Chemoenzymatic Synthesis and Chemical Recycling of Poly(ester-urethane)s

Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Measurements
2.3. Preparation of Diol-Diacid-Type Poly(ester-ether-urethane) (PEEU) and Poly(ester-urethane) (PEU)
Preparation of tetramethylene bis(phenyl carbonate) (1)
Preparation of diurethane-containg dicarboxylic acid (2)
Preparation of PEEU(6) by direct-polycondensation of 2 and hexaethylene glycol
Preparation of PEU(12) by direct-polycondensation of 2 and 1,12-dodecanediol
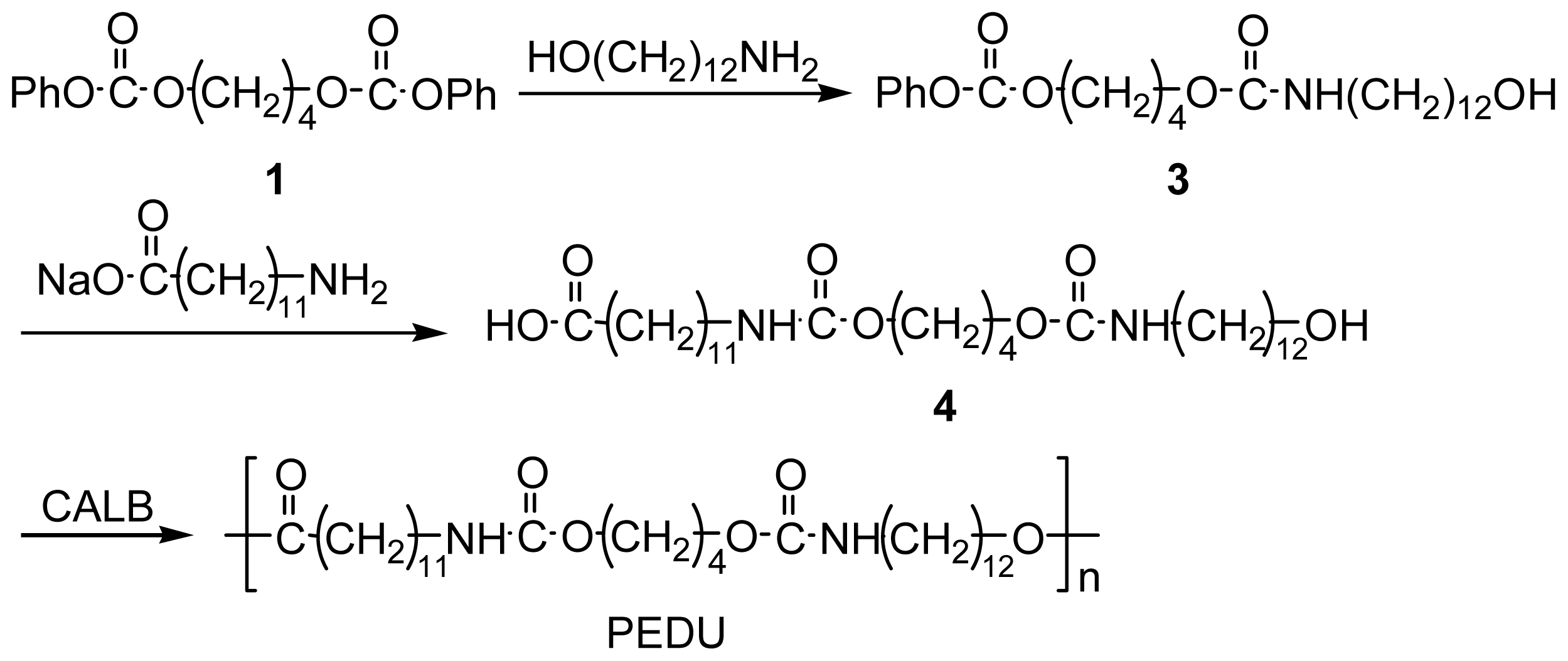
2.4. Preparation of Hydroxy Acid Type Poly(ester-diurethane) (PEDU)
Preparation of urethane-containing hydroxy phenyl carbonate (3)
Preparation of diurethane-containing hydroxy acid (4)
Preparation of PEDU by polycondensation of 4
2.5. Chemical Recycling of PEEU(6) and PEU(12)
Enzymatic degradation of PEEU(6) and PEU(12) into cyclic monomer
3. Results and Discussion
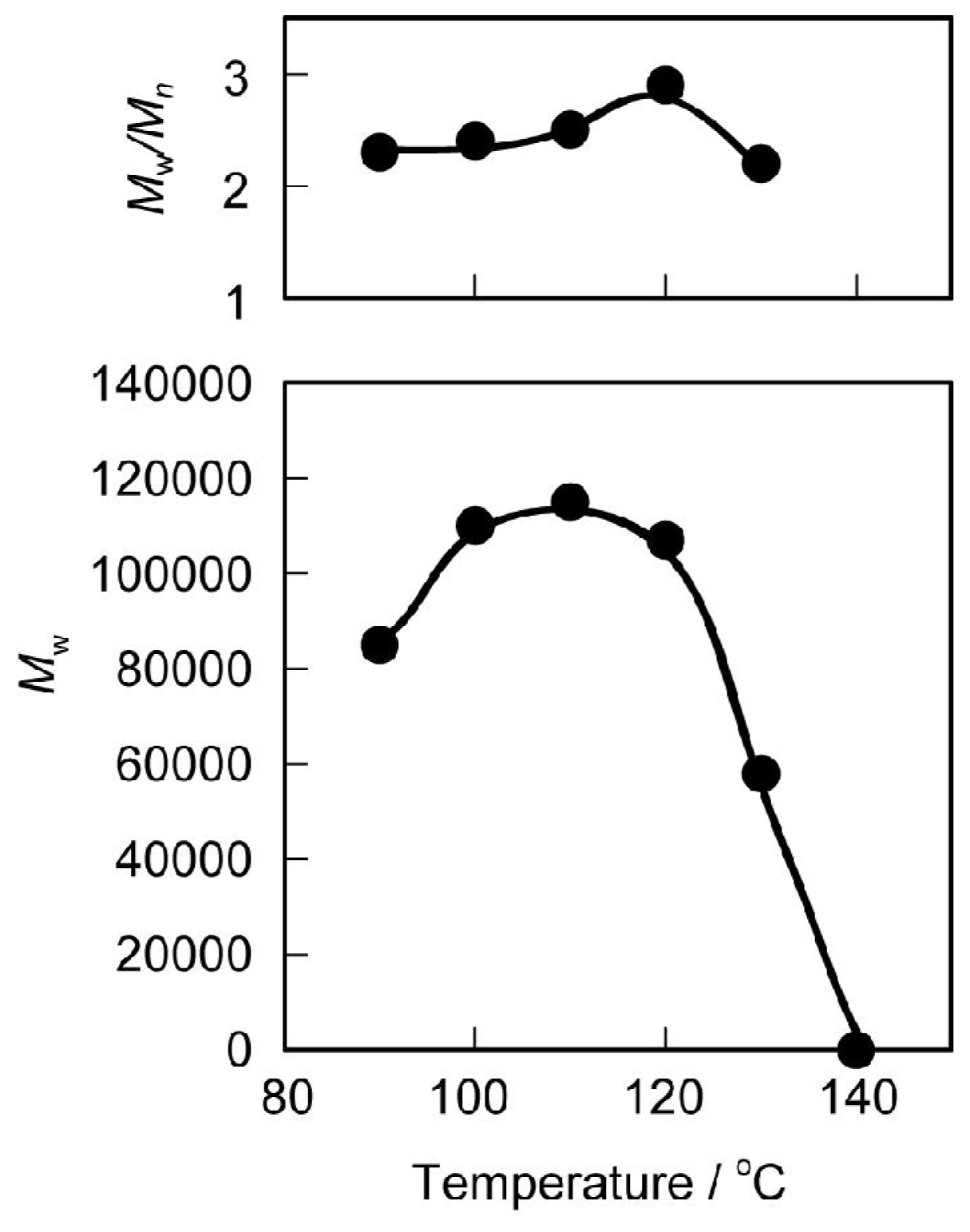
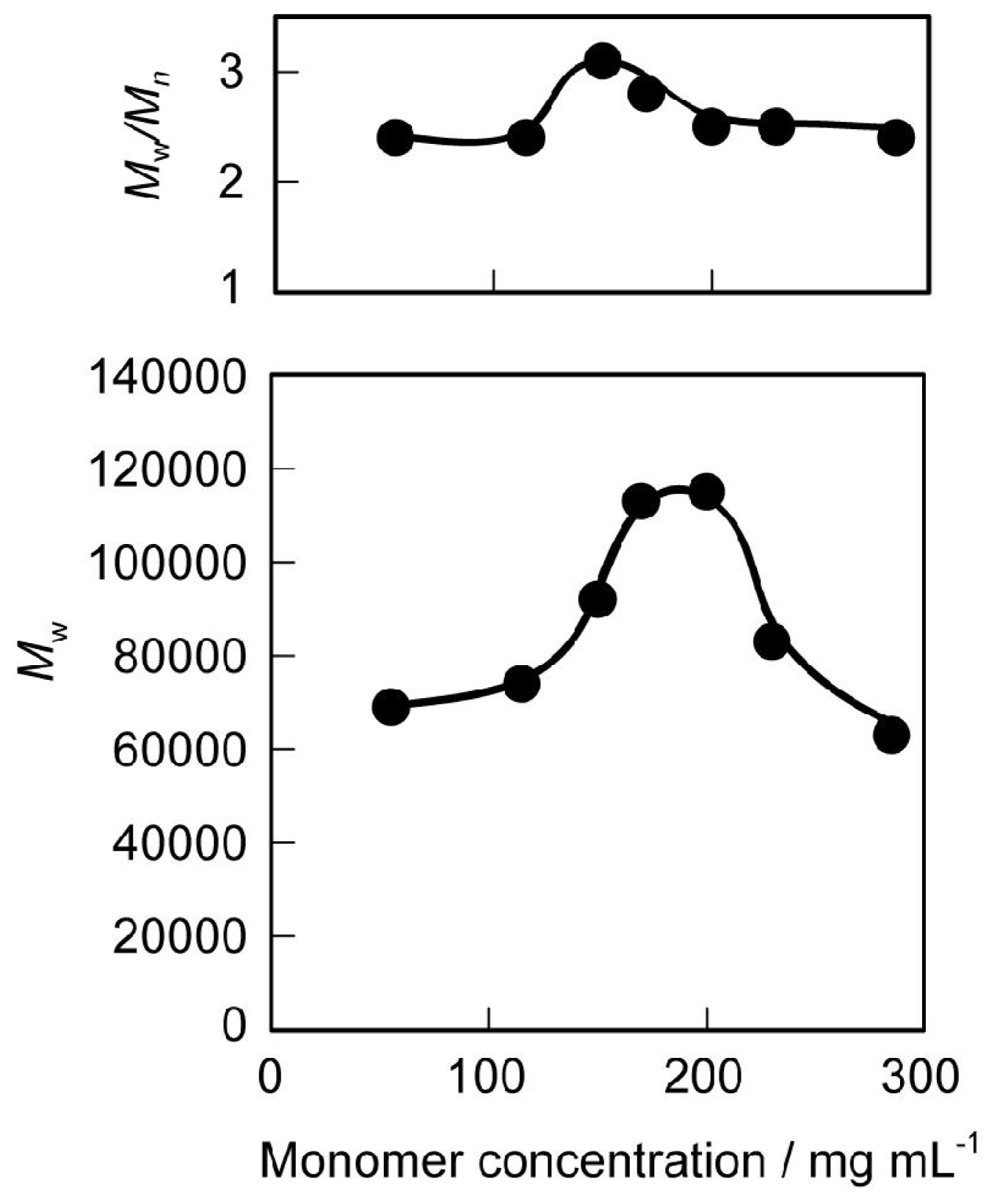
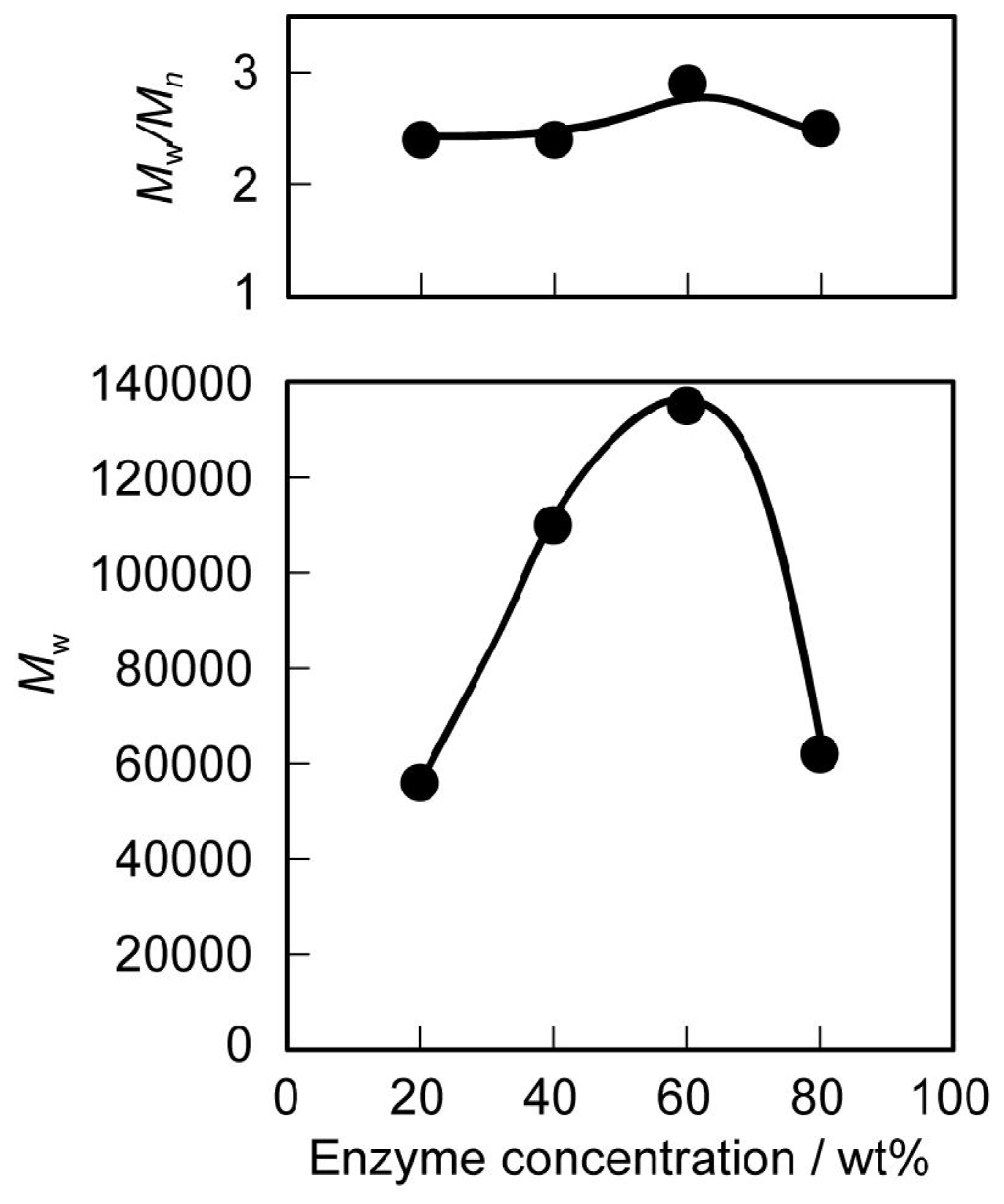
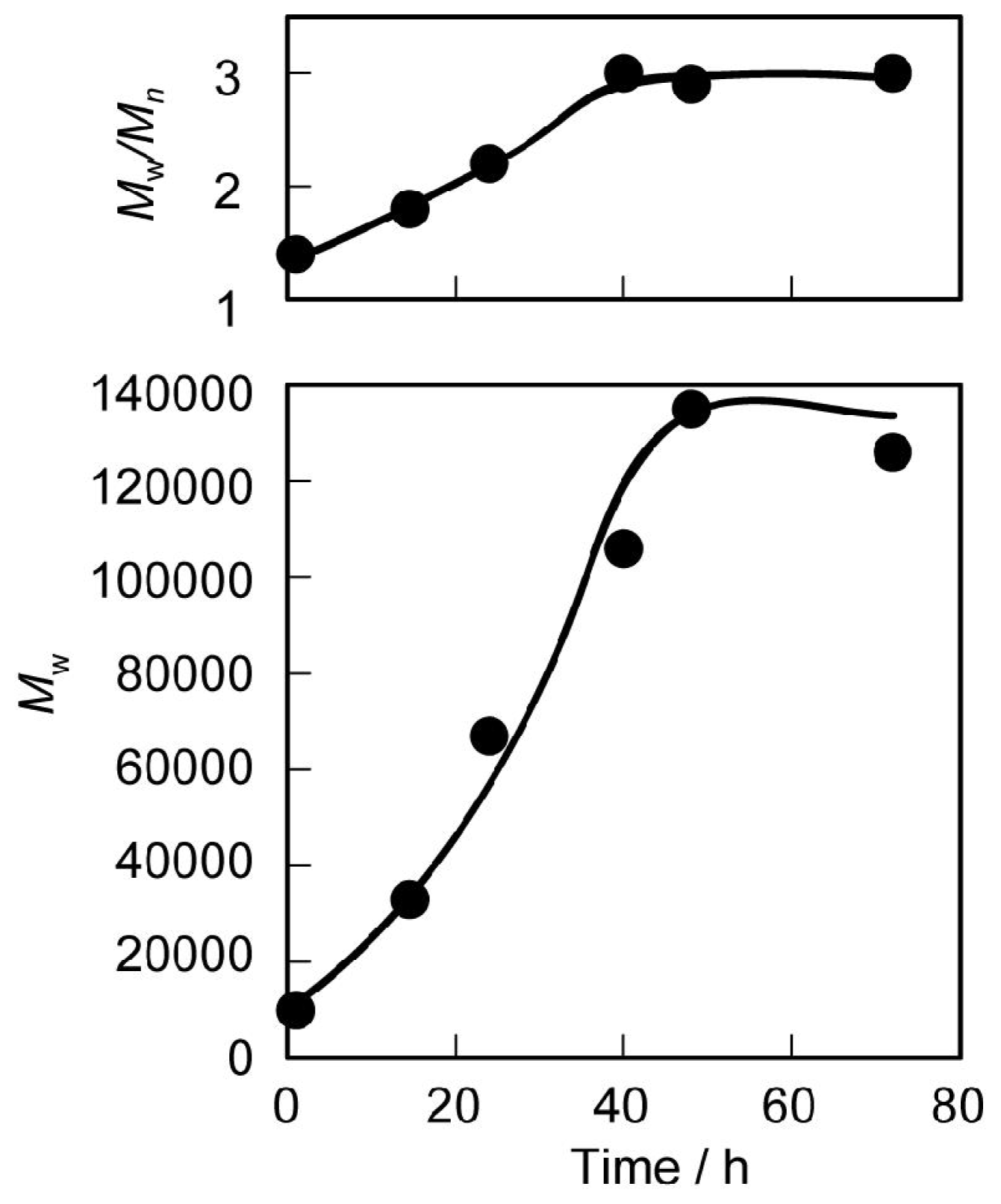
