A Quantitative Structure-Property Relationship (QSPR) Study of Aliphatic Alcohols by the Method of Dividing the Molecular Structure into Substructure

Abstract
:1. Introduction
2. Methodology
2.1. Data Set
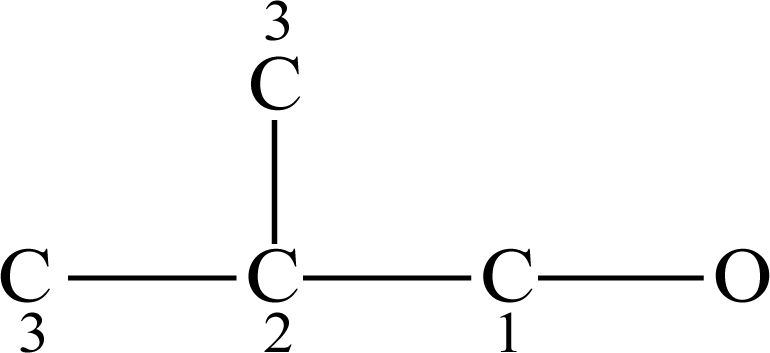
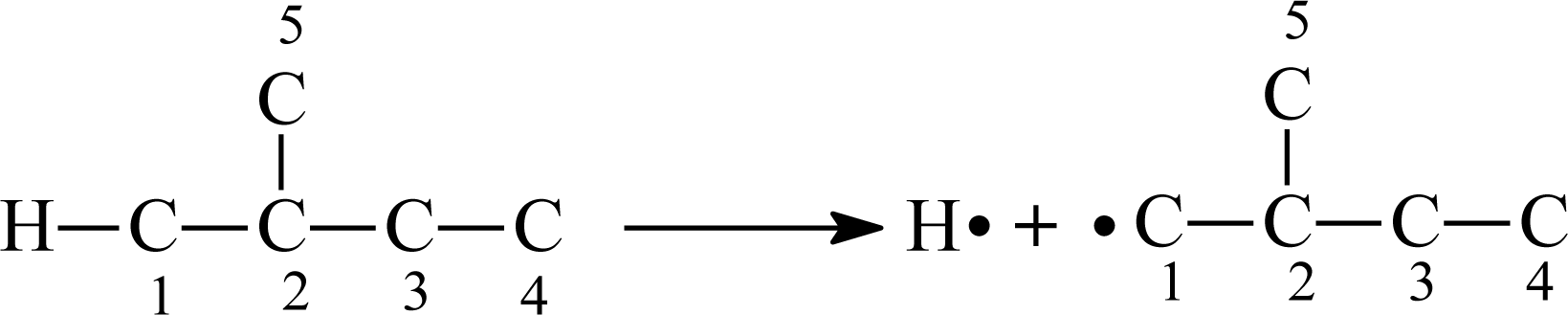
2.2. Definition of the Topological Indices
2.2.1. The Odd–Even Index OEI
2.2.2. The Molecular Polarizability Effect Index MPEI
2.2.3. Eigenvalues of Bond-Connecting Matrix (SX1CH)
- H: PEIH = 0
- R1: PEI1 = 1.2122+ 0.0481= 1.2603
3. Results and Discussion
3.1. Quantitative Structure-Retention Relationship (QSRR) Model for Alcohols on Stationary Phases of Different Polarity
3.2. Quantitative Structure-Property Relationship (QSPR) Model for BP of the Alcohols
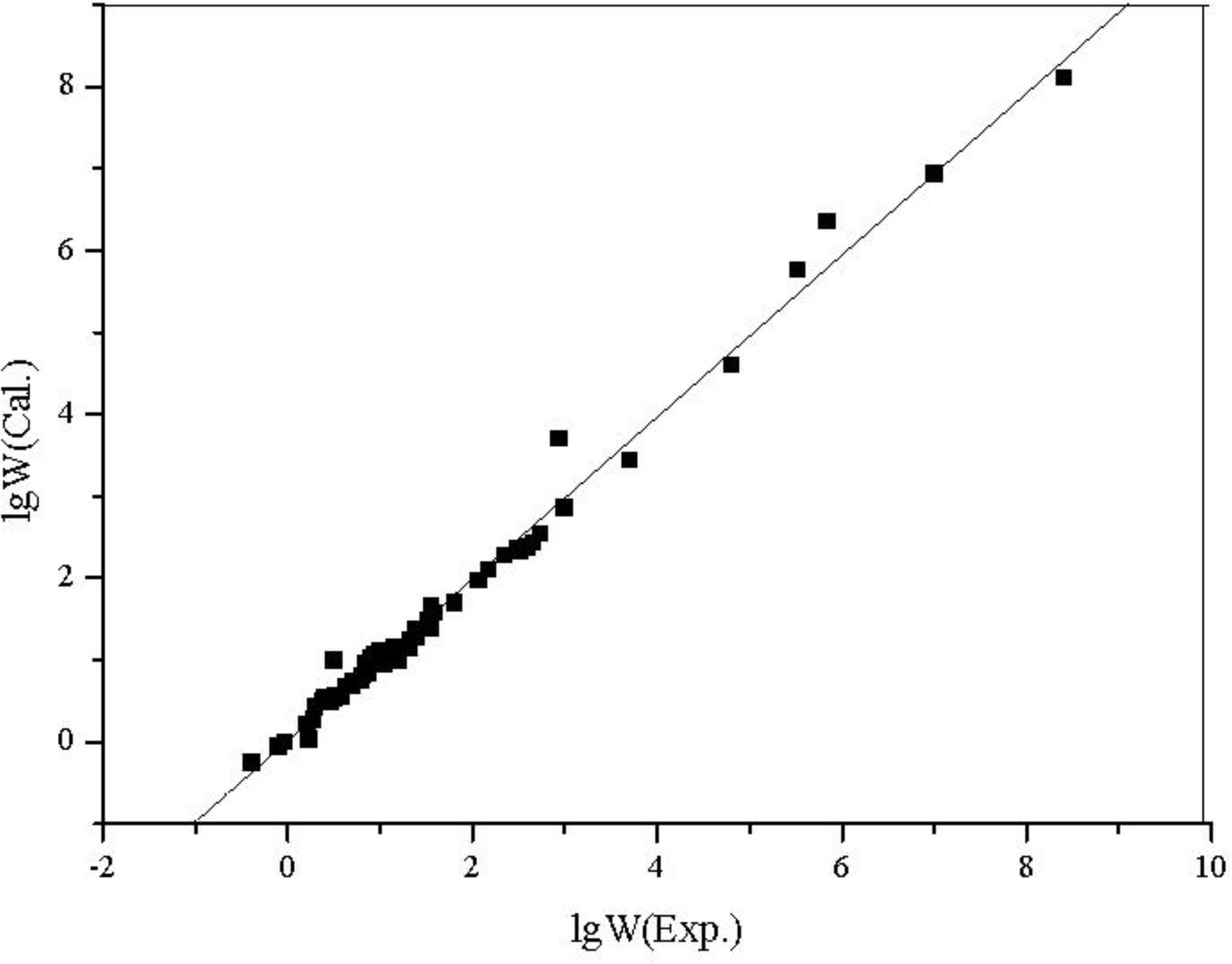
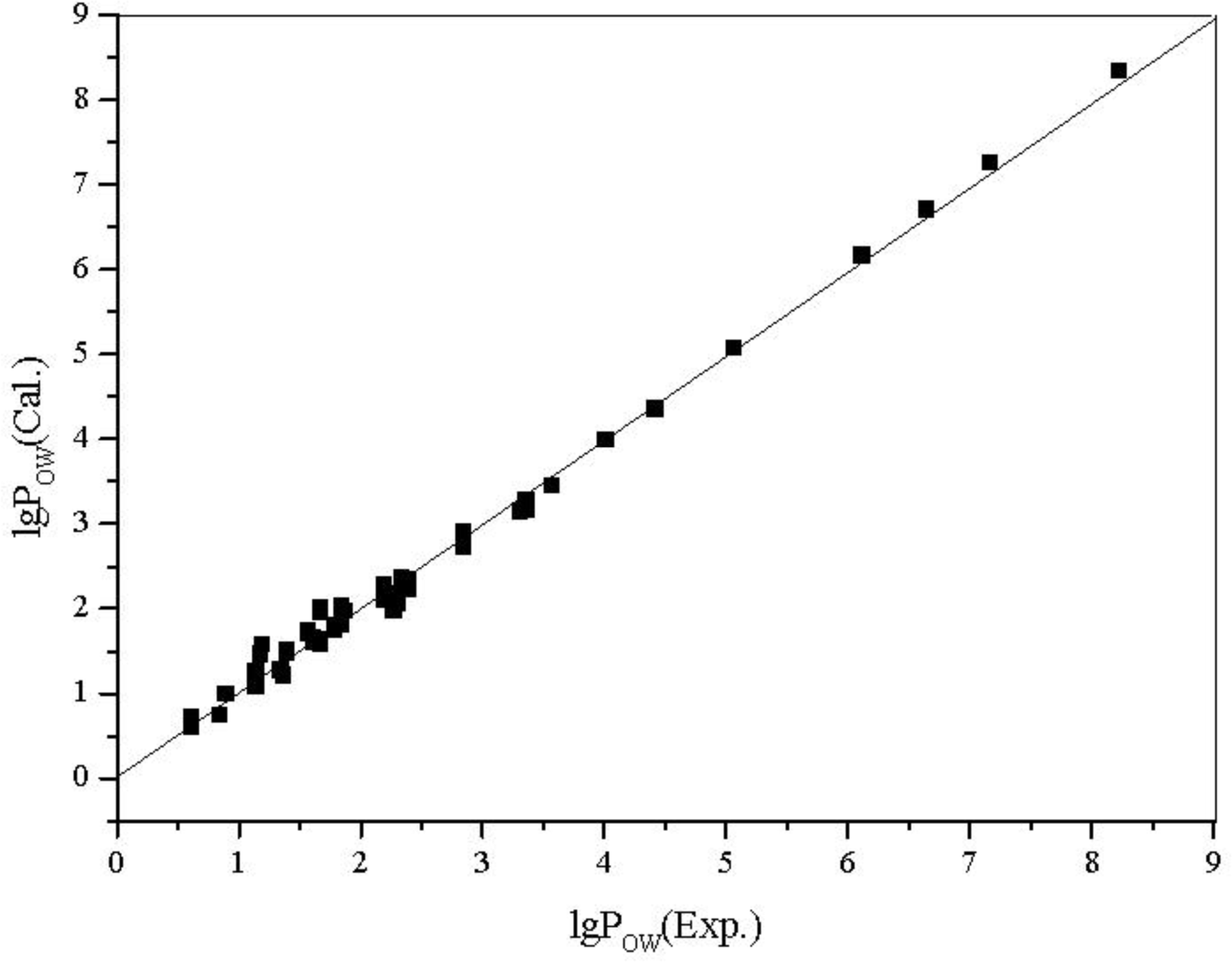
3.3. Quantitative Structure-Property Relationship (QSPR) Models for Water Solubility (lg W), n-Octanol/Water Partition Coefficients (lg POW) of the Alcohols
4. Conclusion
Acknowledgments
References
- Katritzky, AR; Petrukhin, R; Tatham, D. Interpretation of quantitative structure-property and activity relationships. J. Chem. Inf. Comput. Sci 2001, 41, 679–685. [Google Scholar]
- Katritzky, AR; Dobchev, DA; Slavov, S; Karelson, M. Legitimate utilization of large descriptor pools for QSPR/QSAR models. J. Chem. Inf. Model 2008, 48, 2207–2213. [Google Scholar]
- Delgrado, EJ; Alderete, JB; Gonzalo, AJ. A simple QSPR model for predicting soil sorption coefficients of polar and nonpolar organic compounds from molecular formula. J. Chem. Inf. Comput. Sci 2003, 43, 1928–1932. [Google Scholar]
- Katritzky, AR; Slavov, S; Dobchev, D; Karelson, M. Rapid QSPR model development technique for prediction of vapor pressure of organic compounds. Comput. Chem. Eng 2007, 31, 1123–1130. [Google Scholar]
- Souza, ES; Kuhen, CU; Junkes, BS; Yunes, RS; Heinzen, VEF. Modeling the semi-empirical electrotopological index in QSPR studies for aldehydes and ketones. J. Chemom 2009, 23, 229–235. [Google Scholar]
- Katritzky, AR; Fara, D; Karelson, M. QSPR of 3-aryloxazolidin-2-one antibacterials. Bioorg. Med. Chem 2004, 12, 3027–3035. [Google Scholar]
- Gramatica, P; Giani, E; Papa, E. Statistical external validation and consensus modeling: A QSPR case study for Koc prediction. J Mol Graph Model 2007, 25, 755–766. [Google Scholar]
