Sirt3, Mitochondrial ROS, Ageing, and Carcinogenesis



{kind=link}
Abstract
:1. Introduction
2. Sirtuins as Fidelity or Tumor Suppressor Proteins
3. Sirtuins Monitor and Direct Cellular Metabolism and Stress
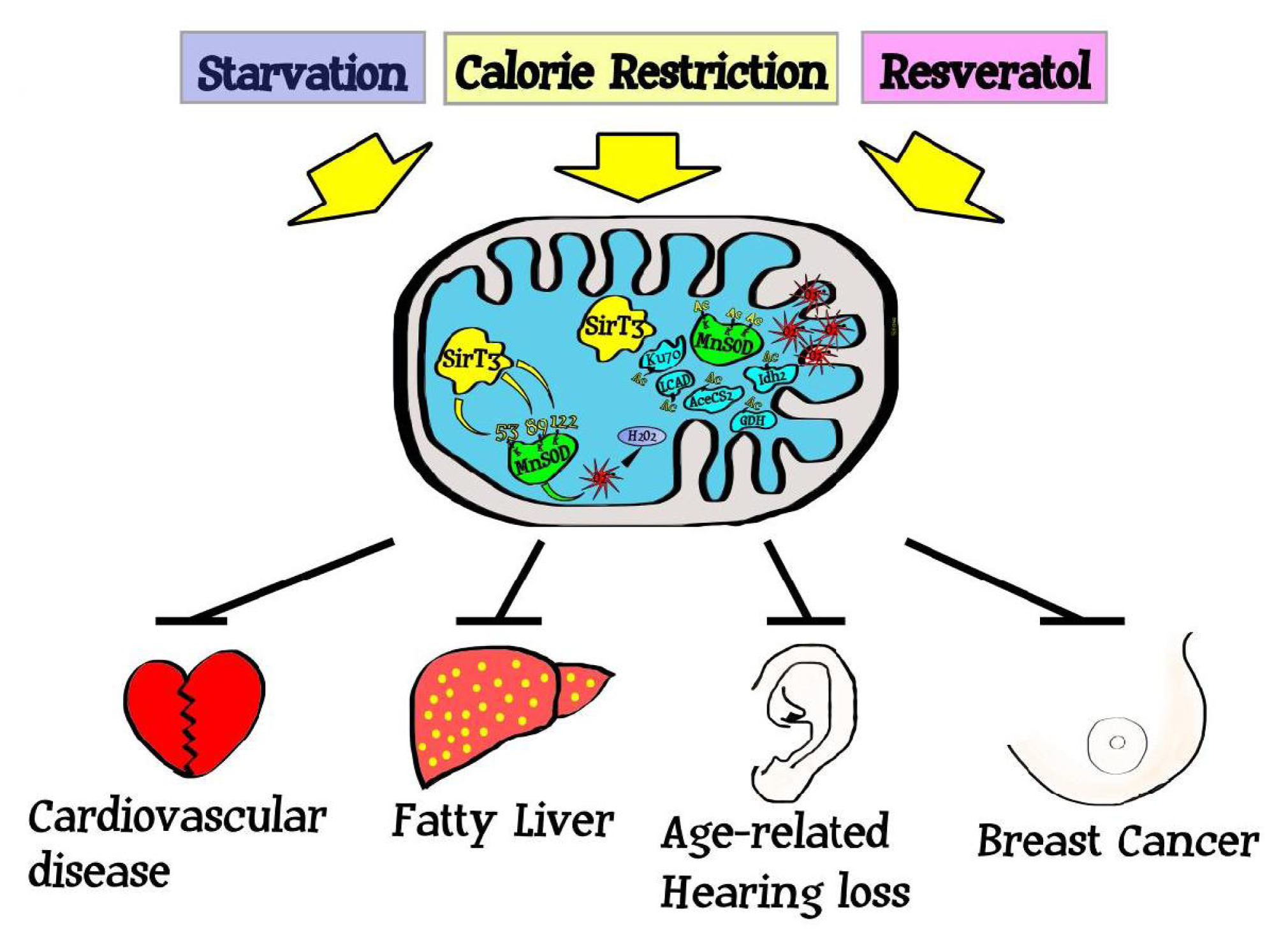
4. Sirtuins and Regulation of the Mitochondrial Acetylome
5. Sirtuins, Mitochondrial ROS, and Carcinogenesis
6. SIRT3 Is a Genomically Expressed, Mitochondrial Localized TS Protein
7. Conclusions
Acknowledgments
References
- Slane, BG; Aykin-Burns, N; Smith, BJ; Kalen, AL; Goswami, PC; Domann, FE; Spitz, DR. Mutation of succinate dehydrogenase subunit C results in increased O 2 .−, oxidative stress, and genomic instability. Cancer Res 2006, 66, 7615–7620. [Google Scholar]
- Aykin-Burns, N; Ahmad, IM; Zhu, Y; Oberley, LW; Spitz, DR. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem J 2009, 418, 29–37. [Google Scholar]
- Du, C; Gao, Z; Venkatesha, VA; Kalen, AL; Chaudhuri, L; Spitz, DR; Cullen, JJ; Oberley, LW; Goswami, PC. Mitochondrial ROS and radiation induced transformation in mouse embryonic fibroblasts. Cancer Biol Ther 2009, 8, 1962–1971. [Google Scholar]
- Mallakin, A; Sugiyama, T; Taneja, P; Matise, LA; Frazier, DP; Choudhary, M; Hawkins, GA; D’Agostino, RB, Jr; Willingham, MC; Inoue, K. Mutually Exclusive Inactivation of DMP1 and ARF/p53 in Lung Cancer. Cancer Cell 2007, 12, 381–394. [Google Scholar]
- Uren, AG; Kool, J; Matentzoglu, K; de Ridder, J; Mattison, J; van Uitert, M; Lagcher, W; Sie, D; Tanger, E; Cox, T; et al. Large-scale mutagenesis in p19ARF-and p53-deficient mice identifies cancer genes and their collaborative networks. Cell 2008, 133, 727–741. [Google Scholar]
- Vousden, KH; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar]
- Greger, V; Passarge, E; Hopping, W; Messmer, E; Horsthemke, B. Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum Genet 1989, 83, 155–158. [Google Scholar]
- Baker, SJ; Markowitz, S; Fearon, ER; Willson, JK; Vogelstein, B. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science 1990, 249, 912–915. [Google Scholar]
- Feinberg, AP; Johnson, LA; Law, DJ; Kuehn, SE; Steenman, M; Williams, BR; Thomas, G; Boland, CR; Rainier, S; Koi, M. Multiple tumor suppressor genes in multistep carcinogenesis. Tohoku J Exp Med 1992, 168, 149–152. [Google Scholar]
- Sherr, CJ; McCormick, F. The Rb and p53 pathways in cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar]
- Hunter, T. Oncoprotein networks. Cell 1997, 88, 333–346. [Google Scholar]
- Clutton, SM; Townsend, KM; Walker, C; Ansell, JD; Wright, EG. Radiation induced genomic instability and persisting oxidative stress in primary bone marrow cultures. Carcinogenesis 1996, 17, 1633–1639. [Google Scholar]
- Spitz, DR; Azzam, EI; Li, JJ; Gius, D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: a unifying concept in stress response biology. Cancer Metastasis Rev 2004, 23, 311–322. [Google Scholar]
