Dolan, E.; Dlanty, N. Acetazolamide: Old drug, new evidence? Epilepsia Open 2022, 7, 371–546. [Google Scholar] [CrossRef] [PubMed]
- Kandeda, A.K.; Taiwe, G.S.; Ayissi, R.E.M.; Moutchida, C. An aqueous extract of Canarium schweinfurthii attenuates seizures and potentiates sleep in mice: Evidence for involvement of GABA Pathway. Biomed. Pharmacother. 2021, 142, 111973. [Google Scholar] [CrossRef] [PubMed]
- Abuelizz, H.A.; El Dib, R.; Marzouk, M.; Anouar, E.H.; Maklad, Y.A.; Attia, H.N.; Al-Salahi, R. Molecular Docking and Anticonvulsant Activity of Newly Synthesized Quinazoline Derivatives. Molecules 2017, 22, 1094. [Google Scholar] [CrossRef] [PubMed]
- Saldıvar-González, A.; Gomez, C.; Martınez-Lomelı, I.; Arias, C. Effect of Flumazenil and Diazepam on Transient Actions in Defensive Burying Elicited by the Social Interaction Experience in Rats. Pharmacol. Biochem. Behav. 2000, 66, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Cheke, R.S.; Shinde, S.D.; Ambhore, J.P.; Chaudhari, S.R.; Bari, S.B. Quinazoline: An update on current status against convulsions. J. Mol. Struct. 2022, 1248, 131384. [Google Scholar] [CrossRef]
- Antonie, D.; Michielin, O.; Zoete, V. iLOGP: A Simple, Robust, and Efficient Description of n-Octanol/Water Partition Coefficient for Drug Design Using the GB/SA Approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar]
- Rybka, S.; Obniska, J.; Rapacz, A.; Filipek, B.; Kamiński, K. Synthesis, physicochemical, and anticonvulsant properties of new N-Mannich bases derived from pyrrolidine-2,5-dione and its 3-methyl analog. Arch. Pharm. 2014, 347, 768–776. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Borlan, R.; Tatar, A.-S.; Soritau, O.; Maniu, D.; Marc, G.; Florea, A.; Focsan, M.; Astilean, S. Design of fluorophore-loaded human serum albumin nanoparticles for specific targeting of NIH:OVCAR3 ovarian cancer cells. Nanotechnology 2020, 31, 315102. [Google Scholar] [CrossRef]
- Oniga, S.; Palage, M.; Araniciu, C.; Marc, G.; Oniga, O.; Vlase, L.; Prisăcari, V.; Valica, V.; Curlat, S.; Uncu, L. Design, synthesis, molecular docking, and antibacterial activity evaluation of some novel norfloxacin analogues. Farmacia 2018, 66, 1048–1058. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Santos-Martins, D.; Forli, S.; Ramos, M.J.; Olson, A.J. AutoDock4 Zn: An Improved AutoDock Force Field for Small-Molecule Docking to Zinc Metalloproteins. J. Chem. Inf. Model. 2014, 54, 2371–2379. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. AMDock: A versatile graphical tool for assisting molecular docking with Autodock Vina and Autodock4. Biol. Direct 2020, 15, 12. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Stoica, C.I.; Marc, G.; Pîrnău, A.; Vlase, L.; Araniciu, C.; Oniga, S.; Palage, M.; Oniga, O. Thiazolyl-oxadiazole derivatives targeting lanosterol 14α-demethylase as potential antifungal agents: Design, synthesis and molecular docking studies. Farmacia 2016, 64, 390–397. [Google Scholar]
- Crișan, O.; Marc, G.; Nastasă, C.; Oniga, S.D.; Vlase, L.; Pîrnău, A.; Oniga, O. Synthesis and In Silico Approaches of New Symmetric Bis-Thiazolidine-2,4-Diones As Ras And Raf Oncoproteins Inhibitors. Farmacia 2023, 71, 254–263. [Google Scholar] [CrossRef]
- Vieira, T.F.; Sousa, S.F. Comparing AutoDock and Vina in Ligand/Decoy Discrimination for Virtual Screening. Appl. Sci. 2019, 9, 4538. [Google Scholar] [CrossRef]
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of Molecular Docking Programs for Virtual Screening against Dihydropteroate Synthase. J. Chem. Inf. Model. 2009, 49, 444–460. [Google Scholar] [CrossRef]