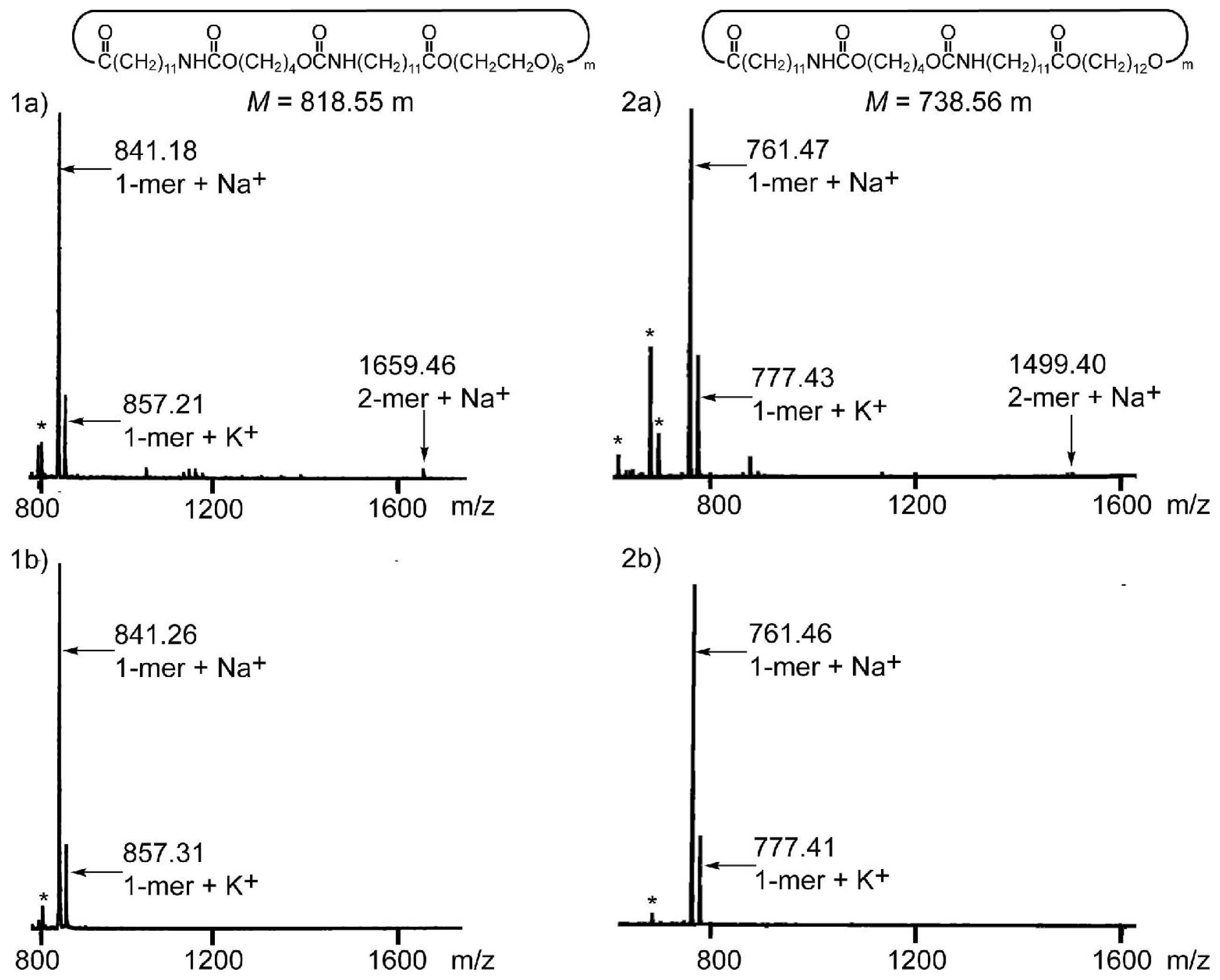
3.1. Synthesis and Characterization
3.2. Thermal and Mechanical Properties of the Polyurethanes
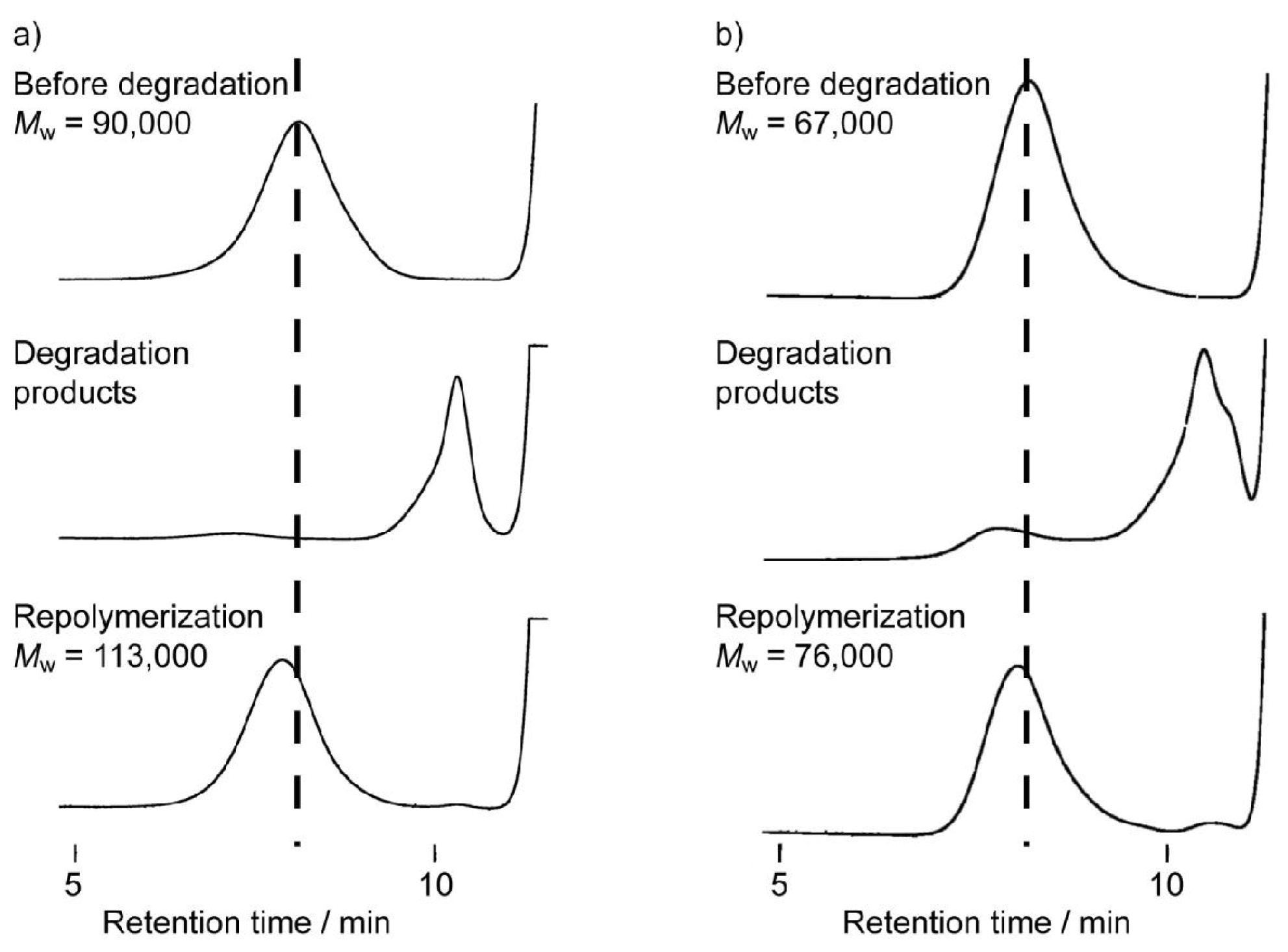
3.3. Chemical Recycling of PEEU and PEU
4. Conclusions
Acknowledgments
Appendix NMR Analysis
References
- Kihara, N; Endo, T. Synthesis and properties of poly(hydroxyurethane)s. J Polym Sci A Polym Chem 1993, 31, 2765–2773. [Google Scholar]
- Rokicki, G; Piotrowska, A. A new route to polyurethanes from ethylene carbonate, diamines and diols. Polymer 2002, 43, 2927–2935. [Google Scholar]
- Neffgen, S; Keul, H; Höcker, H. Cationic ring-opening polymerization of trimethylene urethane: A mechanistic study. Macromolecules 1997, 30, 1286–1297. [Google Scholar]
- Kusan, J; Keul, H; Höcker, H. New routes to [n]-polyurethanes. [3]-polyurethane: Synthesis, characterization, and polymerization-depolymerization equilibrium. Macromol Symp 2001, 165, 63–72. [Google Scholar]
- Kusan, J; Keul, H; Höcker, H. Cationic ring-opening polymerization of tetramethylene urethane. Macromolecules 2001, 34, 389–395. [Google Scholar]
