Introgression Between Cultivars and Wild Populations of Momordica charantia L. (Cucurbitaceae) in Taiwan

Abstract
:1. Introduction
2. Results
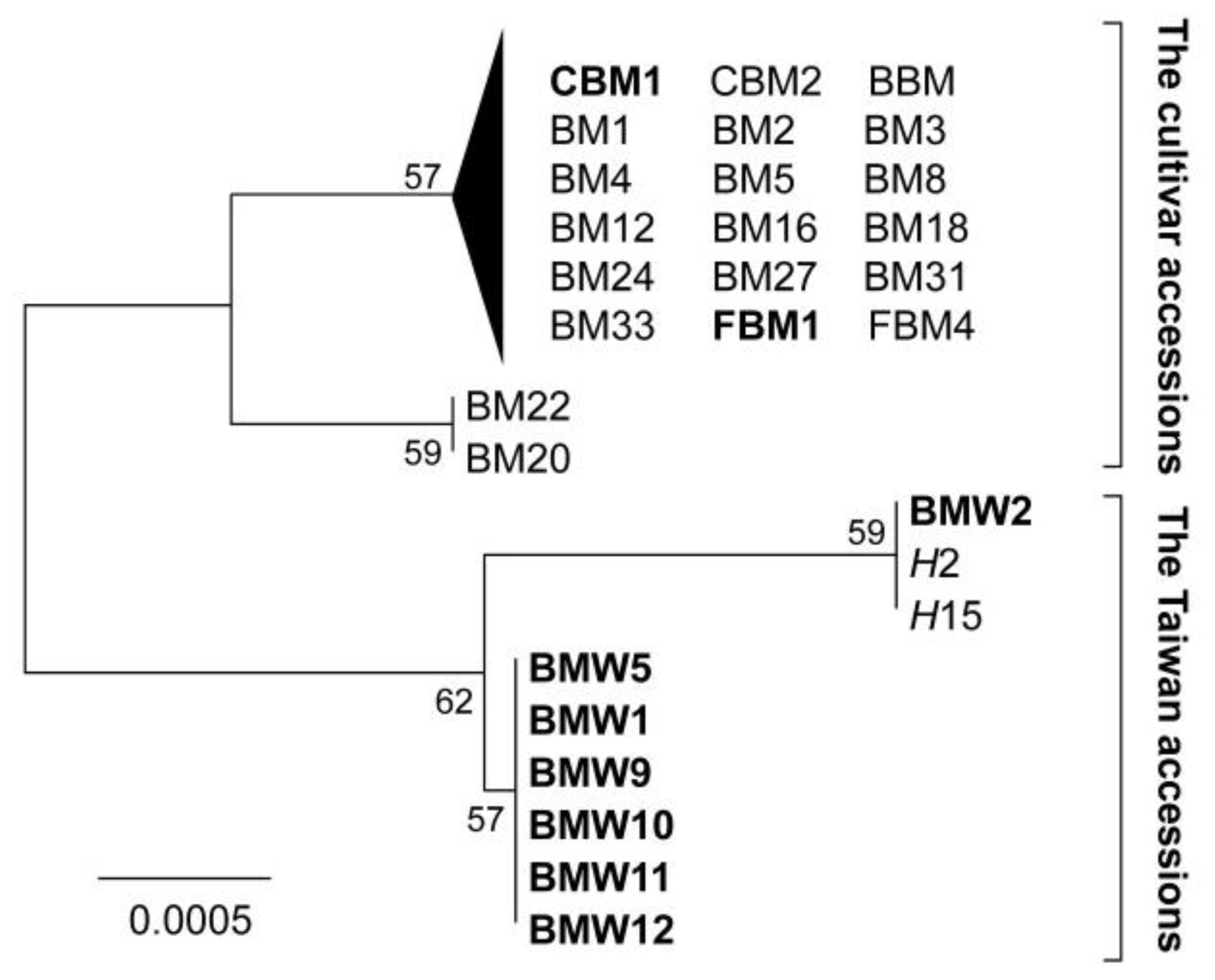
2.1. CpDNA Polymorphisms and Neighbour Joining Tree
2.2. Nuclear Microsatellite Polymorphisms
