Impact of Cyanidin-3-Glucoside on Glycated LDL-Induced NADPH Oxidase Activation, Mitochondrial Dysfunction and Cell Viability in Cultured Vascular Endothelial Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
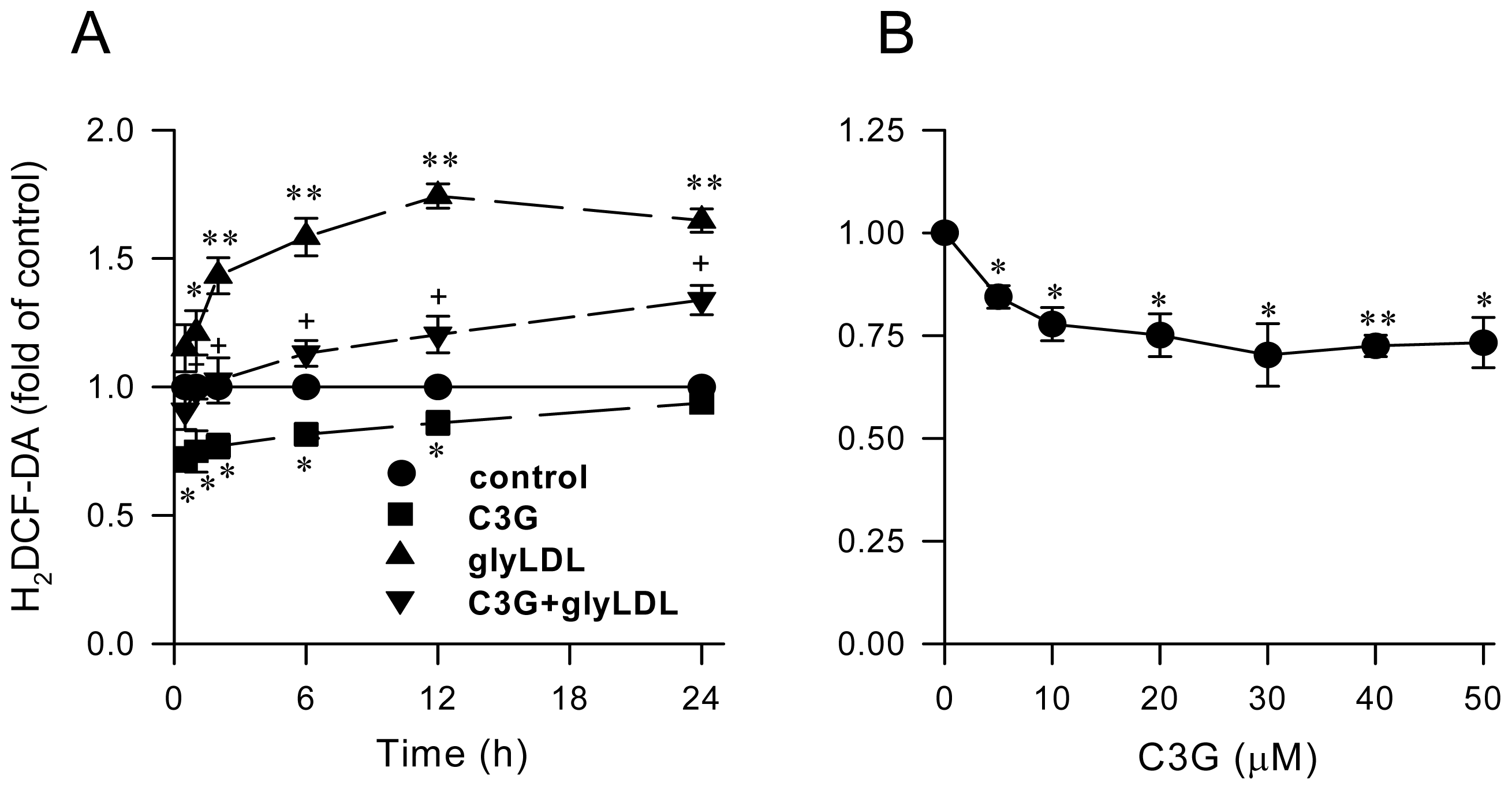
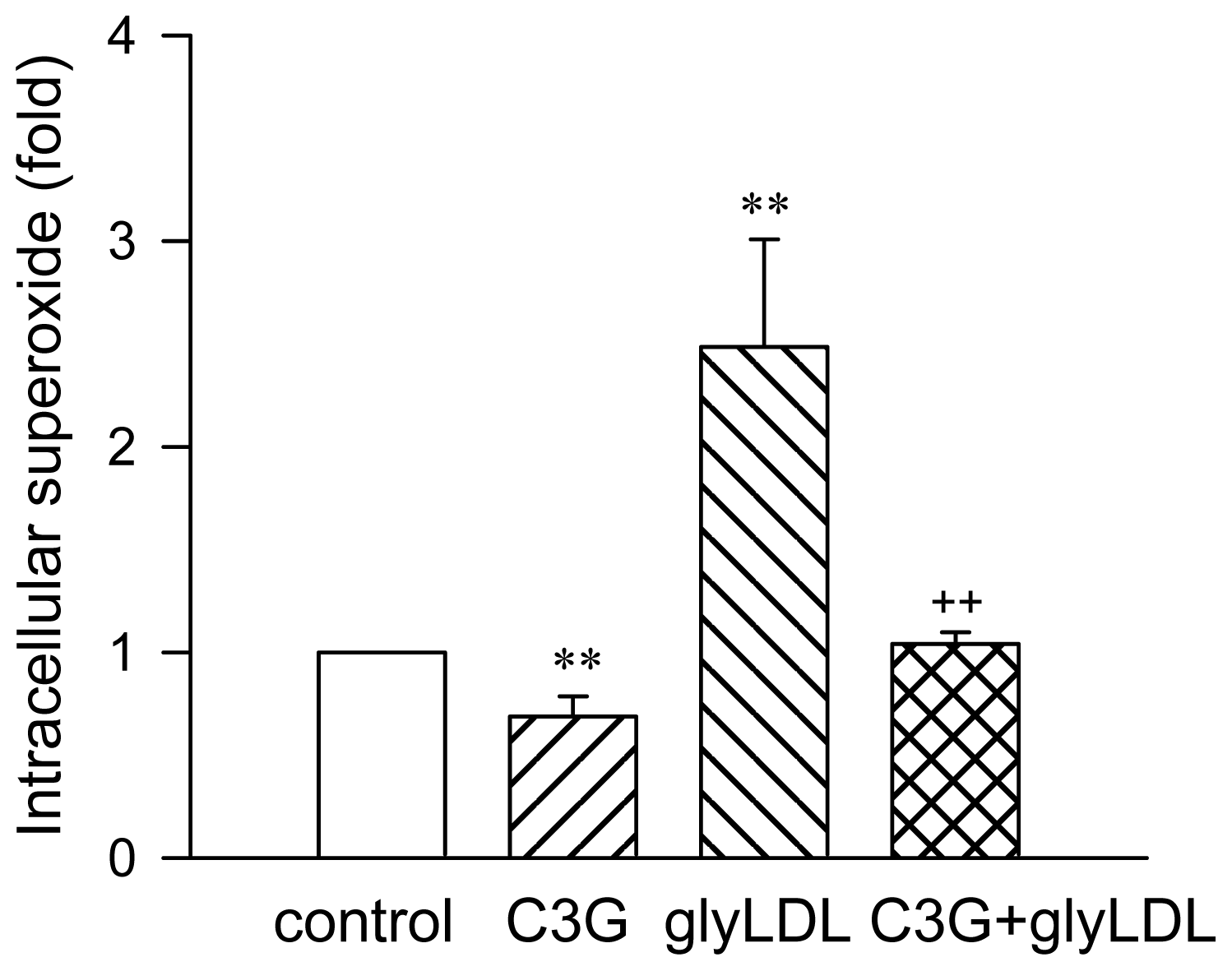
2.1. Effect of C3G on GlyLDL-Induced ROS Production
2.2. Effects of C3G on the Activities of mETC Enzymes in GlyLDL-Treated EC
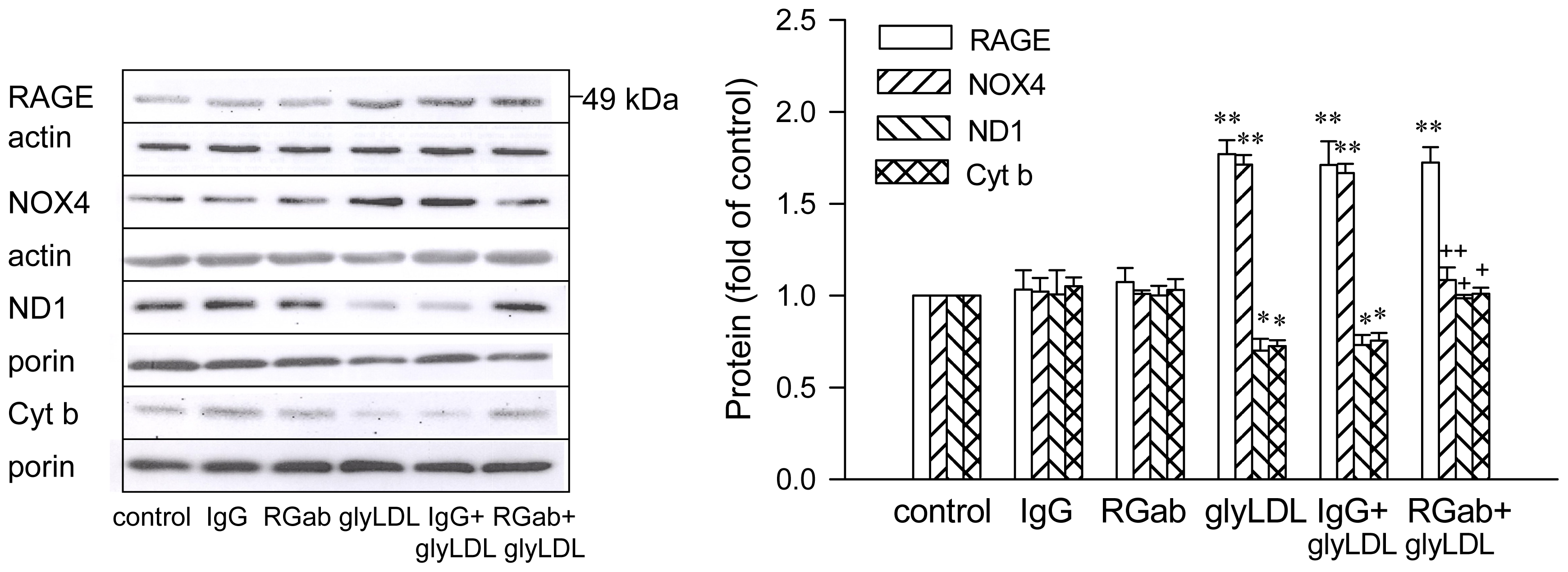
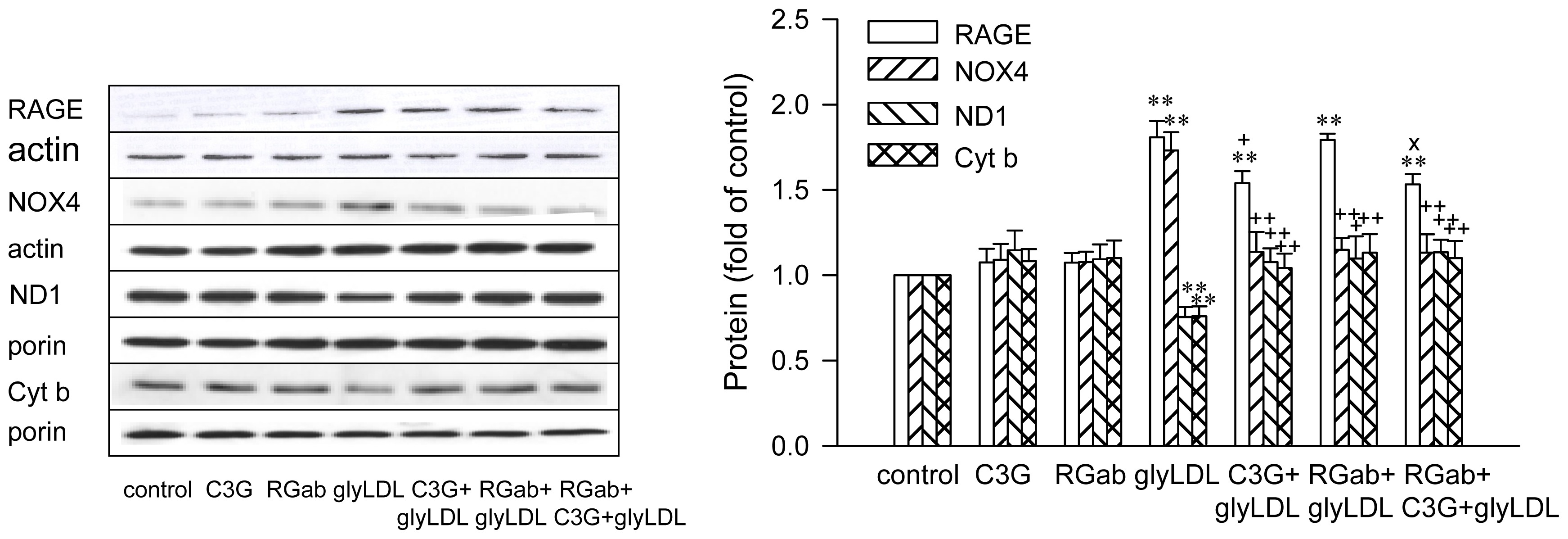
2.3. Effects of C3G on GlyLDL-Induced Changes in NOX4, ND1 and Cyt b
2.4. Effect of RAGE Blockage and C3G on GlyLDL-Induced Changes in RAGE, NOX and mETC Enzymes
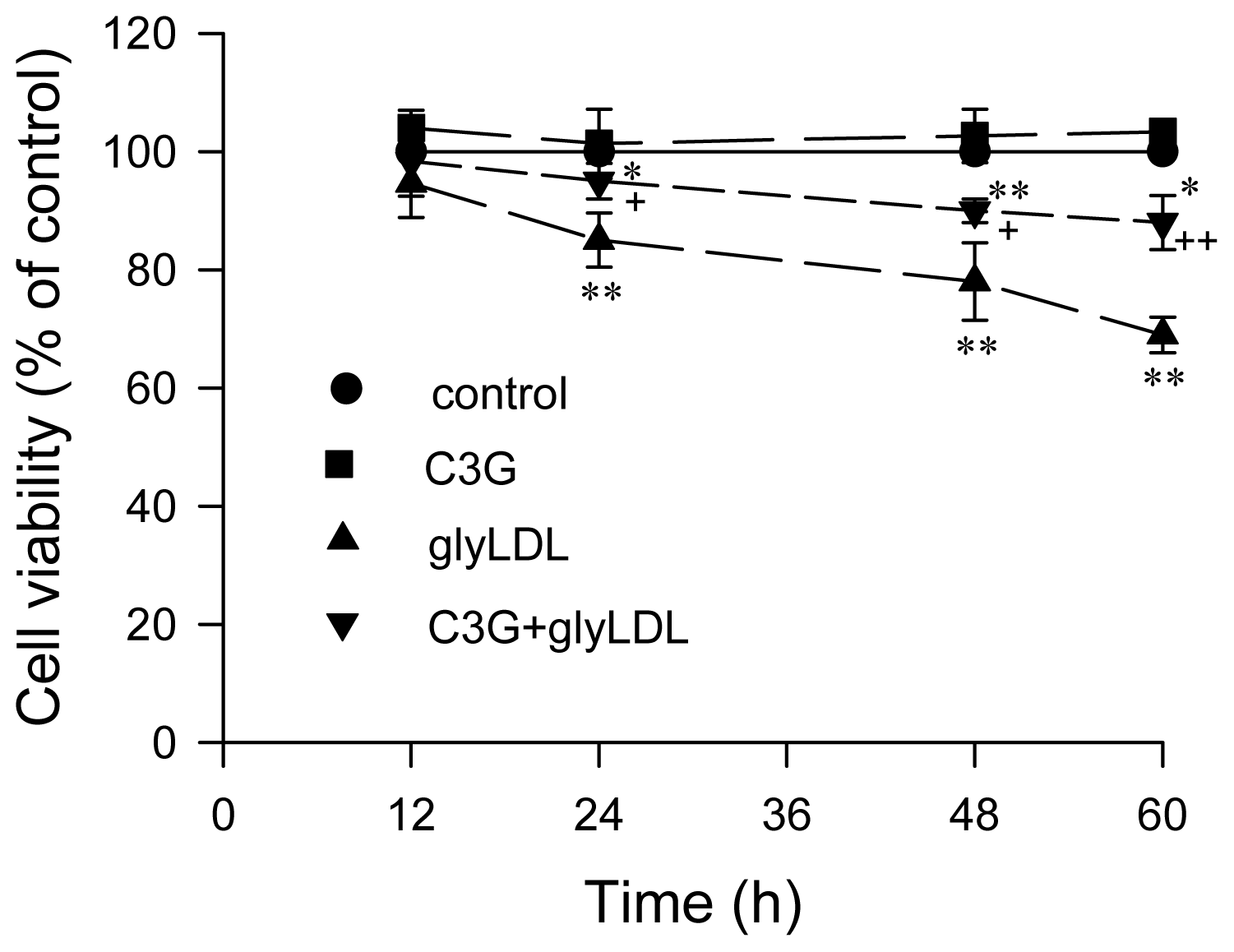
2.5. Effect of GlyLDL on Cell Viability
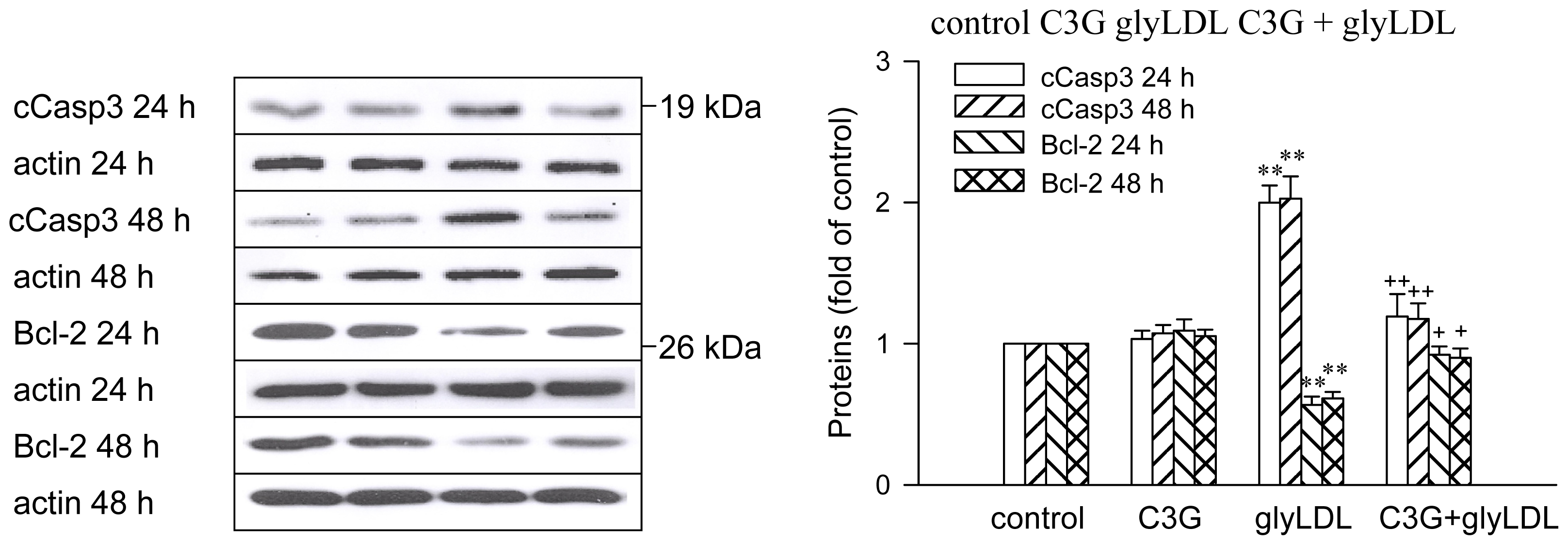
2.6. Effects of GlyLDL on Regulators for Cell Viability
2.7. Discussion
3. Experimental Sections
3.1. Isolation and Modification of Lipoproteins
3.2. Cell Culture
3.3. Experimental Incubation
3.4. Assessment of Redox Status
3.5. Intracellular Superoxide Measurement
3.6. Western Blotting Assay
3.7. ND Activity
3.8. Succinate Cytochrome C Reductase (SCCR) Activity
3.9. Ubiquinol Cytochrome C Reductase (UCCR) Activity
3.10. Cytochrome C Oxidase (COX) Activity
3.11. Citrate Synthase (CS) Activity
3.12. Cell Viability Assay