- Helou, M; Carpentier, J-F; Guillaume, SM. Poly(carbonate-urethane): An isocyanate-free procedure from alpha,omega-di(cyclic carbonate) telechelic poly(trimethylenecarbonate)s. Green Chem 2011, 13, 266–271. [Google Scholar]
- Campbell, GA; Melunch, WC. Polyurethane waste disposal process development: Amine recovery. J Appl Polym Sci 1977, 21, 581–584. [Google Scholar]
- Simioni, F; Bisello, S; Tavan, M. Polyol recovery from rigid polyurethane waste. Cell Polym 1983, 2, 281–293. [Google Scholar]
- Simioni, F; Modesti, M; Rienzi, SA. Polyol recovery from elastomer polyurethane waste. Cell Polym 1987, 6, 27–41. [Google Scholar]
- Simioni, F; Modesti, M; Tavan, M. Controlled degradation of polyurethane for recycling. Mater Eng 1991, 2, 127–144. [Google Scholar]
- Kanaya, K; Takahashi, S. Decomposition of polyurethane foams by alkanolamines. J Appl Polym Sci 1994, 51, 675–682. [Google Scholar]
- Matsumura, S. Enzymatic synthesis of polyesters via ring-opening polymerization. Adv Polym Sci 2006, 194, 95–132. [Google Scholar]