- Duesberg, P. Does aneuploidy or mutation start cancer? Science 2005, 307, 41. [Google Scholar]
- Deng, CX. BRCA1: Cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res 2006, 34, 1416–1426. [Google Scholar]
- Wang, RH; Sengupta, K; Li, C; Kim, HS; Cao, L; Xiao, C; Kim, S; Xu, X; Zheng, Y; Chilton, B; et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 2008, 14, 312–323. [Google Scholar]
- Parada, LF; Land, H; Weinberg, RA; Wolf, D; Rotter, V. Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature 1984, 312, 649–651. [Google Scholar]
- Taylor, WR; Egan, SE; Mowat, M; Greenberg, AH; Wright, JA. Evidence for synergistic interactions between ras, myc and a mutant form of p53 in cellular transformation and tumor dissemination. Oncogene 1992, 7, 1383–1390. [Google Scholar]
- Saunders, LR; Verdin, E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene 2007, 26, 5489–5504. [Google Scholar]
- Kim, HS; Patel, K; Muldoon-Jacobs, K; Bisht, KS; Aykin-Burns, N; Pennington, JD; van der Meer, R; Nguyen, P; Savage, J; Owens, KM; et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 2010, 17, 41–52. [Google Scholar]
- Gius, D; Botero, A; Shah, S; Curry, HA. Intracellular oxidation/reduction status in the regulation of transcription factors NF-[kappa] B and AP-1. Toxicol Lett 1999, 106, 93–106. [Google Scholar]
- Pelicano, H; Carney, D; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat 2004, 7, 97–110. [Google Scholar]
- Valko, M; Izakovic, M; Mazur, M; Rhodes, CJ; Telser, J. Role of oxygen radicals in DNA damage and cancer incidence. Mol Cell Biochem 2004, 266, 37–56. [Google Scholar]
- Spitz, DR; Li, GC. Heat-induced cytotoxicity in H2O2-resistant Chinese hamster fibroblasts. J Cell Physiol 1990, 142, 255–260. [Google Scholar]
- Sies, H. Role of reactive oxygen species in biological processes. Klin Wochenschr 1991, 69, 965–968. [Google Scholar]
- Singh, KK. Mitochondria damage checkpoint, aging, and cancer. Ann NY Acad Sci 2006, 1067, 182–190. [Google Scholar]
- Valko, M; Rhodes, CJ; Moncol, J; Izakovic, M; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact 2006, 160, 1–40. [Google Scholar]
- Chen, J; Kadlubar, FF; Chen, JZ. DNA supercoiling suppresses real-time PCR: a new approach to the quantification of mitochondrial DNA damage and repair. Nucleic Acids Res 2007, 35, 1377–1388. [Google Scholar]
- Tanny, JC; Dowd, GJ; Huang, J; Hilz, H; Moazed, D. An Enzymatic Activity in the Yeast Sir2 Protein that Is Essential for Gene Silencing. Cell 1999, 99, 735–745. [Google Scholar]
- Kaeberlein, M; McVey, M; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 1999, 13, 2570–2580. [Google Scholar]
- Imai, S; Armstrong, CM; Kaeberlein, M; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar]
