Results and Discussion
Despite the availability of a number of methods for the synthesis of various phenazine derivatives [
8], direct nucleophilic substitution of the hydrogen atom, which is characteristic of azine quaternary salts [
9] and usually proceeds under mild conditions, is the most interesting. The reactivity of phenazine derivatives in this reaction can be estimated by comparing the charge electron density (kinetic control) and nucleophilic localization energies (thermodynamic control) [
10]. According to the literature data on the distribution of the electron density on an N-methylphenazinium salt, substitution of the hydrogen atom should occur at positions 2 or 4 [
11]. However, the substitution at position 4 is thermodynamically unfavorable and hence only the hydrogen atom in position 2 should be substituted, which fully agrees with the experimental results [
7,
8].
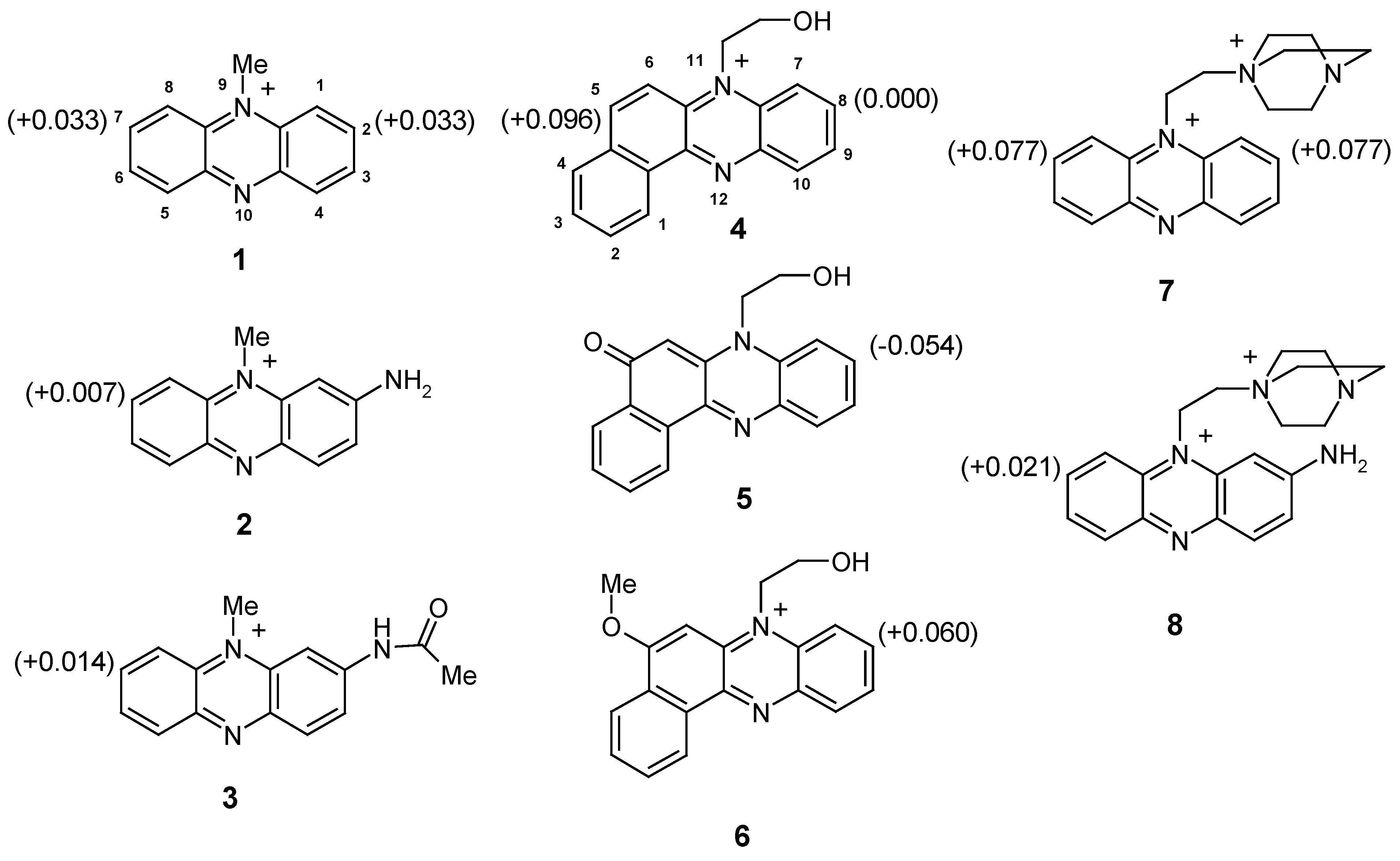
To estimate the relative reactivity of substituted quaternary phenazinium salts, we calculated the charge distribution for positions 2 and 7 in compounds
1-3,
7 and
8 and for positions 5 and 8 in benzo[a]phenazinium derivatives
4-6 (
Figure 1).
These PM3 quantum-mechanical calculations were performed for free cations in vacuo because, under real conditions of nucleophilic substitution of hydrogen, a nucleophile attacks either a solvated cation or an ionic pair separated by solvent molecules. The contribution of solvation to the charge distribution may be taken, in a first approximation, as the same for all quaternary phenazinium salts.
The calculations show that the introduction of the electron-donor amino group at position 2 of a quaternary salt (compound
2) significantly decreases the positive charge in the symmetrical position 7, thus precluding introduction of a second substituent. This suggests that the phenazine ring can be activated for nucleophilic substitution of hydrogen in 2-amino derivatives by acylating the amino group (compound
3). Accordingly, in benzo[a]phenazinium salts the 8 position can be activated by alkylating the oxo group at position 5 (compound
6). This approach, based on a decrease in the donor effects of a substituent, has now been realized by us experimentally (
Scheme 1 and
Scheme 2).
The interaction of 9-methylphenazinium methylsulfate (
1) with methylamine and the methyl ester of
β-alanine easily gives 2-methylamino- and 2-[2-(methoxycarbonyl)ethyl]amino-9-methyl-phenazinium perchlorates (
9 and
10) in high yields (
Scheme 1). The electronic spectra of compounds
9 and
10 are similar to those characteristic of 2-aminophenazinium salts (cf.
