Identification and Characterization of MicroRNAs from Barley (Hordeum vulgare L.) by High-Throughput Sequencing


 ,
,
Abstract
:1. Introduction
2. Results and Discussion
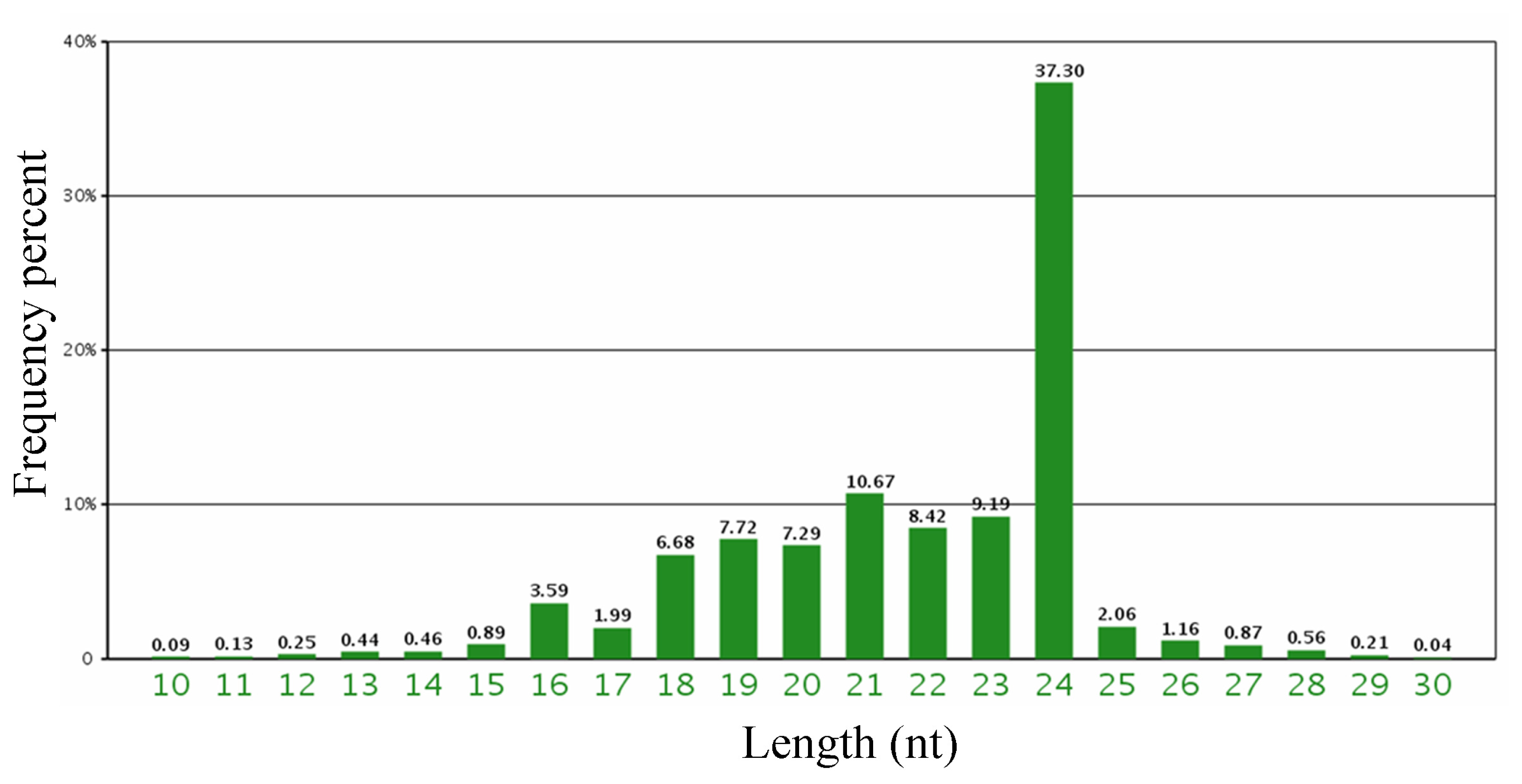
2.1. Sequencing Barley Small RNA Library
2.2. Identification of Conserved Barley miRNA and Evolutionary Conservation
2.3. Identification of Novel miRNAs
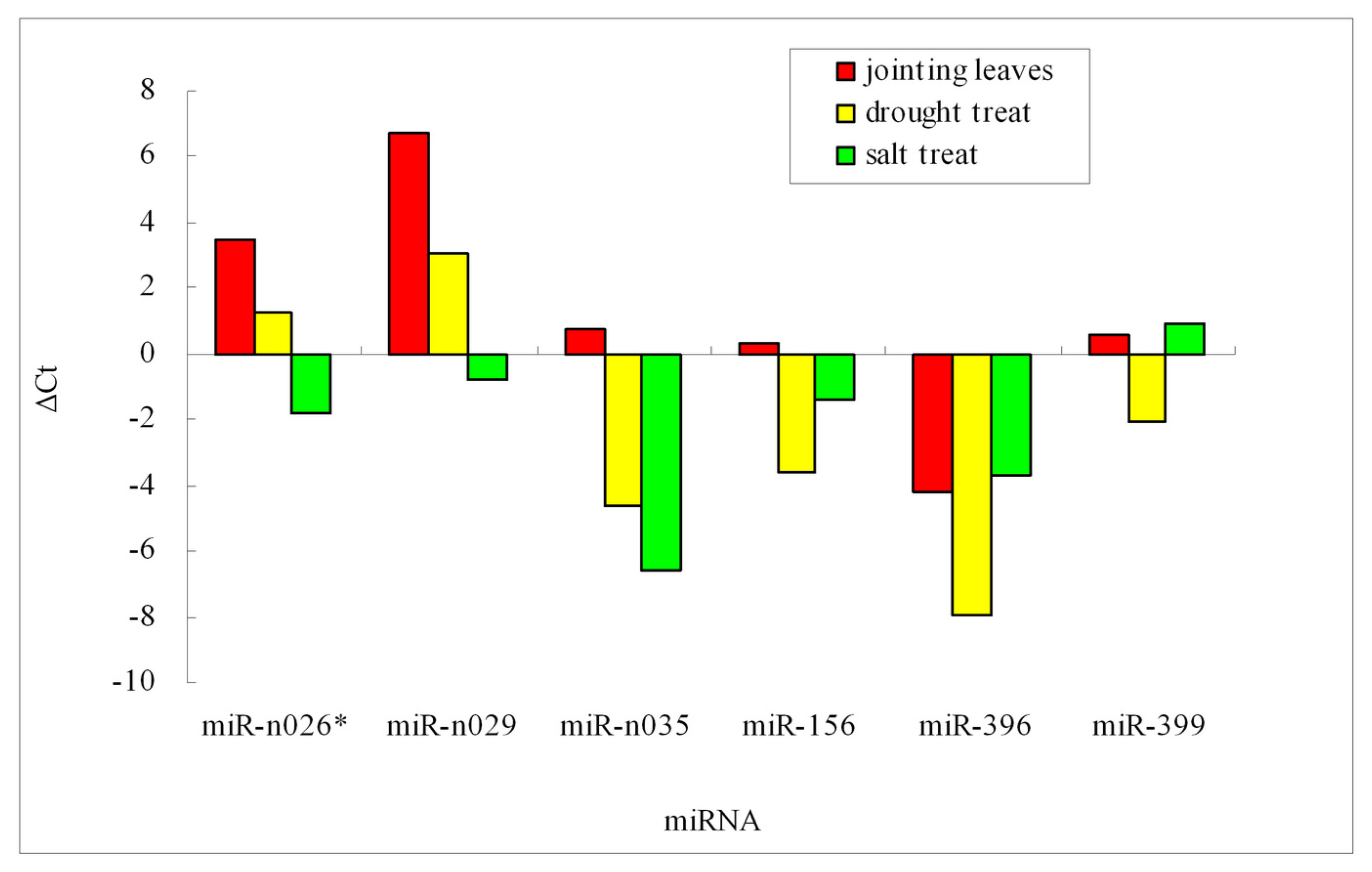
2.4. Validation of Barley miRNA
2.5. miRNA and Stress Tolerance
2.6. Target Gene Prediction
3. Experimental Section
3.1. Plant Materials
3.2. Small RNA Library Preparation and Sequencing
3.3. Bioinformatic Analysis for miRNA Identification
3.4. Validation of Barley miRNAs
3.5. Target Gene Prediction
4. Conclusions
Acknowledgments
References
- Bartel, D.P. MicroRNAs: Genomics biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar]
- Carrington, J.C.; Ambros, V. Role of microRNAs in plant and animal development. Science 2003, 301, 336–338. [Google Scholar]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarily to lin-14. Cell 1993, 75, 843–854. [Google Scholar]
- Cui, Q.H.; Yu, Z.B.; Purisima, E.O.; Wang, E. Principles of microRNA regulation of a human cellular signaling network. Mol. Syst. Biol 2006, 2. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Weinstein, E.G.; Rhoades, M.W.; Bartel, B.; Bartel, D.P. MicroRNAs in plants. Genes Dev 2002, 16, 1616–1626. [Google Scholar]
- Zhang, B.H.; Pan, X.P.; Wang, Q.L.; Cobb, G.P.; Anderson, T.A. Identification and characterization of new plant microRNAs using EST analysis. Cell Res 2005, 15, 336–360. [Google Scholar]
- Lu, C.; Kulkarni, K.; Souret, F.F.; MuthuValliappan, R.; Tej, S.S.; Poethig, R.S.; Henderson, I.R.; Jacobsen, S.E.; Wang, W.; Green, P.J.; et al. MicroRNAs and other small RNAs enriched in the Arabidopsis RNA-dependent RNA polymerase-2 mutant. Genome Res 2006, 16, 1276–1288. [Google Scholar]