- Laura, DH; David, SP; Florian, N; John, BO. Why are some properties more difficult to predict than others? A study of QSPR models of solubility, melting point, and Log P. J. Chem. Inf. Model 2008, 48, 220–232. [Google Scholar]
- Souza, ES; Kuhnen, CA; Junkes, BS; Yunes, RA; Heinzen, VEF. Development of semi-empirical electrotopological index using the atomic charge in QSPR/QSRR models for alcohols. J. Chemom 2010, 24, 149–157. [Google Scholar]
- Junkes, BS; Amboni, RDMC; Yunes, RA; Heinzen, VEF. Prediction of chromatographic retention of saturated alcohols on stationary phases of different polarity applying the novel semi-empirical topological index. Anal. Chim. Acta 2003, 477, 29–39. [Google Scholar]
- Beteringhe, A; Radutiu, AC; Culita, DC; Mischie, A; Spafiu, F. Quantitative structure-retention relationship (QSRR) study for predicting gas chromatographic retention times for some stationary phases. QSAR Comb. Sci 2008, 27, 996–1005. [Google Scholar]
- Ren, B. Novel atom-type AI indices for QSPR studies of alcohols. Comput. Chem 2002, 26, 223–235. [Google Scholar]
- Huuskonen, J. Prediction of soil sorption coefficient of organic pesticides from the atom-type electrotopological state indices. Environ. Toxicol. Chem 2003, 22, 816–820. [Google Scholar]
- Cao, CZ; Yuan, H. Topological indices based on vertex, distance, and ring: on the boiling points of paraffins and cycloalkanes. J. Chem. Inf. Comput. Sci 2001, 41, 867–877. [Google Scholar]
- Cao, CZ; Yuan, H. A new approach of evaluating bond dissociation energy from eigenvalue of bonding orbital-connection matrix for C–C and C–H bonds in alkane. J. Chem. Inf. Comput. Sci 2003, 43, 600–608. [Google Scholar]
- Liu, FP; Liang, YZ; Cao, CZ. QSPR modeling of thermal conductivity detection response factors for diverse organic compound. Chemom. Intell. Lab. Syst 2006, 81, 120–126. [Google Scholar]
- Liu, FP; Liang, YZ; Cao, CZ; Zhou, N. QSPR study of GC retention indices for saturated esters on seven stationary phases based on novel topological indices. Talanta 2007, 72, 1307–1315. [Google Scholar]
- Kabinyi, H. QSAR: Hansch Analysis and Related Approaches; Mannhold, R, Gaad-Larsen, PK, Timmerman, H, Eds.; VCH: Weinheim, Germany, 1993. [Google Scholar]
