
meso-Tetrahexyl-7,8-dihydroxychlorin and Its Conversion to ß-Modified Derivatives

 and
and
Abstract
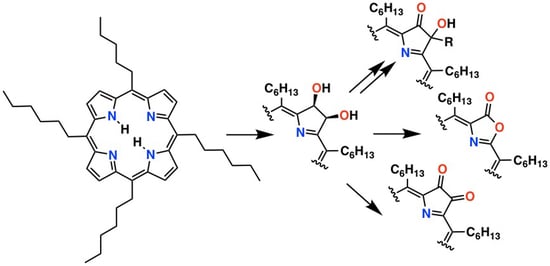
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. The Osmylation of meso-Tetrahexylporphyrin 7
2.2. The Transformations of meso-Tetrahexyl-7,8-dihydroxychlorin 8
2.3. Direct Oxidations of meso-Tetrahexylporphyrin
2.4. The Transformations of meso-Tetrahexylchlorin-7,8-dione 10
3. Materials and Methods
3.1. Materials
3.2. Instruments
3.3. General Procedures
3.3.1. General Procedure A: Hydride Reduction
3.3.2. General Procedure B: CTAP Oxidation
3.3.3. General Procedure C: DMP Oxidation
3.4. Osmium Tetroxide-Mediated Dihydroxylation of meso-Tetrahexylporphyrin (7): Formation of meso-Tetrahexyl-7,8-cis-dihydroxychlorin (8) and meso-Tetrahexyl-7,8,7,18-cis-tetrahydroxybacteriochlorin (9)
3.5. DMP Oxidation of meso-Tetrahexyl-7,8-cis-dihydroxychlorin (8) or meso-Tetrahexylchlorin-7-one (11): Formation of meso-Tetrahexylporphyrin-7,8-dione (10)
3.6. Dehydration of meso-Tetrahexyl-7,8-cis-dihydroxychlorin (8): Formation of meso-Tetrahexylchlorin-7-one (11)
3.7. Hydride Reduction of meso-Tetrahexylchlorin-7-one (11): Formation of meso-Tetrahexyl-7-hydroxychlorin (12)
3.8. CTAP Oxidation of meso-Tetrahexylporphyrin (7) or meso-Tetrahexyl-7,8-cis-dihydroxychlorin (8): Formation of meso-Tetrahexylporpholactone (meso-Tetrahexyl-7-oxo-8-oxa-porphyrin) (13)
3.9. DMP Oxidation of meso-Tetrahexylporphyrin (7): Formation of 5-(1′-oxo-hexyl)-10,15,20-trihexylporphyrin (14)
3.10. CTAP Oxidation of 5,15-Dihexylporphyrin (15): Formation of 5,15-Dihexyl-3-oxo-2-oxa-porphyrin) (16A) and 5,15-Dihexyl-7-oxo-8-oxa-porphyrin) (16B)
3.11. Hydride reduction of meso-Tetrahexylporphyrin-7,8-dione (10): Formation of meso-Tetrahexyl-7,8-trans-dihydroxychlorin (17)
3.12. Methyl-Grignard Addition to meso-Tetrahexylporphyrin-7,8-dione (10): Formation of meso-Tetrahexyl-8-hydroxy-8-methyl-chlorin-7-one (18) and meso-Tetrahexyl-7,8-dihydroxy-7,8-dimethyl-chlorin (19)
3.13. Trifluoromethylation of meso-Tetrahexylporphyrin-7,8-dione (10): Formation of meso-Tetrahexyl-8-hydroxy-8-trifluoromethyl-chlorin-7-one (18F)
3.14. Hydride Reduction of meso-Tetrahexyl-8-hydroxy-8-methyl-chlorin-7-one (18): Formation of meso-Tetrahexyl-7,8-dihydroxy-8-methyl-chlorin (20)
3.15. Hydride Reduction of meso-Tetrahexyl-8-hydroxy-8-trifluoromethyl-chlorin-7-one (18F): meso-Tetrahexyl-7,8-dihydroxy-8-trifluoromethyl-chlorin (20F)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scheer, H. An Overview of Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions, and Applications. In Chlorophylls and Bacteriochlorophylls; Grimm, B., Porra, R.J., Rüdiger, W., Scheer, H., Eds.; Springer: Dordrecht, The Netherlands, 2006; pp. 1–26. [Google Scholar]
- Liu, Y.; Zhang, S.; Lindsey, J.S. Total synthesis campaigns toward chlorophylls and related natural hydroporphyrins–diverse macrocycles, unrealized opportunities. Nat. Prod. Rep. 2018, 35, 879–901. [Google Scholar] [CrossRef] [PubMed]