- Landry, J; Sutton, A; Tafrov, ST; Heller, RC; Stebbins, J; Pillus, L; Sternglanz, R. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci USA 2000, 97, 5807–5811. [Google Scholar]
- Lin, SJ; Defossez, PA; Guarente, L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 2000, 289, 2126–2128. [Google Scholar]
- North, BJ; Verdin, E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol 2004, 5, 224. [Google Scholar]
- Rogina, B; Helfand, SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci USA 2004, 101, 15998–16003. [Google Scholar]
- Wood, JG; Rogina, B; Lavu, S; Howitz, K; Helfand, SL; Tatar, M; Sinclair, D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 2004, 430, 686–689. [Google Scholar]
- Guarente, L; Partridge, L; Wallace, DC. Molecular Biology of Aging; Cold Spring Harbor Laboratory Press: Woodbury, NY, USA, 2008. [Google Scholar]
- Haigis, MC; Guarente, LP. Mammalian sirtuins—emerging roles in physiology, aging, and calorie restriction. Genes Dev 2006, 20, 2913–2921. [Google Scholar]
- Finkel, T; Deng, CX; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 460, 587–591. [Google Scholar]
- Kim, EJ; Kho, JH; Kang, MR; Um, SJ. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol Cell 2007, 28, 277–290. [Google Scholar]
- Smith, KT; Workman, JL. Introducing the acetylome. Nat Biotechnol 2009, 27, 917–919. [Google Scholar]
- Hallows, WC; Albaugh, BN; Denu, JM. Where in the cell is SIRT3?—Functional localization of an NAD+-dependent protein deacetylase. J Biochem 2008, 411, e11–e13. [Google Scholar]
- Chua, KF; Mostoslavsky, R; Lombard, DB; Pang, WW; Saito, S; Franco, S; Kaushal, D; Cheng, HL; Fischer, MR; Stokes, N; et al. Mammalian SIRT1 limits replicative life span in response to chronic genotoxic stress. Cell Metab 2005, 2, 67–76. [Google Scholar]
- Droge, W; Schipper, HM. Oxidative stress and aberrant signaling in aging and cognitive decline. Aging Cell 2007, 6, 361–370. [Google Scholar]
- Scher, MB; Vaquero, A; Reinberg, D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev 2007, 21, 920–928. [Google Scholar]
- Harman, D. Aging: a theory based on free radical and radiation chemistry. J Gerontol 1956, 11, 298–300. [Google Scholar]
- Valko, M; Leibfritz, D; Moncol, J; Cronin, MT; Mazur, M; Telser, J. Free radicals and antioxidant in normal physiological function and human disease. Int J Biochem Cell Biol 2007, 39, 44–84. [Google Scholar]
- Buffenstein, R; Edrey, YH; Yang, T; Mele, J. The oxidative stress theory of aging: embattled or invincible? Insights from non-traditional model organisms. Age (Dordr) 2008, 30, 99–109. [Google Scholar]
- Diamond, DA; Parsian, A; Hunt, CR; Lofgren, S; Spitz, DR; Goswami, PC; Gius, D. Redox factor-1 (Ref-1) mediates the activation of AP-1 in HeLa and NIH 3T3 cells in response to heat shock. J Biol Chem 1999, 274, 16959–16964. [Google Scholar]