Table 1 in the Experimental), and the
1H-NMR spectra display signals in the 6.5-6.7 ppm range with a pronounced coupling constant of
J=2 Hz that is characteristic of aromatic protons in the
meta-position.
As expected, acetylation of compounds 9 and 10 occurs with difficulty, and an acid catalyst is necessary to carry out the reaction. The best results were obtained with boron trifluoride etherate. The products, 9-methyl-2-N-methylacetamido- and 2-N-[2-(methoxycarbonyl)-ethyl)acetamido-9-methyl-phenazinium perchlorates (11 and 12) have spectral characteristics corresponding to the structures suggested. For example, in their 1H-NMR spectra, one can observe the downfield shift of the signals for the protons at position 1 by ~1.5 ppm as compared to compounds 9 and 10, which suggests an increase in the acceptor character of the phenazine cycle. However, insufficient stability precluded the isolation of the analytical samples of these compounds, and they were used in subsequent syntheses without additional purification.
The reaction of acetamides
11 and
12 with methylamine, morpholine, and methyl ester of
β-alanine were investigated. The reactivity of acetamides in these reactions was approximately one order of magnitude lower than that of the starting quaternary salt
1 (by TLC). Nevertheless, acetamide
11 is completely consumed in the reaction with methylamine even after 6 h at room temperature, and compounds
14 and
13, the 9-methyl-2,7-
bis-methylamino- and 9-methyl-2-
N-methylacetamido-7-methylaminophenazinium perchlorates, were isolated from the reaction mixture. Their spectral and analytical data correspond well to those expected. The formation of compound
14 in the course of the reaction attests to the fact that
13, the product of direct nucleophilic substitution, undergoes partial deacylation under the conditions chosen, probably, in a reaction with excess methylamine. This fact allows one to avoid an additional deacetylation stage by merely increasing the reaction time, though this leads to a certain decrease in the yield of the target product, which was exemplified with the synthesis of 9-methyl-2-methylamino-7-morpholinophenazinium perchlorate
16 (procedures
A and
B). 7-[2-(Methoxycarbonyl)ethyl]amino-9-methyl-2-methylaminophenazinium perchlorate (
15) was obtained by reaction of methyl ester of
β-alanine with acetamide
11 or of methylamine with acetamide
12 in 51 and 46 % yields, respectively. As was demonstrated by the chromatographic analysis of the reaction mixture, the relatively low yield of diamine
15 is probably due to aminolysis of the ester group and side reactions, which occur upon prolonged exposure of quaternary phenazinium salts to an excess of amines. The direction of these reactions were studied in more detail using the interaction of compound
11 with morpholine as an example. Along with the target compound
16, isolated in 36 % yield (procedure
B), a series of substituted phenazines was obtained, which were identified by mass spectrometry and
1H-NMR spectroscopy (
Table 2 in the Experimental). As can be seen from the table, the main side processes, which occur in the nucleophilic substitution of hydrogen in quaternary phenazinium salts, are dequaternization of the phenazinium salt and substitution of the N-methylacetamido group. When substituents resistant to acid hydrolysis are introduced, the reduction in the reaction time is justified as well as the involvement of an additional stage of acid hydrolysis (procedure
A for compound
16).
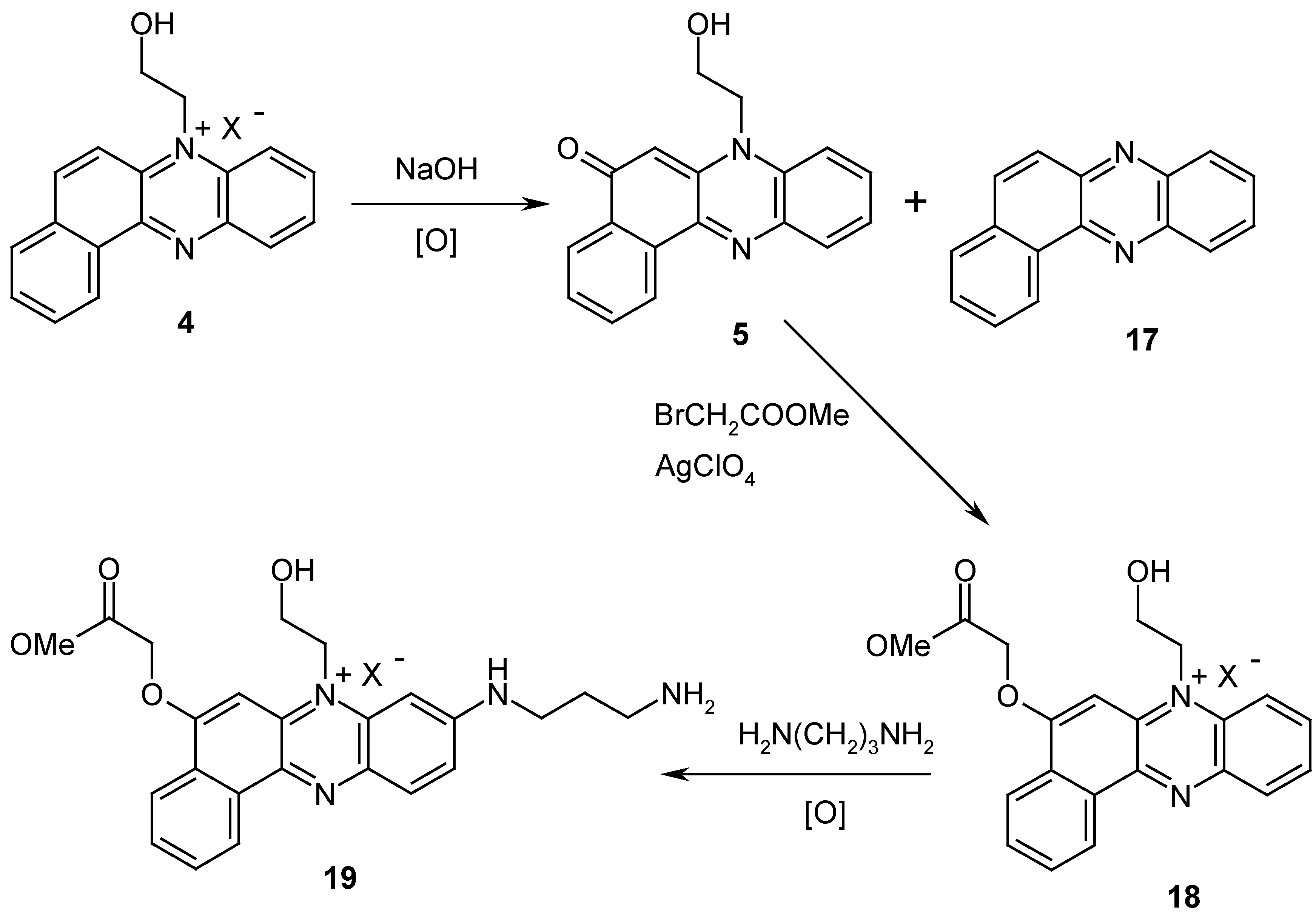
The general synthesis of disubstituted benzo[
a]phenazine derivatives is outlined in
Scheme 2. The interaction of compound
4 with sodium hydroxide gives 5-oxo-11-(2-hydroxyethyl)benzo[
a]phenazine (
5), admixed with benzo[
a]phenazine (
17). Further alkylation of compound
5 by methyl bromoacetate in the presence of silver perchlorate gives product
18. Interaction of
18 with 1,3-diaminopropane at room temperature gives 8-(3-aminopropyl)amino-5-[(methoxycarbonyl)-methyl]hydroxy-11-(2-hydroxyethyl)benzo[a]phenazinium perchlorate (
19) in high yield. Compounds
5 and
18 are stable and can be isolated, but in our case we have used them in subsequent reactions without isolation and purification.