- Yanagishita, Y; Kato, M; Toshima, K; Matsumura, S. Chemoenzymatic synthesis and chemical recycling of sustainable polyurethanes. Chem Sus Chem 2008, 1, 133–142. [Google Scholar]
- Soeda, Y; Toshima, K; Matsumura, S. Synthesis and chemical recycling of novel poly (ester-urethane)s using an enzyme. Macromol Biosci 2005, 5, 277–288. [Google Scholar]
- Al-Hamouz, OCS; Sweileh, BA; Al-Salah, HA. Synthesis and characterization of polycarbonates by melt-phase interchange reactions with alkylene and arylene diphenyl dicarbonates. J Appl Polym Sci 2006, 102, 3597–3609. [Google Scholar]
- Sweileh, BA; A-Hiari, YM; Aiedeh, KM. A new, nonphosgene route to poly(bisphenol a carbonate) by melt-phase interchange reactions of alkylene diphenyl dicarbonates with bisphenol A. J Appl Polym Sci 2008, 110, 2278–2292. [Google Scholar]
- Volkin, DB; Staubli, A; Langer, R; Klibanov, AM. Enzyme thermoinactivation in anhydrous organic-solvents. Biotech Bioeng 1991, 37, 843–853. [Google Scholar]
- Parvaresh, F; Robert, H; Thomas, D; Legoy, MD. Gas-phase transesterification reactions catalyzed by lipolytic enzymes. Biotech Bioeng 1992, 39, 467–473. [Google Scholar]