3.13. Statistical Analysis
4. Conclusions
Acknowledgements
References
- Virella, G.; Lopes-Virella, M.F. The Pathogenic Role of the Adaptive Immune Response to Modified LDL in Diabetes. Front. Endocrinol. (Lausanne) 2012, 3, 76. [Google Scholar]
- Chao, P.C.; Huang, C.N.; Hsu, C.C.; Yin, M.C.; Guo, Y.R. Association of dietary AGEs with circulating AGEs, glycated LDL, IL-1α and MCP-1 levels in type 2 diabetic patients. Eur. J. Nutr 2010, 49, 429–434. [Google Scholar]
- Zhao, R.; Shen, G.X. Functional modulation of antioxidant enzymes in vascular endothelial cells by glycated LDL. Atherosclerosis 2005, 179, 277–284. [Google Scholar]
- Abid, M.R.; Kachra, Z.; Spokes, K.C.; Aird, W.C. NADPH oxidase activity is required for endothelial cell proliferation and migration. FEBS Lett 2000, 4862, 252–256. [Google Scholar]
- Block, K.; Gorinm, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar]
- Hüttemann, M.; Pecina, P.; Rainbolt, M.; Sanderson, T.H.; Kagan, V.E.; Samavati, L.; Doan, J.W.; Lee, I. The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: From respiration to apoptosis. Mitochondrion 2011, 11, 369–378. [Google Scholar]
- Sangle, G.V.; Roy Chowdhury, S.K.; Xie, X.; Halayko, A.J.; Stelmark, G.; Shen, G.X. Impairment of mitochondrial respiratory chain activity in aortic endothelial cells induced by glycated LDL. Free Radic. Biol. Med 2010, 48, 781–790. [Google Scholar]