- Ethirajan, M.; Chen, Y.; Joshi, P.; Pandey, R.K. The role of porphyrin chemistry in tumor imaging and photodynamic therapy. Chem. Soc. Rev. 2011, 40, 340–362. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yu, S.; Wang, X.; Qian, Y.; Wu, W.; Zhang, S.; Zheng, B.; Wei, G.; Gao, S.; Cao, Z.; et al. High Affinity of Chlorin e6 to Immunoglobulin G for Intraoperative Fluorescence Image-Guided Cancer Photodynamic and Checkpoint Blockade Therapy. ACS Nano 2019, 13, 10242–10260. [Google Scholar] [CrossRef] [PubMed]
- Sutton, P.A.; van Dam, M.A.; Cahill, R.A.; Mieog, S.; Polom, K.; Vahrmeijer, A.L.; van der Vorst, J. Fluorescence-guided surgery: Comprehensive review. BJS Open 2023, 7, zrad049. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, G.; Zeng, Z.; Pu, K. Activatable molecular probes for fluorescence-guided surgery, endoscopy and tissue biopsy. Chem. Soc. Rev. 2022, 51, 566–593. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.K. Synthetic strategies in designing porphyrin-based photosensitizers for photodynamic therapy. In CRC Handbook of Organic Photochemistry and Photobiology, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2004; pp. 144/1–144/21. [Google Scholar]
- Zhi, D.; Yang, T.; O’Hagan, J.; Zhang, S.; Donnelly, R.F. Photothermal therapy. J. Control. Release 2020, 325, 52–71. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, M. Photoantimicrobials—So what’s stopping us? Photodiagn. Photodyn. Ther. 2009, 6, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Wiehe, A.; O’Brien, J.M.; Senge, M.O. Trends and targets in antiviral phototherapy. Photochem. Photobiol. Sci. 2019, 18, 2565–2612. [Google Scholar] [CrossRef] [PubMed]
- Panda, M.K.; Ladomenou, K.; Coutsolelos, A.G. Porphyrins in bio-inspired transformations: Light-harvesting to solar cell. Coord. Chem. Rev. 2012, 256, 2601–2627. [Google Scholar] [CrossRef]
- Jiang, J.; Matula, A.J.; Swierk, J.R.; Romano, N.; Wu, Y.; Batista, V.S.; Crabtree, R.H.; Lindsey, J.S.; Wang, H.; Brudvig, G.W. Unusual Stability of a Bacteriochlorin Electrocatalyst under Reductive Conditions. A Case Study on CO2 Conversion to CO. ACS Catal. 2018, 8, 10131–10136. [Google Scholar] [CrossRef]
- Brückner, C.; Samankumara, L.; Ogikubo, J. Syntheses of Bacteriochlorins and Isobacteriochlorins. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: River Edge, NJ, USA, 2012; Volume 17, pp. 1–112. [Google Scholar]
- Lindsey, J.S. De Novo Synthesis of Gem-Dialkyl Chlorophyll Analogues for Probing and Emulating Our Green World. Chem. Rev. 2015, 115, 6534–6620. [Google Scholar] [CrossRef] [PubMed]
- Borbas, K.E. Chlorins. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: River Edge, NY, USA, 2016; Volume 36, pp. 1–149. [Google Scholar]
- Taniguchi, M.; Lindsey, J.S. Synthetic Chlorins, Possible Surrogates for Chlorophylls, Prepared by Derivatization of Porphyrins. Chem. Rev. 2017, 117, 344–535. [Google Scholar] [CrossRef] [PubMed]