- Tao, R; Coleman, MC; Pennington, JD; Ozden, O; Park, SH; Jiang, H; Kim, HS; Flynn, CR; Hill, S; Hayes McDonald, W; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell 40, 893–904.
- Sundaresan, NR; Samant, SA; Pillai, VB; Rajamohan, SB; Gupta, MP. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol Cell Biol 2008, 24, 6384–6401. [Google Scholar]
- Hirschey, MD; Shimazu, T; Goetzman, E; Jing, E; Schwer, B; Lombard, DB; Grueter, CA; Harris, C; Biddinger, S; Ilkayeva, OR; et al. SIRT3 regulates fatty acid oxidation via reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar]
- Qiu, X; Brown, K; Hirschey, MD; Verdin, E; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab 2010, 12, 662–667. [Google Scholar]
- Someya, S; Yu, W; Hallows, WC; Xu, J; Vann, JM; Leeuwenburgh, C; Tanokura, M; Denu, JM; Prolla, TA. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 2010, 143, 802–812. [Google Scholar]
- Lescai, F; Blanche, H; Nebel, A; Beekman, M; Sahbatou, M; Flachsbart, F; Slagboom, E; Schreiber, S; Sorbi, S; Passarino, G; Franceschi, C. Human longevity and 11p15. 5: A study in 1321 centenarians. Eur J Hum Genet 2009, 17, 1515–1519. [Google Scholar]
- Hursting, SD; Lavigne, JA; Berrigan, D; Perkins, SN; Barrett, JC. Calorie Restriction, Aging, and Cancer Prevention: Mechanisms of Action and Applicability to Humans*. Annu Rev Med 2003, 54, 131–152. [Google Scholar]
- Sinclair, DA. Toward a unified theory of caloric restriction and longevity regulation. Mech Ageing Dev 2005, 126, 987–1002. [Google Scholar]
- Ershler, WB; Longo, DL. Aging and cancer: issues of basic and clinical science. J Natl Cancer Inst 1997, 89, 1489–1497. [Google Scholar]
- Nemoto, S; Fergusson, MM; Finkel, T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science 2004, 306, 2105–2108. [Google Scholar]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar]
- Yang, XJ. Lysine acetylation and the bromodomain: a new partnership for signaling. Bioessays 2004, 26, 1076–1087. [Google Scholar]
- Shahbazian, MD; Grunstein, M. of site-specific histone acetylation and deacetylation. Annu Rev Biochem 2007, 76, 75–100. [Google Scholar]
- Schwer, B; Bunkenborg, J; Verdin, RO; Andersen, JS; Verdin, E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci USA 2006, 103, 10224–10229. [Google Scholar]
- Choudhary, C; Kumar, C; Gnad, F; Nielsen, ML; Rehman, M; Walther, TC; Olsen, JV; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar]
- Kim, SC; Sprung, R; Chen, Y; Xu, Y; Ball, H; Pei, J; Cheng, T; Kho, Y; Xiao, H; Xiao, L; et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 2006, 23, 607–618. [Google Scholar]
- Yang, H; Baur, JA; Chen, A; Miller, C; Adams, JK; Kisielewski, A; Howitz, KT; Zipkin, RE; Sinclair, DA. Design and synthesis of compounds that extend yeast replicative lifespan. Aging Cell 2007, 6, 35–43. [Google Scholar]