It should be noted that the substitution in quaternary phenazinium salts proceeds
via an intermediate that is to be oxidized with oxygen. Thus, the preparative synthesis requires the use of low reactant concentrations and intense flow of air. Otherwise, a considerable amount of side products is formed. It should also be noted that according to the calculations, a similar or even more pronounced effect of activation can be attained by introducing an additional positive charge into the substituent at the quaternary nitrogen atom (cf.
Figure 1).
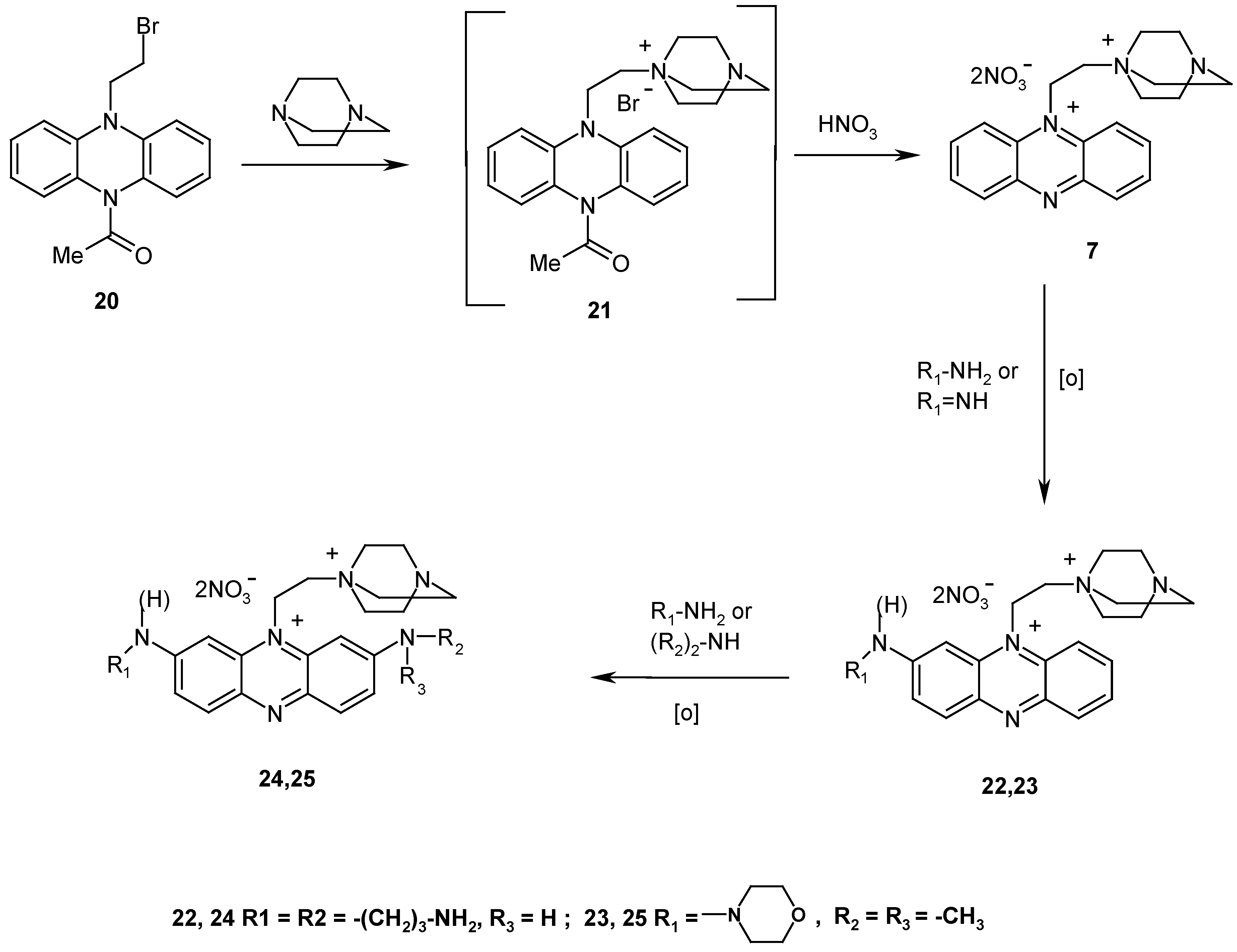
To check this assumption experimentally, we synthesized quaternary phenazinium salt
7, bearing an additional positive charge in the aliphatic fragment of the molecule, and studied its behavior in reactions with aliphatic amines. Salt
7 was synthesized from 9-acetyl-10-(2-bromoethyl)-9,10-dihydrophenazine (
20) [
12] (
Scheme 3). Treatment of compound
20 with an excess of 1,4-diazabicyclo[2.2.2]octane gives compound
21 in quantitative yield (TLC). The latter was treated
in situ with conc. HNO
3 to give dinitrate
7, solvated with two НNО
3 molecules. Prolonged storage of this solvate in vacuum over alkali reduces the content of НNО
3 to 1.2-1.5 acid molecules per molecule of compound
7. In the solid state this compound is stable but its aqueous and alcoholic solutions rapidly darken, especially when exposed to light.