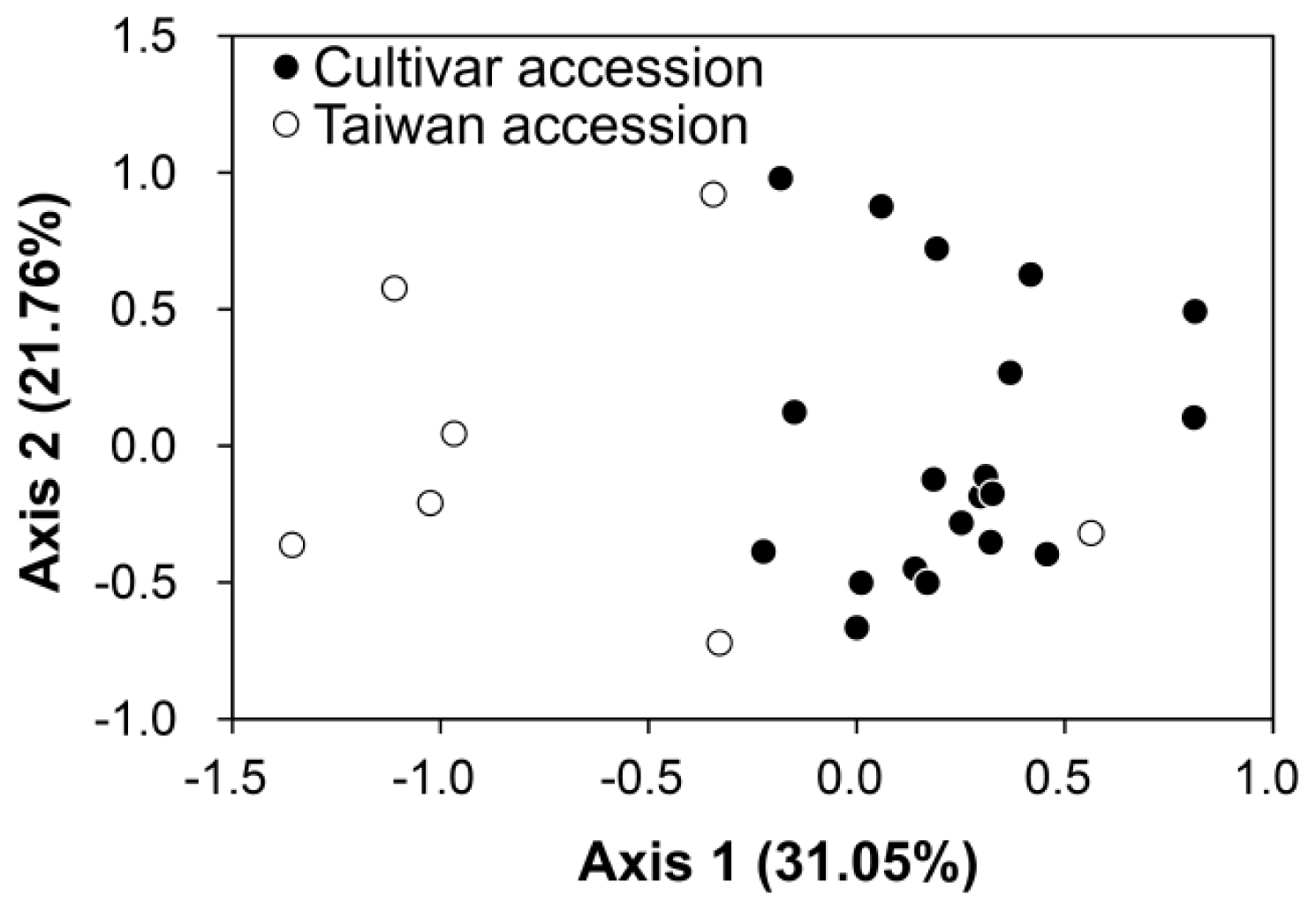
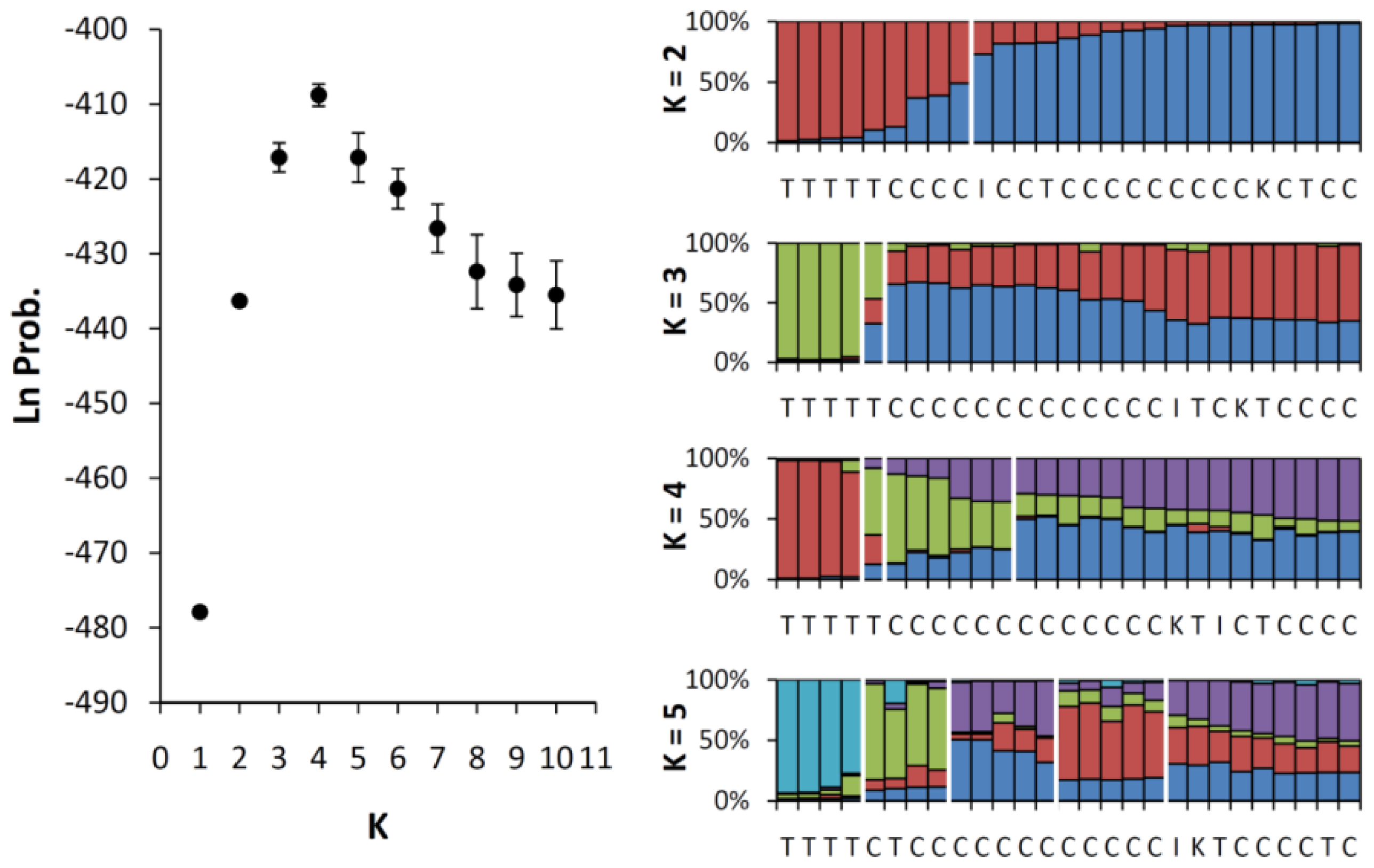
2.3. Genetic Mixing Between the Cultivars and the Wild Population of Taiwan