- Turner, NA; Duchateau, DB; Vulfson, EN. Effect of hydration on thermostability of serine esterases. Biotech Lett 1995, 17, 371–376. [Google Scholar]
- Turner, NA; Vulfson, EN. At what temperature can enzymes maintain their catalytic activity? Enzyme Microbiol Technol 2000, 27, 108–113. [Google Scholar]
- Uyama, H; Takeya, K; Kobayashi, S. Enzymatic ring-opening polymerization of lactones to polyesters by lipase catalyst—Unusually high reativity of macrolides. Bull Chem Soc Jpn 1995, 68, 56–61. [Google Scholar]
- MacDonald, RT; Pulapura, SK; Svirkin, YY; Gross, RA; Kaplan, DL; Akkara, J; Swift, G; Wolk, S. Enzyme-catalyzed ε-caprolactone ring-opening polymerization. Macromolecules 1995, 28, 73–78. [Google Scholar]
- Min, BM; Son, TW; Jo, WH; Choi, SG. Thermal stability of polyacrylonitrile in the melt formed by hydration. J Appl Polym Sci 1992, 46, 1793–1798. [Google Scholar]
- Sarkar, D; Yang, J; Lopina, ST. Structure-property relationship of l-tyrosine-based polyurethanes for biomaterial applications. J Appl Polym Sci 2008, 108, 2345–2355. [Google Scholar]