- Stark, A.; Kheradpour, P.; Parts, L.; Brennecke, J.; Hodges, E.; Hannon, G.J.; Kellis, M. Systematic discovery and characterization of fly microRNAs using 12 Drosophila genomes. Genome Res 2007, 17, 1865–1879. [Google Scholar]
- Xue, L.J.; Zhang, J.J.; Xue, H.W. Characterization and expression profiles of miRNAs in rice seeds. Nucleic Acids Res 2009, 37, 916–930. [Google Scholar]
- Ruan, M.B.; Zhao, Y.T.; Meng, Z.H.; Wang, X.J.; Yang, W.C. Conserved miRNA analysis in Gossypium hirsutum through small RNA sequencing. Genomics 2009, 94, 263–268. [Google Scholar]
- Zhao, C.Z.; Xia, H.; Frazier, T.P.; Yao, Y.Y.; Bi, Y.P.; Li, A.Q.; Li, M.J.; Li, C.S.; Zhang, B.H.; Wang, X.J. Deep sequencing identifies novel and conserved microRNAs in peanuts (Arachis hypogaea L.). BMC Plant Biol 2010, 10. [Google Scholar] [CrossRef]
- Schulte, D.; Timothy, J.C.; Andreas, G.; Langridge, P.; Matsumoto, T.; Muehlbauer, G.; Sato, K.; Schulman, A.H.; Raugh, R.; Roger, P.W.; Stein, N. The international barley sequencing consortium—At the threshold of efficient access to the barley genome. Plant Physiol. 2009, 149, 142–147. [Google Scholar]
- Sreenivasulu, N.; Graner, A.; Wobus, U. Barley genomics: An overview. Int. J. Plant Genomics 2008. [Google Scholar] [CrossRef]
- Colaiacovo, M.; Subacchi, A.; Bagnaresi, P.; Lamontanara, A.; Cattivelli, L.; Faccioli, P. A computational-based update on microRNAs and their targets in barley (Hordeum vulgare L.). BMC Genomics 2010, 11. [Google Scholar] [CrossRef]
- Schreiber, A.W.; Shi, B.J.; Huang, C.Y.; Langridge, P.; Baumann, U. Discovery of barley miRNAs through deep sequencing of short reads. BMC Genomics 2011, 12. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol 1990, 215, 403–410. [Google Scholar]
- Szittya, G.; Moxon, S.; Santos, D.M.; Jing, R.; Fevereiro, M.P.; Moulton, V.; Dalmay, T. High-throughput sequencing of Medicago truncatula short RNAs identifies eight new miRNA families. BMC Genomics 2008, 9. [Google Scholar] [CrossRef]