- Jux, N.; Montforts, F.-P.; Haake, E. Bacteriochlorins and Isobacteriochlorins (Tetrahydroporphyrins), and Hexahydroporphyrins. In Science of Synthesis Knowledge Updates; Georg Thieme Verlag KG: Stuttgart, Germany, 2022; pp. 111–141. [Google Scholar] [CrossRef]
- Fischer, H.; Eckoldt, H. Überführung von Porphyrinen in Dioxy-chlorine durch Einwirkung von Osmiumtetroxyd. Liebigs Ann. Chem. 1940, 544, 138–162. [Google Scholar] [CrossRef]
- Pandey, R.K.; Isaac, M.; MacDonald, I.; Medforth, C.J.; Senge, M.O.; Dougherty, T.J.; Smith, K.M. Pinacol-Pinacolone Rearrangements in vic-Dihydroxychlorins and Bacteriochlorins: Effect of Substituents at the Peripheral Positions. J. Org. Chem. 1997, 62, 1463–1472. [Google Scholar] [CrossRef]
- Adams, K.R.; Bonnett, R.; Burke, P.J.; Salgado, A.; Valles, M.A. Cleavage of (octaethyl-2,3-dihydroxychlorinato)nickel(II) to give the novel 2,3-dioxo-2,3-secochlorin system. J. Chem. Soc. Perkin Trans. 1 1997, 1769–1772. [Google Scholar] [CrossRef]
- Brückner, C.; Rettig, S.J.; Dolphin, D. Formation of a meso-Tetraphenylsecochlorin and a Homoporphyrin with a Twist. J. Org. Chem. 1998, 63, 2094–2098. [Google Scholar] [CrossRef]
- Sutton, J.M.; Fernandez, N.; Boyle, R.W. Functionalized diphenylchlorins and bacteriochlorins: Their synthesis and bioconjugation for targeted photodynamic therapy and tumour cell imaging. J. Porphyr. Phthalocyanines 2000, 4, 655–658. [Google Scholar] [CrossRef]
- Rancan, F.; Wiehe, A.; Nöbel, M.; Senge, M.O.; Omari, S.A.; Böhm, F.; John, M.; Röder, B. Influence of substitutions on asymmetric dihydroxychlorins with regard to intracellular uptake, subcellular localization and photosensitization of Jurkat cells. J. Photochem. Photobiol. B Biol. 2005, 78, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Hyland, M.A.; Morton, M.D.; Brückner, C. meso-Tetra(pentafluorophenyl)porphyrin-derived Chromene-annulated Chlorins. J. Org. Chem. 2012, 77, 3038–3048. [Google Scholar] [CrossRef]
- Bruhn, T.; Brückner, C. Origin of the Regioselective Reduction of Chlorins. J. Org. Chem. 2015, 80, 4861–4868. [Google Scholar] [CrossRef]
- Lalisse, R.F.; Hadad, C.M.; Brückner, C.; Guberman-Pfeffer, M.J. [3 + 2]-Cycloadditions with Porphyrin β,β′-Bonds: Theoretical Basis of the Counterintuitive meso-Aryl Group Influence on the Rates of Reaction. J. Org. Chem. 2022, 87, 16473–16482. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.K.; Sotiriou, C. Migratory aptitudes in pinacol rearrangement of vic-dihydroxychlorins. J. Heterocycl. Chem. 1985, 22, 1739–1741. [Google Scholar] [CrossRef]
- Starnes, S.D.; Rudkevich, D.M.; Rebek Jr., J. Cavitand-Porphyrins. J. Am. Chem. Soc. 2001, 123, 4659–4669. [Google Scholar] [CrossRef] [PubMed]
- Brückner, C. The Breaking and Mending of meso-Tetraarylporphyrins: Transmuting the Pyrrolic Building Blocks. Acc. Chem. Res. 2016, 49, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Luciano, M.P.; Atoyebi, A.O.; Tardie, W.; Zeller, M.; Brückner, C. Pyrrole-Modified Porphyrins Containing Eight-Membered Heterocycles Using a Reversal of the “Breaking and Mending” Strategy. J. Org. Chem. 2020, 85, 15273–15286. [Google Scholar] [CrossRef] [PubMed]