- Pierce, BF; Brown, AH; Sheares, VV. Thermoplastic poly(ester urethane)s with novel soft segments. Macromolecules 2008, 41, 3866–3873. [Google Scholar]
- Stehling, FC; Mandelkern, L. The glass temperature of linear polyethylene. Macromolecules 1970, 3, 242–252. [Google Scholar]
- Luyt, AS; Hato, MJ. Thermal and mechanical properties of linear low-density polyethylene/ low-density polyethylene/wax ternary blends. J Appl Polym Sci 2005, 96, 1748–1755. [Google Scholar]
- Yuan, Z; Chen, H; Zhang, J; Zhao, D; Liu, Y; Zhou, X; Li, S; Shi, P; Tang, J; Chen, X. Preparation and characterization of self-cleaning stable superhydrophobic linear low-density polyethylene. Sci Technol Adv Mater 2008, 9. [Google Scholar] [CrossRef]
- Pricariu, C; Olley, RH; Caraculacu, AA; Bassett, DC; Martin, C. The effect of hard segment ordering in copolyurethane elastomers obtained by using simultaneously two types of diisocyanates. Polymer 2003, 44, 5407–5421. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
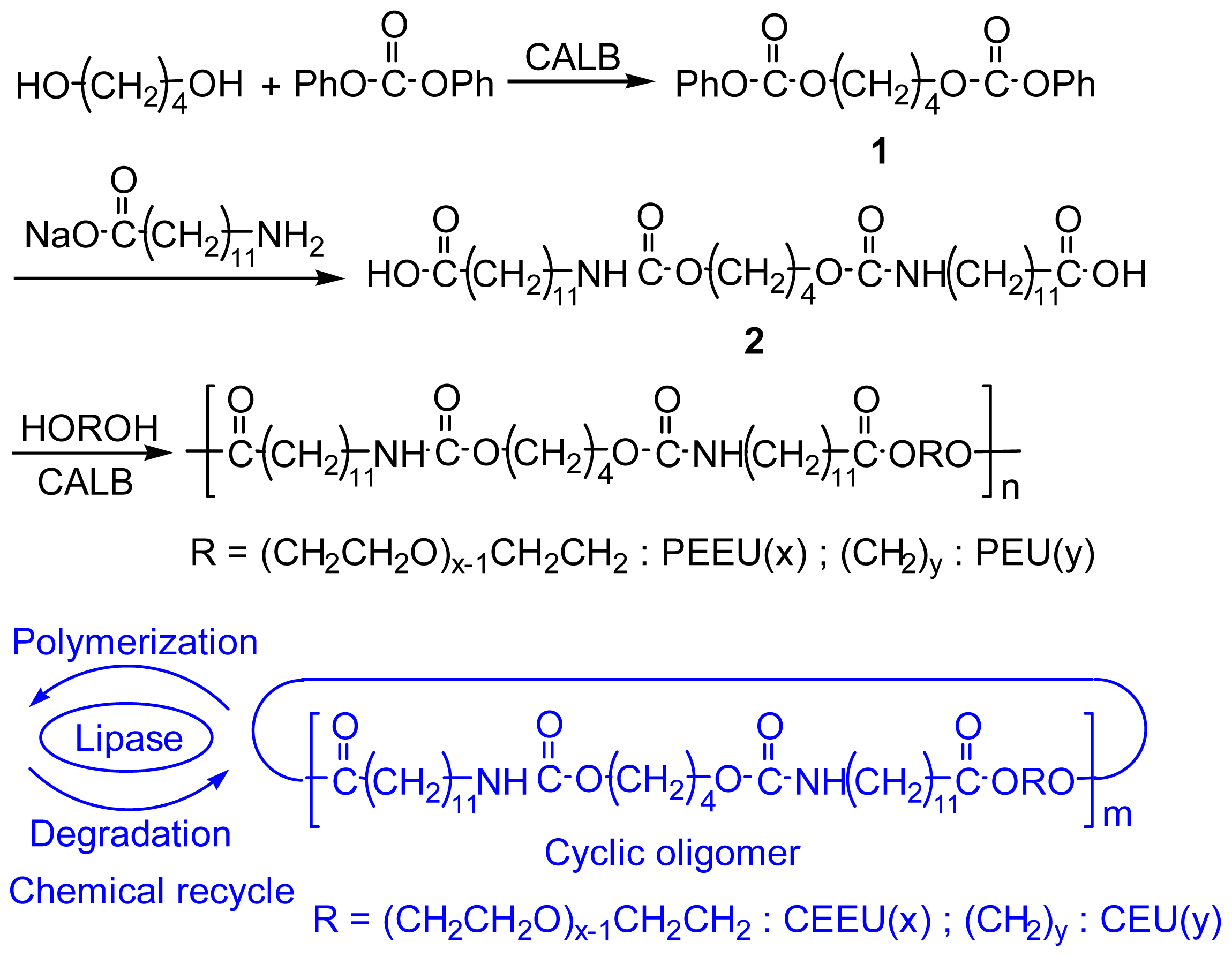{kind=link}
{kind=link}
{kind=link}
| Polymer | Polymerizationa | Polymer
| ||||||