- Goodarzi, M; Freitas, MP. Predicting boiling points of aliphatic alcohols through multivariate image analysis applied to quantitative structure-property relationships. J. Phys. Chem. A 2008, 112, 11263–11265. [Google Scholar]
- Wang, LS; Han, SK. Molecular Structure, Property and Activity; Chemical Industry Press: Beijing, China, 1997; pp. 26–40. [Google Scholar]
- Guo, WQ; Lu, Y; Zheng, XM. The predicting study for chromatographic retention index of saturated alcohols by MLR and ANN. Talanta 2000, 51, 479–488. [Google Scholar]
- Weast, RC. CRC Handbook of Chemistry and Physics, 70th ed; CRC Press Inc: Boca Raton, FL, USA, 1989/1990. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ni | ΔPEI | Ni | ΔPEI | Ni | ΔPEI | Ni | ΔPEI |
|---|---|---|---|---|---|---|---|
| 1 | 1.00000 | 6 | 0.009052 | 11 | 0.002375 | 16 | 0.001073 |
| 2 | 0.140526 | 7 | 0.006388 | 12 | 0.001972 | 17 | 0.000945 |
| 3 | 0.048132 | 8 | 0.004748 | 13 | 0.001628 | 18 | 0.000838 |
| 4 | 0.023503 | 9 | 0.003666 | 14 | 0.001421 | 19 | 0.000749 |
| 5 | 0.013800 | 10 | 0.002196 | 15 | 0.001229 | 20 | 0.000673 |
| Atom | H | C | S | O | F | Cl | Br | I |
|---|---|---|---|---|---|---|---|---|
| αi | 0.6668 | 1.76 | 2.90 | 0.802 | 0.557 | 2.18 | 3.05 | 5.34 |
| Retention Indices | Descriptors | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. | Alcohol | SE-30 | OV-3 | OV-7 | OV-11 | OV-17 | OV-25 | OEI | MPEI | SX1CH |
| 1 | 1-butanol | 650 | 672 | 702 | 725 | 748 | 792 | 5.2222 | 2.5887 | −6.5340 |
| 2 | 1-hexanol | 856 | 881 | 907 | 935 | 959 | 1003 | 8.4967 | 2.6446 | −8.5424 |
| 3 | 1-heptanol | 960 | 985 | 1010 | 1038 | 1062 | 1104 | 10.1183 | 2.6611 | −9.5424 |
| 4 | 2-butanol | 586 | 607 | 633 | 656 | 675 | 711 | 5.2222 | 2.7854 | −6.5407 |
| 5 | 2-pentanol | 689 | 711 | 735 | 756 | 777 | 811 | 6.8194 | 2.8386 | −7.5453 |
| 6 | 3-pentanol | 689 | 708 | 733 | 756 | 777 | 808 | 6.8194 | 2.8850 | −7.5440 |
| 7 | 3-hexanol | 785 | 807 | 830 | 853 | 878 | 904 | 8.4967 | 2.9383 | −8.5434 |
| 8 | 3-heptanol | 886 | 909 | 929 | 955 | 975 | 1008 | 10.1183 | 2.9715 | −9.5414 |
| 9 | 4-heptanol | 880 | 904 | 924 | 946 | 968 | 999 | 10.1183 | 2.9916 | −9.5392 |
| 10 | 2-methyl-2-butanol | 628 | 652 | 674 | 692 | 709 | 738 | 6.4444 | 3.0353 | −7.5706 |
| 11 | 2-methyl-2-hexanol | 822 | 848 | 862 | 884 | 904 | 930 | 9.6739 | 3.1217 | −9.5480 |