The reactions of bisquaternary salt
7 with nucleophiles were studied using 1,3-diaminopropane, morpholine, and dimethylamine as examples. Reaction mixtures were analyzed by HPLC.
Figure 2 shows kinetics of the reaction of compound
7 with a tenfold excess of 1,3-diaminopropane. It can be seen that the content of a monosubstituted product (compound
22) in the reaction mixture (~95%) is a maximum after 30 min. The reaction rate at position 7 of product
22 is much lower, so that the percentage of the disubstituted product
24 does not exceed 4-5% in that time.
The reaction can be stopped in this stage to isolate the monosubstituted product and introduce another nucleophile, which allows one to obtain a product containing different substituents in position 2 and 7. The possibility of synthesizing asymmetrically substituted phenazine derivatives was exemplified in successive introduction of the morpholine and dimethylamine residues (compound
25,
Scheme 3).
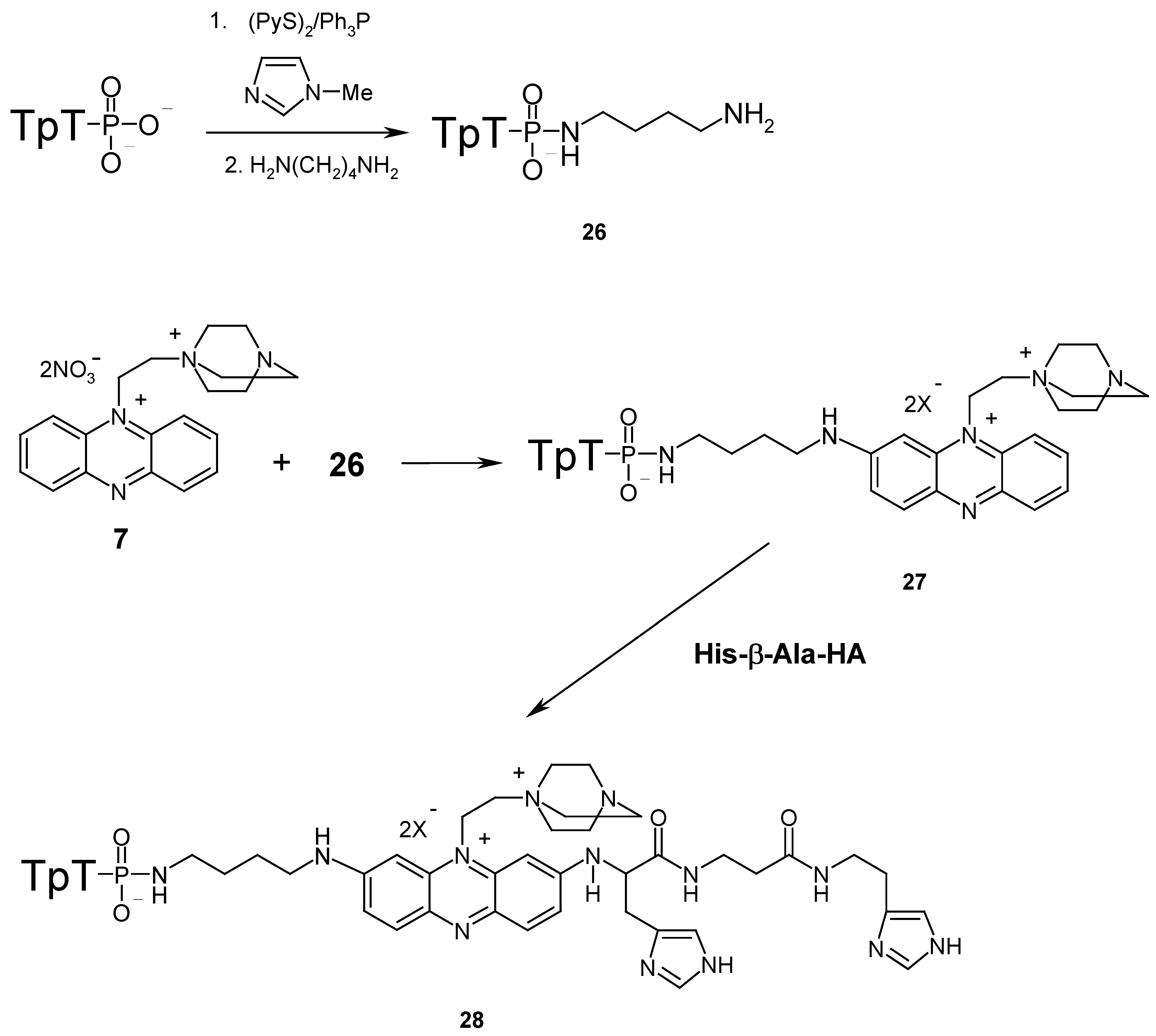
Here we also demonstrated the possibility of these compounds to react with different biopolymers. A dinucleotide (thymidylyl(3’→5’)thymidine phosphate tetrabutylammonium salt; pTpT) and tripeptide (histidyl-β-alanyl-histamine; His-β-Ala-HA) were used as models of oligonucleotides and peptides respectively (
Scheme 4). At the first stage of synthesis a dinucleotide derivative with a primary amino group was prepared. For this purpose a 1,4-diaminobutane residue was attached to the terminal phosphate group of pTpT which had been preliminary activated by triphenylphosphine and 4,4’-dipyridyl disulfide in the presence of 1-methylimidazole [
13]. The yield of this reaction was almost quantitative. Treatment of 26, the amino derivative of pTpT, with an excess of the phenazinium salt
7 gives compound
27 with a high yield (>95% HPLC). The electronic spectra of
27 have absorption maxima corresponding to the dinucleotide part at 267 nm and to the phenazinium part at 236, 293, 397 and 524 nm. His-β-Ala-HA was synthesized by condensation of histamine with the activated pentafluorophenyl esters of the corresponding BOC-protected amino acids using standard methods [
14].