|---|---|---|---|---|---|---|---|---|
| Monomer 1 | Monomer 2b | Solventc | Temp./°C | Purificationd | Mw/g·mol−1 | Mw/Mn | Yield/% | |
| PEEU(6) | 2 | HEG | DMB | 100 | A | 136,000 | 2.9 | 78.9 |
| PEEU(8) | 2 | OEG | DMB | 100 | A | 102,000 | 2.5 | 81.9 |
| PEU(8) | 2 | 1,8-OD | DMB | 100 | B | 100,000 | 2.4 | 88.0 |
| PEU(10) | 2 | 1,10-DD | DMB | 100 | B | 94,000 | 2.1 | 87.8 |
| PEU(12) | 2 | 1,12-DoD | DMB | 100 | B | 101,000 | 2.6 | 86.3 |
| PEDU | 4 | --- | anisole | 110 | C | 109,000 | 1.6 | 90.9 |
| Entry | Polymer | Tg/°C | Tc/°C | Tm/°C | Young’sm odulus/MPa b | Elongation at break /% b | Tensile strength/MPa b | Water contact angle/° c |
|---|---|---|---|---|---|---|---|---|
| 1 | PEEU(6) | - | 70 a | 87 | 87 | 527 | 17.8 | 75.6 |
| 2 | PEEU(8) | - | 60 a | 79 | 56 | 404 | 13.2 | 71.7 |
| 3 | PEU(8) | - | 87 | 116 | 90 | 677 | 26.8 | 83.8 |
| 4 | PEU(10) | - | 85 | 114 | 87 | 738 | 28.1 | 84.0 |
| 5 | PEU(12) | - | 86 | 112 | 91 | 860 | 33.2 | 84.2 |
| 6 | PEDU | - | 123 | 132 | 147 | 639 | 35.1 | 85.9 |
| 7 | PDEU [13] | −33 | - | 42 | 29 | 1,383 | 11.1 | 85.0 |