| 12 | 2-methyl-2-heptanol | 920 | 944 | 961 | 982 | 1001 | 1026 | 11.2400 | 3.1444 | −10.5425 |
| 13 | 2-methyl-3-hexanol | 858 | 876 | 897 | 920 | 939 | 969 | 9.6739 | 3.0379 | −9.5407 |
| 14 | 3-methyl-1-butanol | 725 | 747 | 771 | 798 | 817 | 855 | 6.4444 | 2.6420 | −7.5453 |
| 15 | 4-methyl-1-pentanol | 827 | 849 | 876 | 902 | 923 | 960 | 7.9167 | 2.6551 | −8.5469 |
| 16 | 2-ethyl-1-hexanol | 1019 | 1046 | 1067 | 1092 | 1116 | 1156 | 11.5178 | 2.7975 | −10.5296 |
| 17 | 3-ethyl-3-pentanol | 853 | 876 | 898 | 920 | 939 | 974 | 9.9583 | 3.2345 | −9.5358 |
| 18 | 2,2-dimethyl-3-pentanol | 814 | 834 | 855 | 874 | 890 | 919 | 8.5139 | 3.0843 | −9.5556 |
| 19 | 2,2-dimethyl-3-hexanol | 906 | 926 | 944 | 962 | 977 | 1004 | 10.3511 | 3.1375 | −10.5326 |
| 20 | 1-propanol | 544 | 574 | 3.5000 | 2.5354 | −5.5244 | ||||
| 21 | 1-pentanol | 751 | 777 | 806 | 856 | 900 | 6.8194 | 2.6219 | −7.5404 | |
| 22 | 2-pexanol | 787 | 811 | 835 | 878 | 914 | 8.4967 | 2.8718 | −8.5469 | |
| 23 | 2-methyl-1-propanol | 612 | 641 | 654 | 680 | 740 | 4.5000 | 2.6351 | −6.5407 | |
| 24 | 2-methyl-2-pentanol | 726 | 748 | 767 | 801 | 827 | 7.9167 | 3.0886 | −8.5515 | |
| 25 | 2-ethyl-1-butanol | 834 | 857 | 907 | 928 | 8.2639 | 2.7417 | −8.5400 | ||
| Stationary Phase | Regression Equation | Statistics | |||||
|---|---|---|---|---|---|---|---|
| R | s | F | Rcv | scv | n | ||
| SE-30 | RI = 714.1971 – 53.1823SX1CH | 0.9963 | 11.2 | 942.1 | 0.9943 | 12.8 | 25 |
| –231.145MPEI + 34.62949OEI | |||||||
| OV-3 | RI =756.8884 – 52.1502 SX1CH | 0.9963 | 11.2 | 936.4 | 0.9942 | 12.8 | 25 |
| –236.867MPEI + 35.3456OEI | |||||||
| OV-7 | RI = 798.1506 – 47.8311SX1CH | 0.9953 | 12.3 | 638.7 | 0.9922 | 14.3 | 22 |
| –238.579MPEI + 37.97237OEI | |||||||
| OV-11 | RI = 858.8273 – 43.7851SX1CH | 0.9938 | 13.8 | 453.1 | 0.9891 | 16.4 | 21 |
| –249.092MPEI + 41.39177OEI | |||||||
| OV-17 | RI = 941.0954 – 35.5304SX1CH | 0.9940 | 13.6 | 547.6 | 0.9899 | 16.1 | 24 |
| –263.948MPEI + 47.63748OEI | |||||||
| OV-25 | RI = 1053.736 – 37.8516 SX1CH | 0.9922 | 15.6 | 402.5 | 0.9871 | 18.3 | 23 |
| –292.817MPEI + 45.8317OEI | |||||||
| No. | Alcohol | OEI | MPEI | BP (Exp.) | BP (Cal.) | ΔBP |
|---|---|---|---|---|---|---|
| 1 | methanol | 0.0000 | 2.1859 | 64.7 | 70.1 | −5.4 |