- Martin, S.; Giannone, G.; Andriantsitohaina, R.; Martinez, M.C. Delphinidin, an active compound of red wine, inhibits endothelial cell apoptosis via nitric oxide pathway and regulation of calcium homeostasis. Br. J. Pharmacol 2003, 139, 1095–1102. [Google Scholar]
- Chen, C.Y.; Yi, L.; Jin, X.; Mi, M.T.; Zhang, T.; Ling, W.H.; Yu, B. Delphinidin attenuates stress injury induced by oxidized low-density lipoprotein in human umbilical vein endothelial cells. Chem. Biol. Interact 2010, 183, 105–112. [Google Scholar]
- Hossenian, F.S.; Beta, T. Saskatoon and wild blueberries have higher anthocyanin contents than other Manitoba berries. J. Agric. Food Chem 2007, 55, 10832–10838. [Google Scholar]
- Chen, G.; Bower, K.A.; Xu, M.; Ding, M.; Shi, X.; Ke, Z.J.; Luo, J. Cyanidin-3-glucoside reverses ethanol-induced inhibition of neurite outgrowth: role of glycogen synthase kinase 3 beta. Neurotox. Res 2009, 15, 321–331. [Google Scholar]
- Roy Chowdhury, S.K.; Sangle, G.V.; Xie, X.; Halayko, A.J.; Stelmark, G.; Shen, G.X. Effect of extensively oxidized low-density lipoprotein on mitochondrial function and reactive oxygen species in porcine aortic endothelial cells. Am. J. Physiol. Endocrinol. Metab 2010, 298, E89–E98. [Google Scholar]