- Egorov, G.D.; Solov’ev, V.N.; Shul’ga, A.M. PMR spectra of symmetrical meso-substituted porphyrins and chlorine. Theor. Exp. Chem. 1975, 11, 77–86. [Google Scholar]
- Ulman, A.; Gallicci, J.; Fisher, D.; Ibers, J.A. Facile Synthesis of Tetraalkylchlorin and Tetraalkylporphyrin Complexes and Comparison to the Structures of the Tetramethylchlorin and Tetramethylporphyrin Complexes of Ni(II). J. Am. Chem. Soc. 1980, 102, 6852–6854. [Google Scholar] [CrossRef]
- Frydman, L.; Olivieri, A.C.; Diaz, L.E.; Frydman, B.; Morin, F.G.; Mayne, C.L.; Grant, D.M.; Adler, A.D. High-Resolution Solid-State 13C NMR Spectra of Porphine and 5,10,15,20-Tetraalkylporphyrins: Implications for the N-H Tautomerization Process. J. Am. Chem. Soc. 1988, 110, 336–342. [Google Scholar] [CrossRef]
- Geier, G.R.; Lindsey, J.S. Effects of aldehyde or dipyrromethane substituents on the reaction course leading to meso-substituted porphyrins. Tetrahedron 2004, 60, 11435–11444. [Google Scholar] [CrossRef]
- Smith, B.M.; Kean, S.D.; Wyatt, M.F.; Graham, A.E. Indium Triflate Mediated Synthesis of meso-Substituted Porphyrins. Synlett 2008, 1953–1956. [Google Scholar] [CrossRef]
- DiMagno, S.G.; Williams, R.A.; Therien, M.J. Facile Synthesis of meso-Tetrakis(perfluoroalkyl)porphyrins: Spectroscopic Properties and X-ray Crystal Structure of Highly Electron-Deficient 5,10,15,20-Tetrakis(heptafluoropropyl)porphyrin. J. Org. Chem. 1994, 59, 6943–6948. [Google Scholar] [CrossRef]
- Senge, M.O.; Bischoff, I.; Nelson, N.Y.; Smith, K.M. Synthesis, reactivity and structural chemistry of 5,10,15,20-tetraalkylporphyrins. J. Porphyr. Phthalocyanines 1999, 3, 99–116. [Google Scholar] [CrossRef]
- Feng, X.; Senge, M.O. One-pot synthesis of functionalized asymmetric 5,10,15,20-substituted porphyrins from 5,15-diaryl- or -dialkyl-porphyrins. Tetrahedron 2000, 56, 587–590. [Google Scholar] [CrossRef]
- Feng, X.; Senge, M.O. An efficient synthesis of highly functionalized asymmetric porphyrins with organolithium reagents. J. Chem. Soc. Perkin Trans. 1 2001, 1, 1030–1038. [Google Scholar] [CrossRef]
- Nam, D.T.; Ivanova, Y.B.; Puhovskaya, S.G.; Kruk, M.M.; Syrbu, S.A. Acid–base equilibria and coordination chemistry of the 5,10,15,20-tetraalkyl-porphyrins: Implications for metalloporphyrin synthesis. RSC Adv. 2015, 5, 26125–26131. [Google Scholar] [CrossRef]
- Hiroto, S.; Osuka, A. Meso-Alkyl-Substituted meso-meso Linked Diporphyrins and meso-Alkyl-Substituted meso-meso, b-b, b-b Triply Linked Diporphyrins. J. Org. Chem. 2005, 70, 4054–4058. [Google Scholar] [CrossRef]
- Paliteiro, C.; Sobral, A. Electrochemical and spectroelectrochemical characterization of meso-tetra-alkyl porphyrins. Electrochim. Acta 2005, 50, 2445–2451. [Google Scholar] [CrossRef]