- Morin, R.D.; Aksay, G.; Dolgosheina, E.; Ebhardt, H.A.; Magrini, V.; Mardis, E.R.; Sahinalp, S.C.; Unrau, P.J. Comparative analysis of the small RNA transcriptomes of Pinus contorta and Oryza sativa. Genome Res 2008, 18, 571–584. [Google Scholar]
- Rajagopalan, R.; Vaucheret, H.; Trejo, J.; Bartel, D.P. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev 2006, 20, 3407–3425. [Google Scholar]
- Herr, A.J. Pathways through the small RNA world of plants. FEBS Lett 2005, 579, 5879–5888. [Google Scholar]
- Vazquez, F. Arabidopsis endogenous small RNAs: Highways and byways. Trends Plant Sci 2006, 11, 460–468. [Google Scholar]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.F; Carrington, J.C.; Chen, X.M.; Green, P.J.; et al. Criteria for annotation of plant microRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar]
- Song, C.N.; Wang, C.; Zhang, C.Q.; Korir, N.K.; Yu, H.P.; Ma, Z.Q.; Fang, J.G. Deep sequencing discovery of novel and conserved microRNAs in trifoliate orange (Citrus trifoliata). BMC Genomics 2010, 11. [Google Scholar] [CrossRef]
- Ruan, M.B.; Zhao, Y.T.; Meng, Z.H.; Wang, X.J.; Yang, W.C. Conserved miRNA analysis in Gossypium hirsutum through small RNA sequencing. Genomics 2009, 94, 263–268. [Google Scholar]
- Rodriguez, R.E.; Mecchia, M.A.; Debernardi, J.M.; Schommer, C.; Weigel, D.; Palatnik, J.F. Control of cell proliferation in Arabidopsis thaliana by microRNA miR396. Development 2010, 137, 103–112. [Google Scholar]
- Jones-Rhoades, M.W.; Bartel, D.P. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell 2004, 14, 787–799. [Google Scholar]
- Xie, K.; Wu, C.; Xiong, L. Genomic organization, differential expression, and interaction of SQUAMOSA promoter-binding-like transcription factors and microRNA156 in rice. Plant Physiol 2006, 142, 280–293. [Google Scholar]