| 8 | LLDPE [26–28] | −128 | 111 | 126 | 129 | 1,104 | 19.1 | 102.0 |
| 9 | TPUMDI-PEA-EG [29] | - | - | 195–248 | - | 425 | 47 | - |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hayashi, H.; Yanagishita, Y.; Matsumura, S. Chemoenzymatic Synthesis and Chemical Recycling of Poly(ester-urethane)s. Int. J. Mol. Sci. 2011, 12, 5490-5507. https://doi.org/10.3390/ijms12095490
Hayashi H, Yanagishita Y, Matsumura S. Chemoenzymatic Synthesis and Chemical Recycling of Poly(ester-urethane)s. International Journal of Molecular Sciences. 2011; 12(9):5490-5507. https://doi.org/10.3390/ijms12095490
Chicago/Turabian StyleHayashi, Hiroto, Yoshio Yanagishita, and Shuichi Matsumura. 2011. "Chemoenzymatic Synthesis and Chemical Recycling of Poly(ester-urethane)s" International Journal of Molecular Sciences 12, no. 9: 5490-5507. https://doi.org/10.3390/ijms12095490
APA StyleHayashi, H., Yanagishita, Y., & Matsumura, S. (2011). Chemoenzymatic Synthesis and Chemical Recycling of Poly(ester-urethane)s. International Journal of Molecular Sciences, 12(9), 5490-5507. https://doi.org/10.3390/ijms12095490