- Wiehe, A.; Shaker, Y.M.; Brandt, J.C.; Mebs, S.; Senge, M.O. Lead structures for applications in photodynamic therapy. Part 1: Synthesis and variation of m-THPC (Temoporfin) related amphiphilic A2BC-type porphyrins. Tetrahedron 2005, 61, 5535–5564. [Google Scholar] [CrossRef]
- Dahms, K.; Senge, M.O.; Bakri Bakar, M. Exploration of meso-substituted formyl porphyrins and their Grignard and Wittig reactions. Eur. J. Org. Chem. 2007, 2007, 3833–3848. [Google Scholar] [CrossRef]
- Senge, M.O.; Shaker, Y.M.; Pintea, M.; Ryppa, C.; Hatscher, S.S.; Ryan, A.; Sergeeva, Y. Synthesis of meso-Substituted ABCD-Type Porphyrins by Functionalization Reactions. Eur. J. Org. Chem. 2010, 2010, 237–258. [Google Scholar] [CrossRef]
- Plamont, R.; Kikkawa, Y.; Takahashi, M.; Kanesato, M.; Giorgi, M.; Chan Kam Shun, A.; Roussel, C.; Balaban, T.S. Nanoscopic Imaging of meso-Tetraalkylporphyrins Prepared in High Yields Enabled by Montmorrilonite K10 and 3Å Molecular Sieves. Chem.–Eur. J. 2013, 19, 11293–11300. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, H.; Osuka, A.; Yamamoto, Y.; Tokuji, S.; Tanaka, T. Palladium-Catalyzed Tetraarylation of 5,15-Dialkylporphyrins with Aryl Bromides. Heterocycles 2014, 88, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Król, A.; Plamont, R.; Canard, G.; Edzang, J.A.; Gryko, D.T.; Balaban, T.S. An Efficient Synthesis of Porphyrins with Different meso Substituents that Avoids Scrambling in Aqueous Media. Chem.–Eur. J. 2015, 21, 1488–1498. [Google Scholar] [CrossRef] [PubMed]
- Reimers, J.R.; Panduwinata, D.; Visser, J.; Chin, Y.; Tang, C.; Goerigk, L.; Ford, M.J.; Baker, M.; Sum, T.J.; Coenen, M.J.J.; et al. From Chaos to Order: Chain-Length Dependence of the Free Energy of Formation of meso-Tetraalkylporphyrin Self-Assembled Monolayer Polymorphs. J. Phys. Chem. C 2016, 120, 1739–1748. [Google Scholar] [CrossRef]
- Jiang, X.-L.; Damunupola, D.; Brückner, C. meso-Tetra(dioxanyl)porphyrins: Neutral, low molecular weight, and chiral porphyrins with solubility in aqueous solutions. J. Porphyr. Phthalocyanines 2021, 25, 734–740. [Google Scholar] [CrossRef]
- Aicher, D.; Wiehe, A.; Stark, C.B.W.; Albrecht, V.; Gräfe, S. Preparation of β-Functionalized Dihydroxy-Chlorins for Photodynamic Therapy. WO 2012012809, 26 January 2012. [Google Scholar]
- Aicher, D.; Wiehe, A.; Stark, C.B.W.; Albrecht, V.; Gräfe, S. Application of Beta-Functionalized Dihydroxy-Chlorins for PDT. U.S. Patent 20130041307, 24 April 2013. [Google Scholar]
- Gallucci, J.C.; Swepston, P.N.; Ibers, J.A. The Structures of (5,10,15,20-Tetramethylporphyrinato)nickel(II) and (5,10,15,20-Tetramethylchlorinato)nickel(II). Acta Cryst. Sect. B 1982, 38, 2134–2139. [Google Scholar] [CrossRef]
- Ulman, A.; Fisher, D.; Ibers, J.A. Synthesis of some 5,10,15,20-tetraalkylchlorin and tetraalkylporphyrin complexes of transition metals. J. Heterocycl. Chem. 1982, 19, 409–413. [Google Scholar] [CrossRef]
- Gouterman, M. Optical spectra and electronic structure of porphyrins and related rings. In The Porphyrins; Dolphin, D., Ed.; Academic Press: New York, NY, USA, 1978; Volume 3, pp. 1–165. [Google Scholar]