- Chuck, G.; Candela, H.; Hake, S. Big impacts by small RNAs in plant development. Curr. Opin. Plant Biol 2009, 12, 81–86. [Google Scholar]
- Wei, L.Y.; Zhang, D.F.; Xiang, F.; Zhang, Z.X. Differentially expressed miRNAs potentially involved in the regulation of defense mechanism to drought stress in maize seedlings. Int. J. Plant Sci 2009, 170, 979–989. [Google Scholar]
- Song, Q.X.; Liu, Y.F.; Hu, X.F.; Zhang, W.K.; Ma, B.; Chen, S.Y.; Zhang, J.S. Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant Biology 2011, 11. [Google Scholar] [CrossRef]
- Yin, Z.J.; Shen, F.F. Identification and characterization of conserved microRNAs and their target genes in wheat (Triticum aestivum). Genet. Mol. Res 2010, 9, 1186–1196. [Google Scholar]
- Ueda, A.; Kathiresan, A.; Inada, M.; Narita, Y.; Nakamura, T.; Shi, W.M.; Takabe, T.; Bennett, J. Osmotic stress in barley regulates expression of a different set of genes than salt stress does. J. Exp. Bot 2004, 55, 2213–2218. [Google Scholar]
- Yao, Y.Y.; Ni, Z.F.; Peng, H.R.; Sun, F.L.; Xin, M.M.; Sunkar, R.; Zhu, J.K.; Sun, Q.X. Non-coding small RNAs responsive to abiotic stress in wheat (Triticum aestivum L.). Funct. Integr. Genomics 2010, 10, 187–190. [Google Scholar]
- Sunkar, R.; Girke, T.; Zhu, J.K. Identification and characterization of endogenous small interfering RNAs from rice. Nucleic Acids Res 2005, 33, 4443–4454. [Google Scholar]
- Griffiths, J.S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res 2005, 33, D121–D124. [Google Scholar]
- Griffiths, J.S.; Saini, H.K.; Dongen, V.S.; Enright, A.J. miRBase: Tool for microRNA genomics. Nucleic Acids Res 2008, 36, D154–D158. [Google Scholar]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003, 31, 3406–3415. [Google Scholar]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res 2011, 39, W155–W159. [Google Scholar]