Interaction of 27 with small excess of His-β-Ala-HA give conjugate 28 in good yield (64 % after HPLC). In the electronic spectra of 28 the maximums at 397 and 524 nm have disappeared and the new absorption maximum corresponding of disubstituted phenazinium salt (540 nm) has appeared. The mass spectrum (MALDI) of compound 28 contains peak [M-Li]+ (C58H77LiN17O16P2; 1336.68).
General
UV spectra were recorded on a Specord M-40 spectrophotometer. 1H-NMR spectra were recorded on a Bruker WR-200 spectrometer with HMDS as the internal standard. Mass spectra were run on Vision 2000 (MALDI) mass spectrometers. The course of the reactions was monitored and the purity of compounds was checked by HPLC on a Milikhrom-4 analytical liquid chromatograph (Lichrosorb RP-18 column, gradient 0-80% MeOH, 0.1% CF3COOH). Chromatograms were processed with CHROM software (A. S. Zenkov, Novosibirsk Institute of Bioorganic Chemistry, Siberian Branch of the Russian Academy of Sciences) by comparing peak areas at analytical wavelengths of 250 and 300 nm, with consideration of data obtained from chromatographing artificial mixtures of compounds under study. The following concentrations were used in kinetic experiments: 10-3 mol L-1 (derivatives of quaternary phenazinium salts) and 10-2 mol L-1 (propanediamine, methylamine, and morpholine) in MeOH. TLC was carried out on HPLC-Alufolien Kieselgel 60 plates in the system (v/v) t-BuOH-MeCOEt-HCOOH-H2O = 8:6:3:3. Melting points were measured in sealed capillaries on a Kristall setup, heating rate near the melting range 0.3 oC/min. Quantum-mechanical computations were performed for the ground state of cations by the PM3 method with geometry optimization (the PoIak-Ribiere algorithm, RHF calculation) with the HYPERCHEM program (version 4.5).
Table 1.
The electronic spectra of mono-, di- and unsubstituted phenazinium derivatives.
Table 1.
The electronic spectra of mono-, di- and unsubstituted phenazinium derivatives.
| Compound | λmax / nm (ε); EtOH |
|---|
| 4 | 223 (23400), 268 (16200), 295 (16200), 438 (10000) |
| 7 | 210 (31000), 262 (34000), 392 (10800), 450 (2300) |
| 9 | 238 (42500), 290 (39500), 365 (8000), 515 (19000) |
| 10 | 234 (32500), 290 (33000), 386 (11500), 518 (16500) |
| 11 | 214 (10200), 262 (25200), 410 (10600) |
| 12 | 212 (10000), 262 (23100), 415 (11100) |
| 13 | 240 (26500), 294 (27500), 385 (10000), 510 (16400) |
| 14 | 235 (5300), 270 (22800), 540 (27200) |
| 15 | 235 (5700), 271 (24700), 540 (27700) |
| 16 | 233 (6670), 280 (23100), 543 (28200) |
| 19 | 230 (16700), 274 (21900), 320 (13100), 390 (4600), 494 (9600) |
| 23 | 236 (15800), 294 (13800), 401 (4450), 540 (8110). |
| 24 | 231 (5160), 290 (25200), 543 (21400). |
| 25 | 231 (6170), 278 (23200), 543 (25400). |
9-Methyl-2-methylaminophenazinium perchlorate (9). 9-Methylphenazinium methylsulfate (1 g, 3.3 mmol) was dissolved in a solution of 10% methylamine in dried methanol (50 mL). The reaction mixture was vigorous stirred for 6 h. The product was precipitated with ester. The residue was dissolved in a minimum amount of water, and precipitated with a solution of LiClO4 (2 g) in 0.05 M HClO4 (10 mL). Yield 0.90 g (78 %); m.p. 177-180 °C; Rf 0.56; 1H-NMR (СD3СN) δ: 3.18 (d, 3 H, NНСН3, J= 5 Hz), 4.28 (s, 3 H, N+-CH3), 6.56 (d, 1 H, H(1), J =2 Hz), 7.50 (dd, 1 H, H(3), J1,3 = 2 Hz, J3,4 = 10 Hz); 7.65 (br.s, 1 H, NH), 7.90 (m. 2 H, H arom.), 8.12 (m, 3 H, H arom.); Anal. Found (%): C, 51.90; H, 4.33; N, 12.96; Calculated for C14H14ClN3O4 (%): C, 51.94; H, 4.36; N. 12.98.
2-[2-(Methoxycarbonyl)ethyl]amino-9-methylphenazinium perchlorate (10). Compound 10 was obtained from 9-methylphenazinium methylsulfate (1 g, 3.3 mmol) and the methyl ester of β-alanine (1.03 g, 10 mmol) as is described above, yield 0.80 g (62 %); m.p. > 220 °C (decomp.); Rf 0.64; 1H-NMR (СD3СN) δ: 2.81 (t, 2 H, СОСН2СН2, J= 6.5 Hz), 3.68 (s, 3 H, OCH3), 3.82 (t, 2 H, СОСН2СН2), 4.29 (s, 3 H. N+-CH3), 6.68 (d, 1 H. H(1), J = 2 Hz), 7.54 (dd. 1 H, H(3) J1,3 = 2 Hz, J3,4 = 10 Hz), 7.63 (br. s, 1 H, NH), 7.88 (m, 2 H, H arom.), 8.15 (m, 3 H, H arom.); Anal. Found (%): C, 51.51; H, 4.45; N, 10.60; Calculated for C17H18ClN3O6 (%): C, 51.59; H. 4.58: N. 10.62.