- Richter, A.M.; Waterfield, E.; Jain, A.K.; Sternberg, E.D.; Dolphin, D.; Levy, J.G. In vitro evaluation of phototoxic properties of four structurally related benzoporphyrin derivatives. Photochem. Photobiol. 1990, 52, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Boyle, R.W.; Dolphin, D. Structure and biodistribution relationships of photodynamic sensitizers. Photochem. Photobiol. 1996, 64, 469–485. [Google Scholar] [CrossRef]
- Samankumara, L.P.; Zeller, M.; Krause, J.A.; Brückner, C. Syntheses, structures, modification, and optical properties of meso-tetraaryl-2,3-dimethoxychlorin, and two isomeric meso-tetraaryl-2,3,12,13-tetrahydroxybacteriochlorins. Org. Biomol. Chem. 2010, 8, 1951–1965. [Google Scholar] [CrossRef]
- Hyland, M.A.; Hewage, N.; Walton, K.; Nimthong Roldan, A.; Zeller, M.; Samaraweera, M.; Gascon, J.A.; Brückner, C. Chromene-annulated Bacteriochlorins. J. Org. Chem. 2016, 81, 3603–3618. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.W.; Williams, S.C.; Jenkins, H.A.; Brückner, C. Oxidation of meso-tetraphenyl-2,3-dihydroxychlorin: Simplified synthesis of ß,ß’-dioxochlorins. Tetrahedron Lett. 2003, 44, 4045–4049. [Google Scholar] [CrossRef]
- Brückner, C.; Dolphin, D. 2,3-vic-Dihydroxy-meso-tetraphenylchlorins from the osmium tetroxide oxidation of meso-tetraphenylporphyrin. Tetrahedron Lett. 1995, 36, 3295–3298. [Google Scholar] [CrossRef]
- Dean, M.L.; Schmink, J.R.; Leadbeater, N.E.; Brückner, C. Microwave-promoted insertion of Group 10 metals into free base porphyrins and chlorins: Scope and limitations. Dalton Trans. 2008, 1341–1345. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Medforth, C.J.; Smith, K.M.; Alderfer, J.; Dougherty, T.J.; Pandey, R.K. Effect of meso-Substituents on the Osmium Tetraoxide Reaction and Pinacol-Pinacolone Rearrangement of the Corresponding vic-Dihydroxyporphyrins. J. Org. Chem. 2001, 66, 3930–3939. [Google Scholar] [CrossRef]
- Crossley, M.J.; Harding, M.M.; Sternhell, S. Tautomerism in 2-substituted 5,10,15,20-tetraphenylporphyrins. J. Am. Chem. Soc. 1986, 108, 3608–3613. [Google Scholar] [CrossRef]
- Crossley, M.J.; Harding, M.M.; Sternhell, S. Tautomerism in 2-hydroxy-5,10,15,20-tetraphenylporphyrin: An equilibrium between enol, keto, and aromatic hydroxyl tautomers. J. Org. Chem. 1988, 53, 1132–1137. [Google Scholar] [CrossRef]
- Burns, D.H.; Li, Y.H.; Shi, D.C.; Delaney, M.O. C-H Bond activation by alumina: Facile hydroxylation of chlorins at their saturated b-carbon by molecular oxygen and alumina. Chem. Commun. 1998, 1677–1678. [Google Scholar] [CrossRef]
- McCarthy, J.R.; Jenkins, H.A.; Brückner, C. Free Base meso-Tetraaryl-morpholinochlorins and Porpholactone from meso-Tetraaryl-2,3-dihydroxy-chlorin. Org. Lett. 2003, 5, 19–22. [Google Scholar] [CrossRef]