- Chi, X.Y.; Yang, Q.L.; Chen, X.P.; Wang, J.Y.; Pan, L.J.; Chen, M.N.; Yang, Z.; He, Y.N.; Liang, X.Q.; Yu, S.L. Identification and characterization of microRNAs from peanut (Arachis hypogaea L.) by high-throughput sequencing. PLoS One 2011, 6. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Category | Unique sRNA | Percent (%) | Total sRNA | Percent (%) |
|---|---|---|---|---|
| Total | 4,045,224 | 100 | 9,540,562 | 100 |
| miRNA | 23,239 | 0.57 | 625,232 | 6.55 |
| rRNA | 83,300 | 2.06 | 1,006,189 | 10.55 |
| repeat | 23,642 | 0.58 | 90,970 | 0.95 |
| snRNA | 3,020 | 0.07 | 15,180 | 0.16 |
| snoRNA | 1,872 | 0.05 | 6,711 | 0.07 |
| tRNA | 16,957 | 0.42 | 1,154,444 | 12.10 |
| Unannotated | 3,893,194 | 96.24 | 6,641,836 | 69.62 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lv, S.; Nie, X.; Wang, L.; Du, X.; Biradar, S.S.; Jia, X.; Weining, S. Identification and Characterization of MicroRNAs from Barley (Hordeum vulgare L.) by High-Throughput Sequencing. Int. J. Mol. Sci. 2012, 13, 2973-2984. https://doi.org/10.3390/ijms13032973
Lv S, Nie X, Wang L, Du X, Biradar SS, Jia X, Weining S. Identification and Characterization of MicroRNAs from Barley (Hordeum vulgare L.) by High-Throughput Sequencing. International Journal of Molecular Sciences. 2012; 13(3):2973-2984. https://doi.org/10.3390/ijms13032973
Chicago/Turabian StyleLv, Shuzuo, Xiaojun Nie, Le Wang, Xianghong Du, Siddanagouda S. Biradar, Xiaoou Jia, and Song Weining. 2012. "Identification and Characterization of MicroRNAs from Barley (Hordeum vulgare L.) by High-Throughput Sequencing" International Journal of Molecular Sciences 13, no. 3: 2973-2984. https://doi.org/10.3390/ijms13032973
APA StyleLv, S., Nie, X., Wang, L., Du, X., Biradar, S. S., Jia, X., & Weining, S. (2012). Identification and Characterization of MicroRNAs from Barley (Hordeum vulgare L.) by High-Throughput Sequencing. International Journal of Molecular Sciences, 13(3), 2973-2984. https://doi.org/10.3390/ijms13032973