| 2 | ethanol | 2.0000 | 2.4358 | 78.3 | 82.3 | −4.0 |
| 3 | 1-propanol | 3.5000 | 2.5354 | 97.2 | 96.2 | 1.0 |
| 4 | 1-butanol | 5.2222 | 2.5887 | 117.0 | 115.5 | 1.5 |
| 5 | 1-pentanol | 6.8194 | 2.6219 | 137.8 | 134.2 | 3.6 |
| 6 | 1-hexanol | 8.4967 | 2.6446 | 157.0 | 154.5 | 2.5 |
| 7 | 1-heptanol | 10.1183 | 2.6611 | 176.3 | 174.5 | 1.8 |
| 8 | 1-octanol | 11.7808 | 2.6736 | 195.2 | 195.1 | 0.1 |
| 9 | 1-nonanol | 13.4120 | 2.6835 | 213.1 | 215.5 | −2.4 |
| 10 | 1-decanol | 15.0680 | 2.6914 | 230.2 | 236.4 | −6.2 |
| 11 | 2-propanol | 3.5000 | 2.6857 | 82.3 | 88.1 | −5.8 |
| 12 | 2-butanol | 5.2222 | 2.7854 | 99.6 | 104.9 | −5.3 |
| 13 | 2-pentanol | 6.8194 | 2.8386 | 119.0 | 122.5 | −3.5 |
| 14 | 2-hexanol | 8.4967 | 2.8718 | 139.9 | 142.3 | −2.4 |
| 15 | 2-octanol | 11.7808 | 2.9110 | 179.8 | 182.4 | −2.6 |
| 16 | 2-nonanol | 13.4120 | 2.9235 | 198.5 | 202.6 | −4.1 |
| 17 | 3-pentanol | 6.8194 | 2.8850 | 115.3 | 120.0 | −4.7 |
| 18 | 3-hexanol | 8.4967 | 2.9383 | 135.4 | 138.7 | −3.3 |
| 19 | 3-heptanol | 10.1183 | 2.9715 | 156.8 | 157.7 | −0.9 |
| 20 | 4-heptanol | 10.1183 | 2.9916 | 155.0 | 156.7 | −1.7 |
| 21 | 3-nonanol | 13.4120 | 3.0106 | 194.7 | 197.9 | −3.2 |
| 22 | 4-nonanol | 13.4120 | 3.0474 | 193.0 | 196.0 | −3.0 |
| 23 | 5-nonanol | 13.4120 | 3.0580 | 195.1 | 195.4 | −0.3 |
| 24 | 2-me-1-propanol | 4.5000 | 2.6351 | 107.9 | 103.7 | 4.2 |
| 25 | 2-me-2-propanol | 4.5000 | 2.9356 | 82.4 | 87.5 | −5.1 |
| 26 | 2-me-1-butanol | 6.4444 | 2.6884 | 128.7 | 125.8 | 2.9 |
| 27 | 2-me-2-butanol | 6.4444 | 3.0353 | 102.0 | 107.1 | −5.1 |
| 28 | 3-me-1-butanol | 6.4444 | 2.6420 | 131.2 | 128.3 | 2.9 |
| 29 | 3-me-2-butanol | 6.4444 | 2.8850 | 111.5 | 115.2 | −3.7 |
| 30 | 2-me-1-pentanol | 7.9167 | 2.7216 | 148.0 | 142.9 | 5.1 |
| 31 | 3-me-1-pentanol | 8.2639 | 2.6752 | 152.4 | 149.9 | 2.5 |
| 32 | 4-me-1-pentanol | 7.9167 | 2.6551 | 151.8 | 146.5 | 5.3 |
| 33 | 2-me-2-pentanol | 7.9167 | 3.0885 | 121.4 | 123.2 | −1.8 |
| 34 | 3-me-2-pentanol | 8.2639 | 2.9383 | 134.2 | 135.7 | −1.5 |
| 35 | 4-me-2-pentanol | 7.9167 | 2.8919 | 131.7 | 133.8 | −2.1 |
| 36 | 2-me-3-pentanol | 7.9167 | 2.9846 | 126.6 | 128.8 | −2.2 |
| 37 | 3-me-3-pentanol | 8.2639 | 3.1349 | 122.4 | 125.1 | −2.7 |
| 38 | 2-me-2-hexanol | 9.6739 | 3.1217 | 142.5 | 144.0 | −1.5 |