- Brückner, C.; Ogikubo, J.; McCarthy, J.R.; Akhigbe, J.; Hyland, M.A.; Daddario, P.; Worlinsky, J.L.; Zeller, M.; Engle, J.T.; Ziegler, C.J.; et al. Oxazolochlorins. 6. meso-Arylporpholactones and their Reduction Products. J. Org. Chem. 2012, 77, 6480–6494. [Google Scholar] [CrossRef]
- Hewage, N.; Daddario, P.; Lau, K.S.F.; Guberman-Pfeffer, M.J.; Gascón, J.A.; Zeller, M.; Lee, C.O.; Khalil, G.E.; Gouterman, M.; Brückner, C. Bacterio- and Isobacteriodilactones by Stepwise or Direct Oxidations of meso-Tetrakis(pentafluorophenyl)porphyrin. J. Org. Chem. 2019, 84, 239–256. [Google Scholar] [CrossRef] [PubMed]
- Thuita, D.; Damunupola, D.; Brückner, C. Oxazolochlorins 21. Most Efficient Access to meso-Tetraphenyl- and meso-Tetrakis(pentafluorophenyl)porpholactones, and Their Zinc(II) and Platinum(II) Complexes. Molecules 2020, 25, 4351. [Google Scholar] [CrossRef] [PubMed]
- Crossley, M.J.; King, L.G. Novel heterocyclic systems from selective oxidation at the β-pyrrolic position of porphyrins. J. Chem. Soc. Chem. Commun. 1984, 920–922. [Google Scholar] [CrossRef]
- Gouterman, M.; Hall, R.J.; Khalil, G.E.; Martin, P.C.; Shankland, E.G.; Cerny, R.L. Tetrakis(pentafluorophenyl)porpholactone. J. Am. Chem. Soc. 1989, 111, 3702–3707. [Google Scholar] [CrossRef]
- Jayaraj, K.; Gold, A.; Austin, R.N.; Ball, L.M.; Terner, J.; Mandon, D.; Weiss, R.; Fischer, J.; DeCian, A.; Bill, E.; et al. Compound I and Compound II Analogues from Porpholactones. Inorg. Chem. 1997, 36, 4555–4566. [Google Scholar] [CrossRef] [PubMed]
- Köpke, T.; Pink, M.; Zaleski, J.M. Elucidation of the extraordinary 4-membered pyrrole ring-contracted azeteoporphyrinoid as an intermediate in chlorin oxidation. Chem. Commun. 2006, 4940–4942. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Lv, H.; Ke, X.; Yang, B.; Zhang, J.-L. Ruthenium-Catalyzed Oxidation of the Porphyrin ß-ß’-Pyrrolic Ring: A General and Efficient Approach to Porpholactones. Adv. Synth. Catal. 2012, 354, 3509–3516. [Google Scholar] [CrossRef]
- Arnold, L.; Müllen, K. Modifying the Porphyrin Core—A Chemist’s Jigsaw. J. Porphyr. Phthalocyanines 2011, 15, 757–779. [Google Scholar] [CrossRef]
- Brückner, C.; Akhigbe, J.; Samankumara, L. Syntheses and Structures of Porphyrin Analogues Containing Non-pyrrolic Heterocycles. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: River Edge, NJ, USA, 2014; Volume 31, pp. 1–276. [Google Scholar]
- Thuita, D.W.; Brückner, C. Metal Complexes of Porphyrinoids Containing Nonpyrrolic Heterocycles. Chem. Rev. 2022, 122, 7990–8052. [Google Scholar] [CrossRef]
- Heravi, M.M.; Momeni, T.; Zadsirjan, V.; Mohammadi, L. Applications of the Dess-Martin Oxidation in Total Synthesis of Natural Products. Curr. Org. Synth. 2021, 18, 125–196. [Google Scholar] [CrossRef]
- Shetgaonkar, S.E.; Jothish, S.; Dohi, T.; Singh, F.V. Iodine(V)-Based Oxidants in Oxidation Reactions. Molecules 2023, 28, 5250. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Jin, G.-Q.; Zhang, J.-L. Porpholactone Chemistry: An Emerging Approach to Bioinspired Photosensitizers with Tunable Near-Infrared Photophysical Properties. Acc. Chem. Res. 2019, 52, 2620–2633. [Google Scholar] [CrossRef] [PubMed]