- Youdim, K.A.; McDonald, J.; Kalt, W.; Joseph, J.A. Potential role of dietary flavonoids in reducing microvascular endothelium vulnerability to oxidative and inflammatory insults (small star, filled). J. Nutr. Biochem 2002, 13, 282–288. [Google Scholar]
- Miranda-Rottmann, S.; Aspillaga, A.A.; Pérez, D.D.; Vasquez, L.; Martinez, A.L.; Leighton, F. Juice and phenolic fractions of the berry Aristotelia chilensis inhibit LDL oxidation in vitro and protect human endothelial cells against oxidative stress. J. Agric. Food Chem 2002, 50, 7542–7547. [Google Scholar]
- Bagchi, D.; Sen, C.K.; Ray, S.D.; Das, D.K.; Bagchi, M.; Preuss, H.G.; Vinson, J.A. Molecular mechanisms of cardioprotection by a novel grape seed proanthocyanidin extract. Mutat. Res. 2003, 523–524, 87–97. [Google Scholar]
- Guo, H.; Ling, W.; Wang, Q.; Liu, C.; Hu, Y.; Xia, M. Cyanidin 3-glucoside protects 3T3-L1 adipocytes against H2O2- or TNF-α-induced insulin resistance by inhibiting c-Jun NH2-terminal kinase activation. Biochem. Pharmacol 2008, 74, 1393–1401. [Google Scholar]
- Sangle, G.V.; Zhao, R.; Misuno, T.; Shen, G.X. Involvement of RAGE, NADPH oxidase and Ras/Raf-1 pathway in glycated LDL-induced expression of heat shock factor-1 and plasminogen activator inhibitor-1 in vascular endothelial cells. Endocrinology 2010, 151, 4455–4466. [Google Scholar]