| 39 | 3-me-3-hexanol | 9.8161 | 3.1882 | 142.4 | 142.2 | 0.2 |
| 40 | 7-me-1-octanol | 12.9433 | 2.6861 | 206.0 | 209.4 | −3.4 |
| 41 | 2-et-1-butanol | 8.2639 | 2.7417 | 146.5 | 146.3 | 0.2 |
| 42 | 3-et-3-pentanol | 9.9583 | 3.2345 | 142.5 | 141.5 | 1.0 |
| 43 | 2-et-1-hexanol | 11.5178 | 2.7975 | 184.6 | 185.1 | −0.5 |
| 44 | 2,2-dime-1-propanol | 5.0000 | 2.7347 | 113.1 | 104.8 | 8.3 |
| 45 | 2,2-dime-1-butanol | 7.1667 | 2.7880 | 136.8 | 129.7 | 7.1 |
| 46 | 2,3-dime-1-butanol | 7.8889 | 2.7417 | 149.0 | 141.5 | 7.5 |
| 47 | 3,3-dime-1-butanol | 7.1667 | 2.6953 | 143.0 | 134.7 | 8.3 |
| 48 | 2,3-dime-2-butanol | 7.8889 | 3.1349 | 118.6 | 120.3 | −1.7 |
| 49 | 3,3-dime-2-butanol | 7.1667 | 2.9846 | 120.0 | 119.1 | 0.9 |
| 50 | 2,3-dime-2-pentanol | 9.5833 | 3.1882 | 139.7 | 139.2 | 0.5 |
| 51 | 3,3-dime-2-pentanol | 9.2083 | 3.0379 | 133.0 | 142.5 | −9.5 |
| 52 | 2,2-dime-3-pentanol | 8.5139 | 3.0843 | 136.0 | 131.1 | 4.9 |
| 53 | 2,4-dime-3-pentanol | 8.8889 | 3.0843 | 138.8 | 135.9 | 2.9 |
| 54 | 2,6-dime-4-heptanol | 12.3061 | 3.0982 | 178.0 | 179.0 | −1.0 |
| 55 | 2,3-dime-3-pentanol | 9.5833 | 3.2345 | 139.0 | 136.7 | 2.3 |
| 56 | 3,5-dime-4-heptanol | 12.7922 | 3.1908 | 187.0 | 180.3 | 6.7 |
| 57 | 2,2,3-trime-3-pentanol | 10.4028 | 3.3342 | 152.2 | 141.9 | 10.3 |
| 58 | 3,5,5-trime-1-hexanol | 11.4206 | 2.7433 | 193.0 | 186.8 | 6.2 |
| No. | Alcohol | MPEI | SX1CH | lg W (Exp.) | lg W (Cal.) | lg POW (Exp.) | lg POW (Cal.) |
|---|---|---|---|---|---|---|---|
| 1 | 1-butanol | 2.5887 | −6.5340 | −0.03 | 0.00 | 0.84 | 0.75 |
| 2 | 2-butanol | 2.7854 | −6.5407 | −0.39 | −0.25 | 0.61 | 0.61 |
| 3 | 2-methyl-1-propanol | 2.6348 | −6.5407 | −0.10 | −0.05 | 0.61 | 0.72 |
| 4 | 1-pentanol | 2.6219 | −7.5404 | 0.59 | 0.56 | 1.34 | 1.28 |
| 5 | 3-methyl-1-butanol | 2.6420 | −7.5453 | 0.51 | 0.54 | 1.14 | 1.27 |
| 6 | 2-methyl-1-butanol | 2.6884 | −7.5440 | 0.46 | 0.48 | 1.14 | 1.23 |
| 7 | 2-pentanol | 2.8386 | −7.5453 | 0.28 | 0.28 | 1.14 | 1.13 |
| 8 | 3-pentanol | 2.8850 | −7.5440 | 0.21 | 0.22 | 1.14 | 1.09 |
| 9 | 3-methyl-2-butanol | 2.8850 | −7.5496 | 0.21 | 0.22 | 1.14 | 1.10 |
| 10 | 2-methyl-2-butanol | 3.0353 | −7.5706 | 0.23 | 0.04 | 0.89 | 1.00 |
| 11 | 2,2-dimethyl-1-propanol | 2.7347 | −7.5706 | 0.30 | 0.43 | 1.36 | 1.22 |
| 12 | 1-hexanol | 2.6446 | −8.5424 | 1.21 | 1.13 | 1.84 | 1.82 |