- Lombard, DB; Alt, FW; Cheng, HL; Bunkenborg, J; Streeper, RS; Mostoslavsky, R; Kim, J; Yancopoulos, G; Valenzuela, D; Murphy, A; et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol 2007, 27, 8807–8814. [Google Scholar]
- Ahn, BH; Kim, HS; Song, S; Lee, IH; Liu, J; Vassilopoulos, A; Deng, CX; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA 2008, 105, 14447–14452. [Google Scholar]
- Ershler, WB; Longo, DL. The biology of aging. Cancer 1997, 80, 1284–1293. [Google Scholar]
- Gemma, C; Vila, J; Bachstetter, A; Bickford, PC. Early inhibition of TNF [alpha] increases 6-hydroxydopamine-induced striatal degeneration. Brain Res 2007, 1147, 240–247. [Google Scholar]
- Dayal, D; Martin, SM; Limoli, CL; Spitz, DR. Hydrogen peroxide mediates the radiation-induced mutator phenotype in mammalian cells. Biochem J 2008, 413, 185–191. [Google Scholar]
- Schwer, B; Eckersdorff, M; Li, Y; Silva, JC; Fermin, D; Kurtev, MV; Giallourakis, C; Comb, MJ; Alt, FW; Lombard, DB. Calorie restriction alters mitochondrial protein acetylation. Aging Cell 2009, 8, 604–606. [Google Scholar]
- Lombard, DB. Sirtuins at the breaking point: SIRT6 in DNA repair. Aging (Albany NY) 2009, 1, 12–16. [Google Scholar]
- Sundaresan, NR; Gupta, M; Kim, G; Rajamohan, SB; Isbatan, A; Gupta, MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest 2009, 119, 2758–2771. [Google Scholar]
- Bell, EL; Emerling, BM; Ricoult, SJ; Guarente, L. SirT3 suppresses hypoxia inducible factor 1α and tumor growth by inhibiting mitochondrial ROS production. Oncogene 2011, 30, 2986–2996. [Google Scholar]
- Finley, LW; Carracedo, A; Lee, J; Souza, A; Egia, A; Zhang, J; Teruya-Feldstein, J; Moreira, PI; Cardoso, SM; Clish, CB; Pandolfi, PP; Haigis, MC. SIRT3 Opposes Reprogramming of Cancer Cell Metabolism through HIF1 a Destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar]
- Kendrick, AA; Choudhury, M; Rahman, SM; McCurdy, CE; Friederich, M; Vanhove, JL; Watson, PA; Birdsey, N; Bao, J; Gius, D; et al. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem J 2011, 433, 505–514. [Google Scholar]
- Sebastian, C; Mostoslavsky, R. SIRT3 in Calorie Restriction: Can You Hear Me Now? Cell 2010, 143, 667–668. [Google Scholar]
- Deng, YT; Huang, HC; Lin, JK. Rotenone induces apoptosis in MCF-7 human breast cancer cell-mediated ROS through JNK and p38 signaling. Mol Carcinog 2010, 49, 141–151. [Google Scholar]
- Pujana, MA; Han, JD; Starita, LM; Stevens, KN; Tewari, M; Ahn, JS; Rennert, G; Moreno, V; Kirchhoff, T; Gold, B; et al. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat Genet 2007, 39, 1338–1349. [Google Scholar]
- Oberley, LW; Oberley, TD. Role of antioxidant enzymes in cell immortalization and transformation. Mol Cell Biochem 1988, 84, 147–153. [Google Scholar]
- Chen, Y; Zhang, J; Lin, Y; Lei, Q; Guan, KL; Zhao, S; Xiong, Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep 12, 534–541.