- Liu, B.; Chen, F.; Bi, C.; Wang, L.; Zhong, X.; Cai, H.; Deng, X.; Niu, X.; Wang, D. Quercitrin, an Inhibitor of Sortase A, Interferes with the Adhesion of Staphylococcal aureus. Molecules 2015, 20, 6533–6543. [Google Scholar] [CrossRef]
- Wang, L.; Shi, S.-H.; Li, H.; Zeng, X.-X.; Liu, S.-Y.; Liu, Z.-Q.; Deng, Y.-F.; Lu, A.-P.; Hou, T.-J.; Cao, D.-S. Reducing false positive rate of docking-based virtual screening by active learning. Brief. Bioinform. 2023, 24, bbac626. [Google Scholar] [CrossRef]
- Khanjiwala, Z.; Khale, A.; Prabhu, A. Docking structurally similar analogues: Dealing with the false-positive. J. Mol. Graph. Model. 2019, 93, 107451. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Gharpure, A.; Teng, J.; Zhuang, Y.; Howard, R.J.; Zhu, S.; Noviello, C.M.; Walsh, R.M.; Lindahl, E.; Hibbs, R.E. Shared structural mechanisms of general anaesthetics and benzodiazepines. Nature 2020, 585, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Maresca, A.; Temperini, C.; Vu, H.; Pham, N.B.; Poulsen, S.-A.; Scozzafava, A.; Quinn, R.J.; Supuran, C.T. Non-Zinc Mediated Inhibition of Carbonic Anhydrases: Coumarins Are a New Class of Suicide Inhibitors. J. Am. Chem. Soc. 2009, 131, 3057–3062. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Watanabe, E.; Kokubo, H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations. J. Comput. Aided. Mol. Des. 2017, 31, 201–211. [Google Scholar] [CrossRef]
- Liu, X.; Shi, D.; Zhou, S.; Liu, H.; Liu, H.; Yao, X. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 2018, 13, 23–37. [Google Scholar] [CrossRef]
- Verma, R.; Boshoff, H.I.M.; Arora, K.; Bairy, I.; Tiwari, M.; Varadaraj, B.G.; Shenoy, G.G. Synthesis, evaluation, molecular docking, and molecular dynamics studies of novel N-(4-[pyridin-2-yloxy]benzyl)arylamine derivatives as potential antitubercular agents. Drug Dev. Res. 2020, 81, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Makeneni, S.; Thieker, D.F.; Woods, R.J. Applying Pose Clustering and MD Simulations to Eliminate False Positives in Molecular Docking. J. Chem. Inf. Model. 2018, 58, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.F.; Jensen, A.A.; Bunch, L. From Methaqualone and Beyond: Structure—Activity Relationship of 6-, 7-, and 8-Substituted 2,3-Diphenyl-quinazolin-4(3H)-ones and in Silico Prediction of Putative Binding Modes of Quinazolin-4(3H)-ones as Positive Allosteric Modulators of GABAA Receptor. ACS Chem. Neurosci. 2020, 11, 4362–4375. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Parra-Cruz, R.; Jäger, C.M.; Lau, P.L.; Gomes, R.L.; Pordea, A. Rational Design of Thermostable Carbonic Anhydrase Mutants Using Molecular Dynamics Simulations. J. Phys. Chem. B 2018, 122, 8526–8536. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Ali, A.; Warsi, M.H.; Rahman, M.A.; Ahsan, M.J.; Azam, F. Toward the Discovery of a Novel Class of Leads for High Altitude Disorders by Virtual Screening and Molecular Dynamics Approaches Targeting Carbonic Anhydrase. Int. J. Mol. Sci. 2022, 23, 5054. [Google Scholar] [CrossRef] [PubMed]
- Shilkar, D.; Mohd Siddique, M.U.; Bua, S.; Yasmin, S.; Patil, M.; Timiri, A.K.; Supuran, C.T.; Jayaprakash, V. Carbonic anhydrase inhibitory activity of phthalimide-capped benzene sulphonamide derivatives. J. Enzym. Inhib. Med. Chem. 2023, 38, 2235089. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Lv, Z.; Wang, H.S.; Niu, X.D. Molecular dynamics simulations reveal insight into key structural elements of aaptamines as sortase inhibitors with free energy calculations. Chem. Phys. Lett. 2013, 585, 171–177. [Google Scholar] [CrossRef]
- Jin, H.; Zhou, Z.; Wang, D.; Guan, S.; Han, W. Molecular Dynamics Simulations of Acylpeptide Hydrolase Bound to Chlorpyrifosmethyl Oxon and Dichlorvos. Int. J. Mol. Sci. 2015, 16, 6217–6234. [Google Scholar] [CrossRef] [PubMed]