9-Methyl-2-N-methylacetamidophenaziniuin perchlorate (11). ВF3-Еt2О (0.5 mL, 5.4 mmol) was added to a solution of compound 9 (0.3 g. 0.9 mmol) in Ac2O (5 mL), and the reaction mixture was stirred at 20 °C for 24 h. Then ether was added until precipitation began, and the reaction mixture was stirred for 2 h. The crystals that formed were filtered off and washed with a small amount of Ac2O and then with ether. Compound 11 was further used without additional purification. Yield 0.25 g (62 %); m.p. > 200 °C (decomp.); Rf 0.25; 1H-NMR (CD3CN) δ: 2.34 (s, 3 H, C(O)CH3), 3.63 (s, 3 H. NCH3), 4.76 (s, 3 H, N+-CH3), 8.21 (m, 2 H, H arom.), 8.37 (d. 1 H, H(1), J = 2 Hz); 8.55 (m, 4 H, H arom.).
2-N-[2-(Methoxycarbony)ethyl]acetamido-9-methylphenazinium perchlorate (12). Compound 12 was obtained from 10 (1 g, 2.5 mmol) as is described above. Yield 0.45 g (40 %); m.p. 133-136 °C (decomp.); Rf 0.22;1H-NMR (CD3CN) δ: 2.27 (s, 3 H, C(O)CH3), 2.70 (t, 2 H, C(O)CH2CH2, J = 6.5 Hz), 3.55 (s, 3 H, OCH3), 4.28 (t, 2 H, C(O)CH2CH2N, J = 6.5 Hz), 4.79 (s, 3 H, N+-CH3), 8.25 (m, 2 H, H arom.), 8.37 (d, 1 H, H(1), J = 2 Hz), 8.57 (m, 4 H, H arom.).
9-Methyl-2,7-bismethylaminophenazinium perchlorate (14) and 9-methyl-2-N-methylacetamido-7-methylaminophenazinium perchlorate (13). Gaseous methylamine was passed through a suspension of compound 11 (0.2 g, 0.6 mmol) in dry methanol (10 mL) for 6 h. The precipitate was filtered off and washed with hot EtOH. The mother liquor was concentrated, dissolved in the minimal amount of water, and applied onto a 2x30 cm column containig a polymeric dextran gel (Molselect G-10). Compounds 13 and 14 were eluted in succession with water. The yield of compound 13 was 0.09 g (42 %); m.p. > 300 °C (decomp.); Rf 0.36; 1H-NMR (CD3CN) δ: 2.18 (s, 3 H, C(O)CH3), 3.29 (s, 3 H, HNCH3), 3.52 (s, 3 H, C(O)NCH3), 4.38 (s, 3 H, N+-CH3), 6.75 (d, 1 H, H(8), J6,8 = 2 Hz), 7.60 (dd, 1 H, H(6), J6,8 = 2 Hz, J6,5 = 10 Hz), 7.82 (dd, 1 H. H(3), J1,3 = 2 Hz, J3,4 = 9 Hz), 8.00 (d, 1 H, H(4), J4,3 =9 Hz), 8.25 (d, 1 H, H(1), J1,3 = 2 Hz), 8.34 (d. 1 H. H(5), J5,6 = 10 Hz); Anal. Found (%): С, 51.58; H, 4.80; N, 14.24; Calculated for C17H19ClN4O5 (%): С, 51.72; H, 4.85; N, 14.19. The yield of compound 14 was 0.07 g (36 %); m.p. 240-246 °C (decomp.); Rf 0.5; 1H-NMR (СD3СN) δ: 3.13 (s, 6 H, HNCH3), 4.20 (s, 3 H, N+-CH3), 6.70 (d, 2 H, H(l) and Н(8), J = 2 Hz), 7.35 (dd, 2 H, H(3) and H(6), J1,3 (6,8) = 2Hz, J4,3 (5,6) = 10 Hz), 7.86 (d, 2 H, H(4) and H(5), J4,3 (5,6) = 10 Hz); Anal. Found (%): С. 51.00; H, 4.82; N, 15.90; Calculated for C15H17ClN4O4 (%): С, 51.07; H, 4.86; N, 15.88.
7-[2-(Methoxycarbonyl)ethyl]amino-9-methyl-2-methylaminophenazinium perchlorate (15).
Method A: Methyl ester of β-alanine hydrochloride (0.74 g, 0.53 mmol) and Et3N (0.1 mL, 0.7 mmol) were added to a suspension of compound 11 (0.1 g, 0.3 mmol) in dry MeOH (5 mL), and the reaction mixture was stirred for 48 h. The precipitate was filtered off and washed with hot methanol, and the solvent was concentrated. The residue was dissolved in methanol, and compound 15 was precipitated with ether. After crystallization from 0.1 M HClO4, the yield of compound 15 was 0.06 g (51 %); m.p. > 170 °C (decomp.); Rf 0.45; 1H-NMR (DMF-d7) δ: 2.35 (t, 2 H, NСН2СH2С(О), J = 6.5 Hz), 2.72 (t, 2 H, NСН2СH2С(О), J = 6.5 Hz), 3.16 (s, 3H, HNCH3), 3.72 (s, 3 H, OCH3), 4.32 (s, 3 H, N+-CH3), 6.56 (d, 1 H, H(1), J1,3 = 2 Hz), 6.76 (d, 1 H, H(8), J8,6 = 1.5 Hz), 7.36 (dd, 1 H, H(3), J1,3 = 2 Hz, J4,3 = 9 Hz), 7.48 (dd, 1 H, H(6), J8,6 = 1.5 Hz, J6,5 =10.2 Hz), 7.92 (m, 2 H, H(4) and H(5)); Anal. Found (%): C, 50.76; H, 4.95; N, 13.21; Calculated for C18H21ClN4O6 (%): C, 50.89; H, 4.98; N, 13.19.
Method B: Gaseous methylamine was passed through a suspension of compound 12 (0.1 g, 0.2 mmol) in anhydrous methanol (10 mL) for 6 h. The reaction mixture was allowed to stand for 72 h. and then processed as in procedure A. The yield of compound 12 was 0.045 g (46 %).
9-Methyl-2-methylamino-7-morpholinophenazinium perchlorate (16).