| 13 | 2-hexanol | 2.8718 | −8.5469 | 0.87 | 0.84 | 1.61 | 1.66 |
| 14 | 3-hexanol | 2.9383 | −8.5434 | 0.80 | 0.75 | 1.61 | 1.61 |
| 15 | 3-methyl-3-pentanol | 3.1028 | −8.5480 | 0.39 | 0.54 | 1.39 | 1.49 |
| 16 | 2-methyl-2-pentanol | 3.0886 | −8.5515 | 0.51 | 0.56 | 1.39 | 1.51 |
| 17 | 2-methyl-3-pentanol | 2.9846 | −8.5454 | 0.70 | 0.69 | 1.67 | 1.58 |
| 18 | 3-methyl-2-pentanol | 2.9383 | −8.5454 | 0.71 | 0.75 | 1.67 | 1.61 |
| 19 | 2,2-dimethyl-1-butanol | 2.7880 | −8.5480 | 1.04 | 0.94 | 1.57 | 1.72 |
| 20 | 2,3-dimethyl-1-butanol | 2.7417 | −8.5454 | 0.50 | 1.00 | 1.57 | 1.75 |
| 21 | 2,3-dimethyl-2-butanol | 3.1349 | −8.5526 | 0.37 | 0.50 | 1.17 | 1.47 |
| 22 | 3,3-dimethyl-2-butanol | 2.9846 | −8.5526 | 0.64 | 0.69 | 1.19 | 1.58 |
| 23 | 2-methyl-1-pentanol | 2.7216 | −8.5434 | 1.05 | 1.03 | 1.78 | 1.76 |
| 24 | 4-methyl-1-pentanol | 2.6551 | −8.5469 | 0.99 | 1.12 | 1.78 | 1.81 |
| 25 | 4-methyl-2-pentanol | 2.8919 | −8.5486 | 0.81 | 0.81 | 1.67 | 1.64 |
| 26 | 2-ethyl-1-butanol | 2.7417 | −8.5400 | 1.21 | 1.00 | 1.78 | 1.75 |
| 27 | 1-heptanol | 2.6611 | −9.5424 | 1.81 | 1.70 | 2.34 | 2.36 |
| 28 | 2-heptanol | 2.8945 | −9.5454 | 1.55 | 1.40 | 2.31 | 2.19 |
| 29 | 3-heptanol | 2.9715 | −9.5414 | 1.39 | 1.30 | 2.31 | 2.14 |
| 30 | 4-heptanol | 2.9916 | −9.5392 | 1.39 | 1.27 | 2.31 | 2.12 |
| 31 | 2-methyl-2-hexanol | 3.1217 | −9.5480 | 1.07 | 1.11 | 1.84 | 2.03 |
| 32 | 5-methyl-2-hexanol | 2.9050 | −9.5482 | 1.38 | 1.39 | 2.19 | 2.19 |
| 33 | 3-methyl-2-hexanol | 3.1882 | −9.5405 | 0.98 | 1.02 | 1.87 | 1.98 |
| 34 | 2-methyl-3-hexanol | 3.0058 | −9.5407 | 1.32 | 1.25 | 2.19 | 2.11 |
| 35 | 2,2-dimethyl-1-pentanol | 2.8212 | −9.5405 | 1.52 | 1.49 | 2.39 | 2.24 |
| 36 | 2,4-dimethyl-1-pentanol | 2.7548 | −9.5432 | 1.60 | 1.58 | 2.19 | 2.29 |
| 37 | 4,4-dimethyl-1-pentanol | 2.6883 | −9.5480 | 1.55 | 1.67 | 2.39 | 2.34 |
| 38 | 2,3-dimethyl-2-pentanol | 3.1882 | −9.5556 | 0.91 | 1.03 | 2.27 | 1.99 |
| 39 | 2,4-dimethyl-2-pentanol | 3.1419 | −9.5487 | 0.93 | 1.08 | 1.67 | 2.02 |
| 40 | 2,2-dimethyl-3-pentanol | 3.0843 | −9.5556 | 1.16 | 1.16 | 2.27 | 2.06 |
| 41 | 2,3-dimethyl-3-pentanol | 3.2345 | −9.5399 | 0.84 | 0.96 | 1.67 | 1.95 |
| 42 | 2,4-dimethyl-3-pentanol | 3.0843 | −9.5409 | 1.32 | 1.15 | 2.31 | 2.06 |