- DiMauro, S.; Andreu, A.L. Mutations in mitochondrial DNA as a cause of exercise intolerance. Ann. Med 2001, 33, 472–476. [Google Scholar]
- Munusamy, S.; Saba, H.; Mitchell, T.; Megyesi, J.K.; Brock, R.W.; Macmillan-Crow, L.A. Alteration of renal respiratory Complex-III during experimental type-1 diabetes. BMC Endocr. Disord 2009, 9, 2. [Google Scholar]
- Bonawitz, N.D.; Clayton, D.A.; Shadel, G.S. Initiation and beyond: multiple functions of the human mitochondrial transcription machinery. Mol. Cell 2006, 24, 813–825. [Google Scholar]
- Ramasamy, R.; Yan, S.F.; Schmidt, A.M. Advanced glycation endproducts: from precursors to RAGE: Round and round we go. Amino Acids 2012, 42, 1151–1161. [Google Scholar]
- Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev 2001, 15, 2922–2933. [Google Scholar]
- Kelly, P.N.; Strasser, A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ 2011, 18, 1414–1424. [Google Scholar]
- Ren, S.; Shatadal, S.; Shen, G.X. Protein Kinase C-β Mediates Lipoprotein-induced Generation of PAI-1 from Vascular Endothelial Cells. Am. J. Physiol.: Endocrinol. Metab 2000, 278, E656–E662. [Google Scholar]
- Zhang, J.Y.; Ren, S.; Sun, D.F.; Shen, G.X. Influence of Glycation on LDL-induced Generation of Fibrinolytic Regulators in Vascular Endothelial Cells. Arterioscler. Thromb. Vasc. Biol 1998, 18, 1140–1148. [Google Scholar]