Method A: Morpholine (0.1 mL, 1.2 mmol) was added to a suspension of compound 11 (0.1 g, 0.3 mmol) in dry methanol (10 mL) and the reaction mixture was stirred for 3 h. The precipitate was filtered off and washed with hot methanol. The filtrate was concentrated, and the residue was dissolved in methanol. A mixture of compound 16 and 9-methyl-2-/V-methylacetamido-7-morpholino-phenazinium perchlorate was precipitated with ether. The sediment was dissolved with heating in 0.1 M HClO4 (5 mL) and refluxed for 2 h. On cooling, compound 16 (0.085 g. 71 %) precipitated, m.p. 205-212 °C (decomp.); Rf 0.29; 1H-NMR (DMF-d7) δ: 3.02 (d. 3 H, НNСН3, J = 4 Hz), 3.40 (t, 4 H, NCH2CH2O, J = 5.5 Hz), 3.69 (t, 4 H, NCH2CH2O, J = 5.5 Hz), 4.13 (s, 3 H, N+-СН3), 6.62 (d. 1 H, J1,3 = 2 Hz), 7.03 (d, 1 H, H(8). J8,6 = 2 Hz), 7.26 (dd, 1 H. H(3) J1,3 = 2 Hz, J4,3 = 9 Hz), 7.57 (dd, I H, H(6), J6,8 = 2 Hz, J5,6 = 10 Hz), 7.65 (br. d, 1 H. NH J = 4 Hz); 7.71 (d, 1 H, H(4), J4,3 = 9 Hz), 7.92 (d. 1 H. H(5), J6,5 = 10 Hz); Anal. Found (%): C, 52.92; H, 5.15; N, 13.72; Calculated for C18H21ClN4O5 (%): C, 52.88; H, 5.18; N, 13.70.
Method B: Morpholine (0.1 mL, 1.2 mmol) was added to a suspension of compound
11 (0.1 g, 0.3 mmol) in dry methanol (10 mL) and the reaction mixture was stirred for 72 h. The precipitate was filtered off and washed with hot methanol. The filtrate was concentrated, and the residue was dissolved in methanol. Compound
16 was precipitated with ether. After crystallization from 0.1
M HClO
4, the yield of compound
16 was 0,04 g (36 %). The mother liquor after precipitation with ether was concentrated, the residue was dissolved in a minimal amount of chloroform and chromatographed on a 1х20 cm column with Silasorb-600 (15—20 μm, detection at 350 nm) using gradient elution СНСl
3 → Cl
3-МеОН, 5:1. The main fractions were concentrated, and phenazine, 2-morpholinophenazine, 2-/V-methylacetamido-7-morpholino-phenazine, 2-/V-methylacetamidophenazine, and 9-methyl-phenazin-2-one were obtained in yields of 6-12 mg. The main characteristics of the compounds obtained are given in
Table 2.
Table 2.
The main by-products obtained in the reaction of compound 11 with morpholine.
Table 2.
The main by-products obtained in the reaction of compound 11 with morpholine.
| Compound | Molecular formula | Mcalc | Mexp | 1H-NMR (CDCl3), δ, J/Hz |
|---|
| Phenazine | C12H8N2 | 180.06847 | 180.06850 | 7.82 (q, 4 H, H arom., J=3); 8.24 (q, 4H, H arom., J=3) |
| 2-Morpholino-phenazine | C16H15N3O | 265.12150 | 265.12129 | 3.48 (t, 4 H, NCH2, J=4), 3.92 (t, 4H, OCH2, J=4), 7.39 (s, 1 H, H(1)), 7.73 (m, 4 H, arom.), 8.34 (m, 3 H, arom.) |
| 2-N-Methyl-acetamido-7- morpholino-phenazine | C19H20N4O2 | 336.15862 | 336.15856 | 2.07 (s, 3 H, COCH3), 3.43 (s, 3 H, NCH3), 3.51 (t, 4 H, NCH2, J=4), 3.92 (t, 4H, OCH2, J=4), 7.34 –8.19 (m, 6 H, arom.) |
| 2-N-Methylacetamido- phenazine | C15H13N3O | 251.10586 | 251.10603 | 2.07 (s, 3 H, COCH3), 3.43 (s, 3 H, NCH3), 7.86 (m, 2 H, arom.), 8.03 (d, 1 H, H(1) J=3), 8.25 (m, 4 H, arom.) |
| 9-methylphenazin-2-one | C13H10N2O | 210.07931 | 210.07971 | 3.84 (s, 3 H, NCH3), 6.07 (d 1 H, H(1) J=2), 6.89 (dd, 1 H, H(3), J=2, J=10), 7.48 (m, 2 H, arom.), 7.85 (m, 3 H, arom.) |
8-(3-Aminopropyl)amino-5-(methoxycarbonyl)methyloxy-11-(2-hydroxyethyl)benz[a]phenazinium perchlorate (19). A solution of 1 M NaOH (15 ml) was added under vigorous stirring to a solution of 11-(2-hydroxyethyl)benzo[a]phenazinium chloride (1 g, 3.2 mmol) in water (50 mL) and stirred for 3 h. The resulting precipitate was filtered and washed with water to pH 7. The precipitate (according to the TLC data, it contained 5-oxo-7-(2-hydroxyethyl)benzo[a]phenazine (5) with an admixture of benzo[a]phenazine) was dried and suspended in toluene (20 mL), methyl bromoacetate (2 mL) and silver perchlorate (20 ml of 0.1 M toluene solution) were added, and the mixture was stirred for 1 h. The precipitate was filtered, washed with ether and dissolved in methanol (50 mL). AgBr was filtered and washed with hot methanol (3x10 mL). The methanol was concentrated and 5-(methoxy-carbonyl)methyloxy-11-(2-hydroxyethyl)benzo[a]phenazinium perchlorate (18) (1.06 g, 2.5 mmol) was precipitated with ether. Then compound 18 was dissolved in methanol (25 mL) and treated with a solution of 1,3-diaminopropane (0.2 mL, 24 mmol) in water (25 mL). The mixture was vigorously stirred for 24 h. The reaction mixture was concentrated, diluted by water (20 mL) and mixed with 10% aqueous LiClO4 (5 mL). Compound 19 was filtered off and washed with cooled water. Yield: 0.80 g (46% overall); Rf 0.70; 1H-NMR (DMF-d7) δ: 2.20 (m, 2 H, СН2СН2СН2), 3.36 (t, 2 Н, СН2СН2NH2, J =7.5 Hz); 3.72 (t, 2 H, NHCH2, J =7.5 Hz); 3.78 (s, 3 H, OCH3); 4.30 (t, 2 H, CH2OH, J =7.5 Hz), 4.92 (s, 2 H, OCH2C(O)), 5.28 (t, 2 H, N+-CH2, J =7.5 Hz), 7.18 (s, 1 H, H(6)), 7.24 (d, 1 H, H(7), J =2 Hz), 7.88-8.42 (m, 5 H, H arom), 9.2 (dd, 1H, H(9), J7,9 = 2 Hz, J9,10 = 9 Hz); Anal. Found (%): C, 53.74; H, 5.15; N, 10.32; Calculated for C24H27ClN4O8 (%): C, 53,89; H, 5.09; N, 10.47.