- Hewage, N.; Damunupola, D.; Zeller, M.; Brückner, C. Direct Oxidations of meso-Tetrakis(pentafluorophenyl)porphyrin: Porphotrilactones and Entry into a Non-biological Porphyrin Degradation Pathway. J. Org. Chem. 2024, 89, 6584–6589. [Google Scholar] [CrossRef] [PubMed]
- Crossley, M.J.; Burn, P.L.; Langford, S.J.; Pyke, S.M.; Stark, A.G. A New Method for the Synthesis of Porphryin-alpha-diones that is Applicable to the Synthesis of Trans-annular Extended Porphyrin Systems. J. Chem. Soc. Chem. Commun. 1991, 1567–1568. [Google Scholar] [CrossRef]
- Akhigbe, J.; Brückner, C. Expansion of a Pyrrole in meso-Tetraphenylporphyrin to a Pyrazine Imide Moiety Using a Beckmann Rearrangement. Eur. J. Org. Chem. 2013, 2013, 3876–3884. [Google Scholar] [CrossRef]
- Banerjee, S.; Zeller, M.; Brückner, C. meso-Tetraphenylporphyrin-derived Oxypyriporphyrin, Oxypyrichlorin, and Thiomorpholinochlorin, as their Ni(II) Complexes. J. Porphyr. Phthalocyanines 2012, 16, 576–588. [Google Scholar] [CrossRef]
- Ruppert, I.; Schlich, K.; Volbach, W. Die ersten CF3-substituierten organyl(chlor)silane. Tetrahedron Lett. 1984, 25, 2195–2198. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Krishnamurti, R.; Olah, G.A. Synthetic methods and reactions. 141. Fluoride-induced trifluoromethylation of carbonyl compounds with trifluoromethyltrimethylsilane (TMS-CF3). A trifluoromethide equivalent. J. Am. Chem. Soc. 1989, 111, 393–395. [Google Scholar] [CrossRef]
- Hewage, N.; Zeller, M.; Brückner, C. Oxidations of chromene-annulated chlorins. Org. Biomol. Chem. 2017, 15, 396–407. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 6th ed.; Butterworth-Heinemann: Burlington, MA, USA, 2009. [Google Scholar]
- Furniss, B.S.; Hannaford, A.J.; Smith, P.W.G.; Tatchell, A.R. Vogel’s Textbook of Practical Organic Chemistry, 5th ed.; Longman: Essex, UK, 1989; p. 549. [Google Scholar]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aicher, D.; Damunupola, D.; Stark, C.B.W.; Wiehe, A.; Brückner, C. meso-Tetrahexyl-7,8-dihydroxychlorin and Its Conversion to ß-Modified Derivatives. Molecules 2024, 29, 2144. https://doi.org/10.3390/molecules29092144
Aicher D, Damunupola D, Stark CBW, Wiehe A, Brückner C. meso-Tetrahexyl-7,8-dihydroxychlorin and Its Conversion to ß-Modified Derivatives. Molecules. 2024; 29(9):2144. https://doi.org/10.3390/molecules29092144
Chicago/Turabian StyleAicher, Daniel, Dinusha Damunupola, Christian B. W. Stark, Arno Wiehe, and Christian Brückner. 2024. "meso-Tetrahexyl-7,8-dihydroxychlorin and Its Conversion to ß-Modified Derivatives" Molecules 29, no. 9: 2144. https://doi.org/10.3390/molecules29092144
APA StyleAicher, D., Damunupola, D., Stark, C. B. W., Wiehe, A., & Brückner, C. (2024). meso-Tetrahexyl-7,8-dihydroxychlorin and Its Conversion to ß-Modified Derivatives. Molecules, 29(9), 2144. https://doi.org/10.3390/molecules29092144