- Duell, P.B.; Oram, J.F.; Bierman, E.L. Nonenzymatic glycosylation of HDL resulting in inhibition of high-affinity binding to cultured human fibroblasts. Diabetes 1990, 39, 1257–1263. [Google Scholar]
- Xanthos, T.; Bassiakou, E.; Koudouna, E.; Tsirikos-Karapanos, N.; Lelovas, P.; Papadimitriou, D.; Dontas, I.; Papadimitriou, L. Baseline hemodynamics in anesthetized landrace-large white swine: Reference values for research in cardiac arrest and cardiopulmonary resuscitation models. J. Am. Assoc. Lab. Anim. Sci 2007, 46, 21–25. [Google Scholar]
- Hou, D.; Tong, X.H.; Terahara, N.; Luo, D.; Fuji, M. Delphinidin 3-sambubioside, a Hibiscus anthocyanin, induces apoptosis in human leukemia cells through reactive oxygen species-mediated mitochondrial pathway. Arch. Biochem. Biophys 2005, 440, 101–109. [Google Scholar]
- Xie, X.; Zhao, R.; Shen, G.X. Influence of delphinidin-3-glucoside on oxidized low density lipoprotein-indued oxidative stress and apoptosis in cultured endothelial cells. J. Agric. Food Chem 2012, 60, 1850–1856. [Google Scholar]
- Gilleron, M.; Marechal, X.; Montaigne, D.; Franczak, J.; Neviere, R.; Lancel, S. NADPH oxidase participates to doxorubicin-induced cardiac myocyte apoptosis. Biochem. Biophys. Res. Commun 2009, 388, 727–731. [Google Scholar]