9-[2-(4-Azaquinuclidino)ethyl]phenazinium dinitrate (7). 1,4-Diazabicyclo[2.2.2]octane (0.45 g, 4.0 mmol) was added to a solution of 9-acetyl-10-(2-bromoethyl)-9,10-dihydro-phenazine (20) (0.13 g, 0.4 mmol) in DMF (4 mL). The reaction mixture was stirred at 20 °C for one day. The solvent was evaporated, and the residue was washed with ether, dried, and suspended in water (5 mL). Conc. HNO3 (1 mL) was added, and the resulting solution was stirred at ~20 °C for 2 h. Then it was concentrated in vacuo, and the residue was dissolved in a minimum amount of water. Methanol was added until precipitation began. The crystals that formed were filtered off, washed with methanol and dried in vacuo to give the solvate, 4•2HNO3 (0.20 g, 88%); m.p. >140 °C (decomp., from water); Rf = 0.20; 1H-NMR (D2O) δ: 3.66 (m, 6 H, N(СН2-)3), 4.24 (m, 8 H, -CH2-N+-(CH2-)), 6.31 (m, 2 H, -CH2-N+phen), 8.30 (m, 2 H, H(4,5)), 8.69 (m, 4 H, Н(2,3,6,7)), 8.79 (m, 2 H, H(1,8)); Anal. Found (%): C, 41.9; H, 4.74; N, 19.5; Calculated for C20H24N6O6•2HNO3 (%): C, 42.11; H. 4.59; N, 19.64.
2,7-Bis(3-aminopropyl)amino-9-[2-(4-azaquinuclidino)ethyl]phenazinium dinitrate (24). 1,3-Diamino-propane (0.17 mL, 2.0 mmol) was added with vigorous stirring to a suspension of compound 4•2HNO3 (0.11 g, 0.2 mmol) in anhydrous MeOH (10 mL), and the mixture was stirred at 20 °C for 18 h. The solvent was evaporated in vacuo, and the residue was dissolved in a minimum amount of methanol. Compound 24 was precipitated with ether. Threefold reprecipitation with ether from methanol was followed by dissolution in a minimum amount of 9:1 H2O-MeOH mixture and chromatography (15х2 cm column, Silasorb S-2, 15 μm, elution with a 0 to 80% MeOH gradient of aqueous methanol). The main fraction with Rf = 0.24 was concentrated and reprecipitated with ether from methanol, yield 0.08 g (69%); m.p. >217 °C (decomp., from methanol); 1H-NMR (D2O) δ: 2.21 (t, 4 H, 2 CH2CH2NH2, J = 8 Hz), 3.23 (m 4 H, 2 CH2CH2CH2NH2,), 3.84 (m, 10 H, 2 NHCH2CH2, +N-(СН2-)3), 4.01 (m, 8 H, -CH2-N+-(CH2-)), 5.3 (m, 2 H, -CH2-N+phen), 6.61 (s, 2 H, H(1,8)), 7.83 (d, 2 H, Н(3,6), J3,4(6,5) = 9 Hz ), 7.88 (d, 2 H, H (4,5), J4,3(5,6) = 9 Hz).
2-Morpholino-9-[2-(4-azaquinuclidino)ethyl]phenazinium dinitrate (23). Morpholine (0.1 mL, 1.2 mmol) was added with vigorous stirring to a suspension of compound 4•2HNO3 (0.11 g, 0.2 mmol) in anhydrous MeOH (10 mL). Stirring was continued under air at 20 °C for 30 min. The reaction mixture was poured into ether. The precipitate that formed was reprecipitated twice with ether from methanol, yield 0.09 g (84%); m.p. >170 °C (decomp., from methanol); Rf. = 0.22; 1H-NMR (D2O) δ: 3.61 (t, 4 H, (-CH2-)2O, J =8 Hz ), 4.04 (m, 10 H, N(-CH2-)2, N(-CH2-)3), 4.16 (m, 8 H, -CH2-N+(-СН2-)3); 5.38 (m, 2 H, CH2-N+ phen); 6.87 (d, 1 H, H(1) J1,3 = 2 Hz); 8.19 (m, 6 H, H arom).
2-Dimethylamino-7-morpholino-9-[2-(4-azaquinuclidino)ethyl]phenazinium dinitrate (25). A 40% aqueous solution of dimethylamine (0.1 mL, 0.8 mmol) was added with vigorous stirring under air to a solution of compound 9 (0.05 g, ~0.1 mmol) in MeOH (5 mL). Stirring was continued at 20 °C for 18 h. The reaction mixture was concentrated in vacuo, and the solid residue was reprecipitated with ether from methanol, dissolved in a minimum amount of 9:1 H2O-MeOH mixture, and chromatographed (15х2 cm column, Silasorb C-2, 15 μm, elution with a 0 to 80% MeOH gradient of aqueous methanol). The main fraction with Rf = 0.36 was concentrated and reprecipitated with ether from methanol, yield 0.04 g, (70%); m.p. >156-162 °C (decomp., from methanol); 1H-NMR (D2O) δ: 3.34 (s, 6 H, -N(CH3)2), 3.56 (t, 4 H, (-СН2-)2О, J = 8 Hz), 3.90 (m, 10 H, -N(-CH2-)2, N(-CH2-)3), 4.27 (m, 8 H, -СН2-N+-(СН2-)3), 5.24 (m 2 H, -CH2-N+phen ), 6.53 (br s, 2 H, H(1,8)), 7.81 (d, 2 H, H(3,6), J3,4(6,5) = 9 Hz); 7.88 (d, 2 H, H(4,5), J4,3(5,6) = 9 Hz).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}