- Abbas, S.; Nasir, H.H.; Zaib, S.; Ali, S.; Mahmood, T.; Ayub, K.; Tahir, M.N.; Iqbal, J. Carbonic anhydrase inhibition of Schiff base derivative of imino-methyl-naphthalen-2-ol: Synthesis, structure elucidation, molecular docking, dynamic simulation and density functional theory calculations. J. Mol. Struct. 2018, 1156, 193–200. [Google Scholar] [CrossRef]
- Mushtaque, M.; Avecilla, F.; Ahmad, I.; Alharbi, A.M.; Khan, P.; Ahamad, S.; Hassan, M.I. 5-Fluorouracil (5-FU)-based Aza-Michael addition product: A selective carbonic anhydrase IX inhibitor. J. Mol. Struct. 2021, 1231, 129977. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | GABAA Receptor | Carbonic Anhydrase II | ||||||
|---|---|---|---|---|---|---|---|---|
| AutoDock | AutoDock Vina | AutoDock | AutoDock Vina | |||||
| Global ΔGmin (kcal/mol) | The Most Populated Cluster | Global ΔGmin (kcal/mol) | Global ΔGmin (kcal/mol) | The Most Populated Cluster | Global ΔGmin (kcal/mol) | |||
| Cluster ΔGmin (kcal/mol) | No. Poses | Cluster ΔGmin (kcal/mol) | No. Poses | |||||
| “a” series | ||||||||
| 1a | −10.07 | −9.59 | 96 | −8.65 | −7.50 | −6.19 | 30 | −6.49 |
| 2a | −9.67 | −9.67 | 47 | −9.17 | −6.69 | −6.42 | 70 | −6.27 |
| 3a | −9.91 | −9.31 | 70 | −9.40 | −7.22 | −6.50 | 31 | −6.50 |
| 4a | −9.95 | −9.35 | 87 | −9.64 | −7.07 | −6.51 | 35 | −6.62 |
| 5a | −10.88 | −10.58 | 68 | −10.16 | −7.99 | −7.75 | 20 | −7.02 |
| 6a | −10.28 | −10.10 | 35 | −9.99 | −7.28 | −6.78 | 67 | −6.04 |
| 7a | −11.43 | −11.43 | 96 | −11.19 | −7.90 | −5.85 | 43 | −6.25 |
| 8a | −9.97 | −9.97 | 22 | −9.72 | −7.48 | −6.84 | 55 | −6.30 |
| 9a | −8.18 | −7.48 | 63 | −8.09 | −6.07 | −5.56 | 115 | −6.09 |
| “b” series | ||||||||
| 1b | −10.29 | −9.55 | 39 | -9.40 | −7.86 | −7.49 | 28 | −7.20 |
| 2b | −10.40 | −10.17 | 77 | −10.11 | −7.53 | −7.27 | 54 | −7.15 |
| 3b | −10.66 | −10.54 | 90 | −10.10 | −8.01 | −6.74 | 25 | −7.33 |
| 4b | −10.70 | −10.50 | 69 | −9.98 | −8.09 | −6.65 | 24 | −7.45 |
| 5b | −11.41 | −11.08 | 59 | −10.22 | −8.62 | −7.81 | 22 | −8.10 |
| 6b | −9.99 | −9.85 | 42 | −9.55 | −7.69 | −7.31 | 30 | −7.15 |
| 7b | −10.21 | −9.12 | 53 | −10.01 | −8.24 | −6.50 | 34 | −7.25 |
| 8b | −11.27 | −10.84 | 51 | −10.55 | −8.70 | −7.39 | 43 | −7.26 |
| 9b | −9.12 | −8.66 | 79 | −7.93 | −6.07 | −5.56 | 115 | −6.09 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pele, R.; Marc, G.; Mogoșan, C.; Apan, A.; Ionuț, I.; Tiperciuc, B.; Moldovan, C.; Araniciu, C.; Oniga, I.; Pîrnău, A.; et al. Synthesis, In Vivo Anticonvulsant Activity Evaluation and In Silico Studies of Some Quinazolin-4(3H)-One Derivatives. Molecules 2024, 29, 1951. https://doi.org/10.3390/molecules29091951
Pele R, Marc G, Mogoșan C, Apan A, Ionuț I, Tiperciuc B, Moldovan C, Araniciu C, Oniga I, Pîrnău A, et al. Synthesis, In Vivo Anticonvulsant Activity Evaluation and In Silico Studies of Some Quinazolin-4(3H)-One Derivatives. Molecules. 2024; 29(9):1951. https://doi.org/10.3390/molecules29091951
Chicago/Turabian StylePele, Raluca, Gabriel Marc, Cristina Mogoșan, Anamaria Apan, Ioana Ionuț, Brîndușa Tiperciuc, Cristina Moldovan, Cătălin Araniciu, Ilioara Oniga, Adrian Pîrnău, and et al. 2024. "Synthesis, In Vivo Anticonvulsant Activity Evaluation and In Silico Studies of Some Quinazolin-4(3H)-One Derivatives" Molecules 29, no. 9: 1951. https://doi.org/10.3390/molecules29091951
APA StylePele, R., Marc, G., Mogoșan, C., Apan, A., Ionuț, I., Tiperciuc, B., Moldovan, C., Araniciu, C., Oniga, I., Pîrnău, A., Vlase, L., & Oniga, O. (2024). Synthesis, In Vivo Anticonvulsant Activity Evaluation and In Silico Studies of Some Quinazolin-4(3H)-One Derivatives. Molecules, 29(9), 1951. https://doi.org/10.3390/molecules29091951