- Zhao, R.; Moghadasian, M.H.; Shen, G.X. Involvement of NADPH oxidase in up-regulation of plasminogen activator-inhibitor-1 and heat shock factor-1 in mouse embryo fibroblasts induced by oxidized LDL and in apolipoprotein E-deficient mice. Free Radic. Res 2011, 45, 1013–1023. [Google Scholar]
- Birch-Machin, M.A.; Briggs, H.L.; Saborido, A.A.; Bindoff, L.A.; Turnbull, D.M. An evaluation of the measurement of the activities of complexes I–IV in the respiratory chain of human skeletal muscle mitochondria. Biochem. Med. Metab. Biol 1994, 51, 35–42. [Google Scholar]
- Chowdhury, S.K.; Drahota, Z.; Floryk, D.; Calda, P.; Houstek, J. Activities of mitochondrial oxidative phosphorylation enzymes in cultured amniocytes. Clin. Chim. Acta 2000, 298, 157–173. [Google Scholar]
- Vondra, K.; Rath, R.; Bass, A.; Slabochová, Z.; Teisinger, J.; Vitek, V. Enzyme activities in quadriceps femoris muscle of obese diabetic male patients. Diabetologia 1977, 13, 527–529. [Google Scholar]
- Kirkland, R.A.; Franklin, J.L. Evidence for redox regulation of cytochrome c release during programmed neuronal death: antioxidant effects of protein synthesis and casepase inhibition. J. Neuroscience 2001, 21, 1949–1963. [Google Scholar]
- Srere, P.A. Citrate synthase. Methods Enzymol 1969, 13, 3–26. [Google Scholar]








© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xie, X.; Zhao, R.; Shen, G.X. Impact of Cyanidin-3-Glucoside on Glycated LDL-Induced NADPH Oxidase Activation, Mitochondrial Dysfunction and Cell Viability in Cultured Vascular Endothelial Cells. Int. J. Mol. Sci. 2012, 13, 15867-15880. https://doi.org/10.3390/ijms131215867
Xie X, Zhao R, Shen GX. Impact of Cyanidin-3-Glucoside on Glycated LDL-Induced NADPH Oxidase Activation, Mitochondrial Dysfunction and Cell Viability in Cultured Vascular Endothelial Cells. International Journal of Molecular Sciences. 2012; 13(12):15867-15880. https://doi.org/10.3390/ijms131215867
Chicago/Turabian StyleXie, Xueping, Ruozhi Zhao, and Garry X. Shen. 2012. "Impact of Cyanidin-3-Glucoside on Glycated LDL-Induced NADPH Oxidase Activation, Mitochondrial Dysfunction and Cell Viability in Cultured Vascular Endothelial Cells" International Journal of Molecular Sciences 13, no. 12: 15867-15880. https://doi.org/10.3390/ijms131215867
APA StyleXie, X., Zhao, R., & Shen, G. X. (2012). Impact of Cyanidin-3-Glucoside on Glycated LDL-Induced NADPH Oxidase Activation, Mitochondrial Dysfunction and Cell Viability in Cultured Vascular Endothelial Cells. International Journal of Molecular Sciences, 13(12), 15867-15880. https://doi.org/10.3390/ijms131215867