| 43 | 1-octanol | 2.6736 | −10.5390 | 2.35 | 2.28 | 2.84 | 2.90 |
| 44 | 2-octanol | 2.9110 | −10.5423 | 2.07 | 1.97 | 2.84 | 2.73 |
| 45 | 2-ethyl-1-hexanol | 2.7975 | −10.5296 | 2.17 | 2.11 | 2.84 | 2.81 |
| 46 | 1-nonanol | 2.6820 | −11.5348 | 3.00 | 2.86 | 3.57 | 3.45 |
| 47 | 2-nonanol | 2.9235 | −11.5372 | 2.74 | 2.55 | 3.36 | 3.28 |
| 48 | 3-nonanol | 3.0106 | −11.5315 | 2.66 | 2.43 | 3.36 | 3.21 |
| 49 | 4-nonanol | 3.0474 | −11.5280 | 2.59 | 2.38 | 3.36 | 3.18 |
| 50 | 5-nonanol | 3.0580 | −11.5268 | 2.49 | 2.37 | 3.36 | 3.17 |
| 51 | 2,6-dimethyl-4-heptanol | 3.0982 | −11.5273 | 2.51 | 2.32 | 3.31 | 3.15 |
| 52 | 1-decanol | 2.6892 | −12.5296 | 3.70 | 3.44 | 4.01 | 3.99 |
| 53 | 2-undcanol | 2.9391 | −13.5220 | 2.94 | 3.71 | 4.42 | 4.36 |
| 54 | 1-dodecanol | 2.7011 | −14.5138 | 4.80 | 4.61 | 5.06 | 5.08 |
| 55 | 1-tetradecanol | 2.7098 | −16.4948 | 5.52 | 5.77 | 6.11 | 6.17 |
| 56 | 1-pentadecanol | 2.7132 | −17.4838 | 5.84 | 6.36 | 6.64 | 6.71 |
| 57 | 1-hexadecanol | 2.7163 | −18.4720 | 7.00 | 6.94 | 7.17 | 7.26 |
| 58 | 1-octadecanol | 2.7214 | −20.4476 | 8.40 | 8.11 | 8.22 | 8.35 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, F.; Cao, C.; Cheng, B. A Quantitative Structure-Property Relationship (QSPR) Study of Aliphatic Alcohols by the Method of Dividing the Molecular Structure into Substructure. Int. J. Mol. Sci. 2011, 12, 2448-2462. https://doi.org/10.3390/ijms12042448
Liu F, Cao C, Cheng B. A Quantitative Structure-Property Relationship (QSPR) Study of Aliphatic Alcohols by the Method of Dividing the Molecular Structure into Substructure. International Journal of Molecular Sciences. 2011; 12(4):2448-2462. https://doi.org/10.3390/ijms12042448
Chicago/Turabian StyleLiu, Fengping, Chenzhong Cao, and Bin Cheng. 2011. "A Quantitative Structure-Property Relationship (QSPR) Study of Aliphatic Alcohols by the Method of Dividing the Molecular Structure into Substructure" International Journal of Molecular Sciences 12, no. 4: 2448-2462. https://doi.org/10.3390/ijms12042448
APA StyleLiu, F., Cao, C., & Cheng, B. (2011). A Quantitative Structure-Property Relationship (QSPR) Study of Aliphatic Alcohols by the Method of Dividing the Molecular Structure into Substructure. International Journal of Molecular Sciences, 12(4), 2448-2462. https://doi.org/10.3390/ijms12042448