- Schumacker, PT. SIRT3 Controls Cancer Metabolic Reprogramming by Regulating ROS and HIF. Cancer Cell 2011, 19, 299–300. [Google Scholar]
- Hallows, WC; Lee, S; Denu, JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci USA 2006, 103, 10230–10235. [Google Scholar]
- Schlicker, C; Gertz, M; Papatheodorou, P; Kachholz, B; Becker, CF; Steegborn, C. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J Mol Biol 2008, 382, 790–801. [Google Scholar]
- Cimen, H; Han, MJ; Yang, Y; Tong, Q; Koc, H; Koc, EC. Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry 2010, 49, 304–311. [Google Scholar]
- Yang, Y; Cimen, H; Han, MJ; Shi, T; Deng, JH; Koc, H; Palacios, OM; Montier, L; Bai, Y; Tong, Q; Koc, EC. NAD+-dependent deacetylase SIRT3 regulates mitochondrial protein synthesis by deacetylation of the ribosomal protein MRPL10. J Biol Chem 2010, 285, 7417–7429. [Google Scholar]
- Marfe, G; Tafani, M; Indelicato, M; Sinibaldi-Salimei, P; Reali, V; Pucci, B; Fini, M; Russo, MA. Kaempferol induces apoptosis in two different cell lines via Akt inactivation, Bax and SIRT3 activation, and mitochondrial dysfunction. J Cell Biochem 2009, 106, 643–650. [Google Scholar]
- Allison, SJ; Milner, J. SIRT3 is pro-apoptotic and participates in distinct basal apoptotic pathways. Cell Cycle 2007, 6, 2669–2677. [Google Scholar]
- Veatch, JR; McMurray, MA; Nelson, ZW; Gottschling, DE. Mitochondrial dysfunction leads to nuclear genome instability via an iron-sulfur cluster defect. Cell 2009, 137, 1247–1258. [Google Scholar]
- Chandel, NS; McClintock, DS; Feliciano, CE; Wood, TM; Melendez, JA; Rodriguez, AM; Schumacker, PT. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1 during hypoxia. A mechanism of O2 sensing. J Biol Chem 2000, 275, 25130–25138. [Google Scholar]
- Han, D; Antunes, F; Canali, R; Rettori, D; Cadenas, E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem 2003, 278, 5557–5563. [Google Scholar]
- Kulisz, A; Chen, N; Chandel, NS; Shao, Z; Schumacker, PT. Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes. Am J Physiol Lung Cell Mol Physiol 2002, 282, L1324–L1329. [Google Scholar]
- Sherr, CJ. Principles of tumor suppression. Cell 2004, 116, 235–246. [Google Scholar]

© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Park, S.-H.; Ozden, O.; Jiang, H.; Cha, Y.I.; Pennington, J.D.; Aykin-Burns, N.; Spitz, D.R.; Gius, D.; Kim, H.-S. Sirt3, Mitochondrial ROS, Ageing, and Carcinogenesis. Int. J. Mol. Sci. 2011, 12, 6226-6239. https://doi.org/10.3390/ijms12096226
Park S-H, Ozden O, Jiang H, Cha YI, Pennington JD, Aykin-Burns N, Spitz DR, Gius D, Kim H-S. Sirt3, Mitochondrial ROS, Ageing, and Carcinogenesis. International Journal of Molecular Sciences. 2011; 12(9):6226-6239. https://doi.org/10.3390/ijms12096226
Chicago/Turabian StylePark, Seong-Hoon, Ozkan Ozden, Haiyan Jiang, Yong I. Cha, J. Daniel Pennington, Nukhet Aykin-Burns, Douglas R. Spitz, David Gius, and Hyun-Seok Kim. 2011. "Sirt3, Mitochondrial ROS, Ageing, and Carcinogenesis" International Journal of Molecular Sciences 12, no. 9: 6226-6239. https://doi.org/10.3390/ijms12096226
APA StylePark, S. -H., Ozden, O., Jiang, H., Cha, Y. I., Pennington, J. D., Aykin-Burns, N., Spitz, D. R., Gius, D., & Kim, H. -S. (2011). Sirt3, Mitochondrial ROS, Ageing, and Carcinogenesis. International Journal of Molecular Sciences, 12(9), 6226-6239. https://doi.org/10.3390/ijms12096226