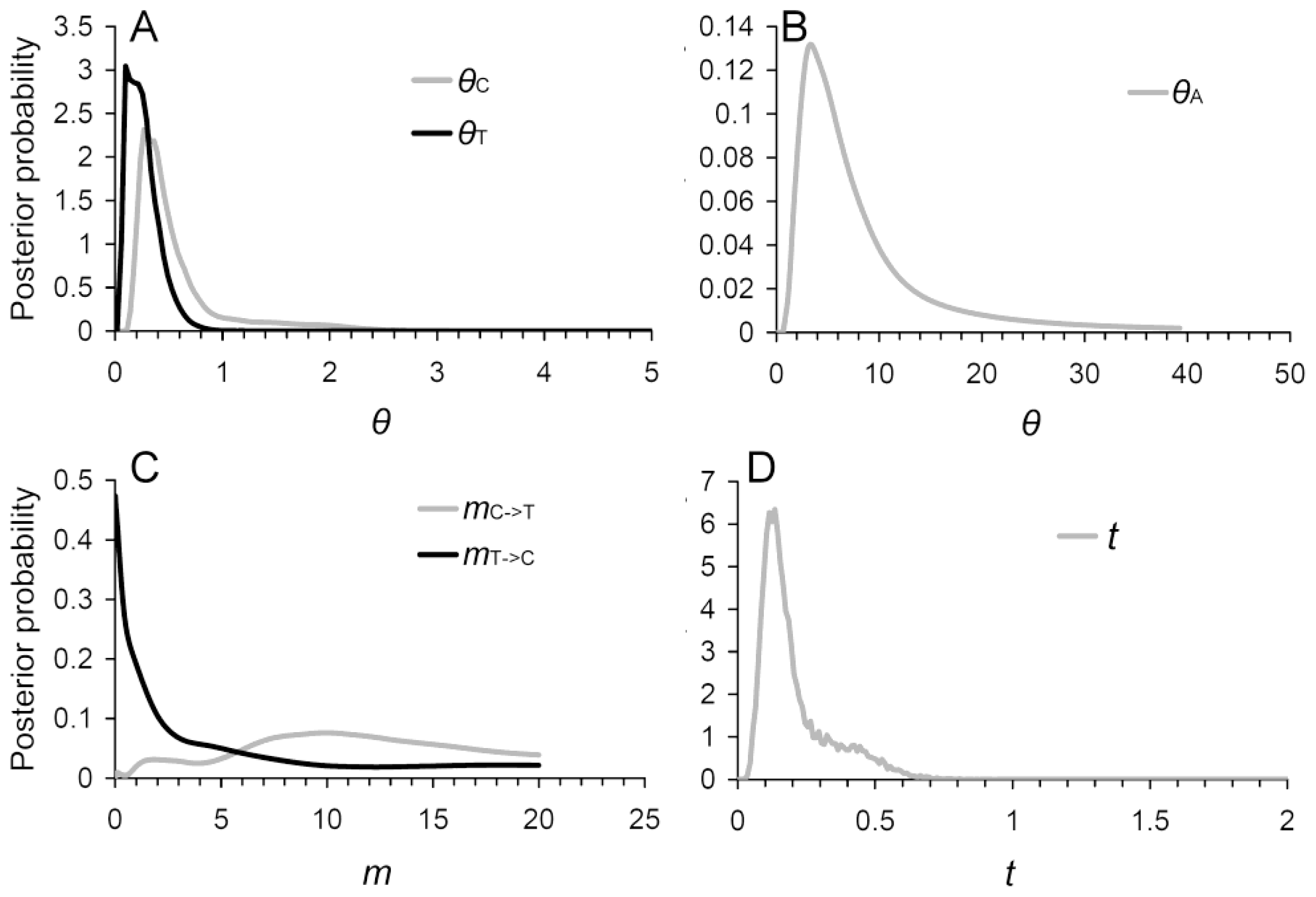
2.4. Evidence of Asymmetric Introgression Inferred by the Isolation-with-Migration Model
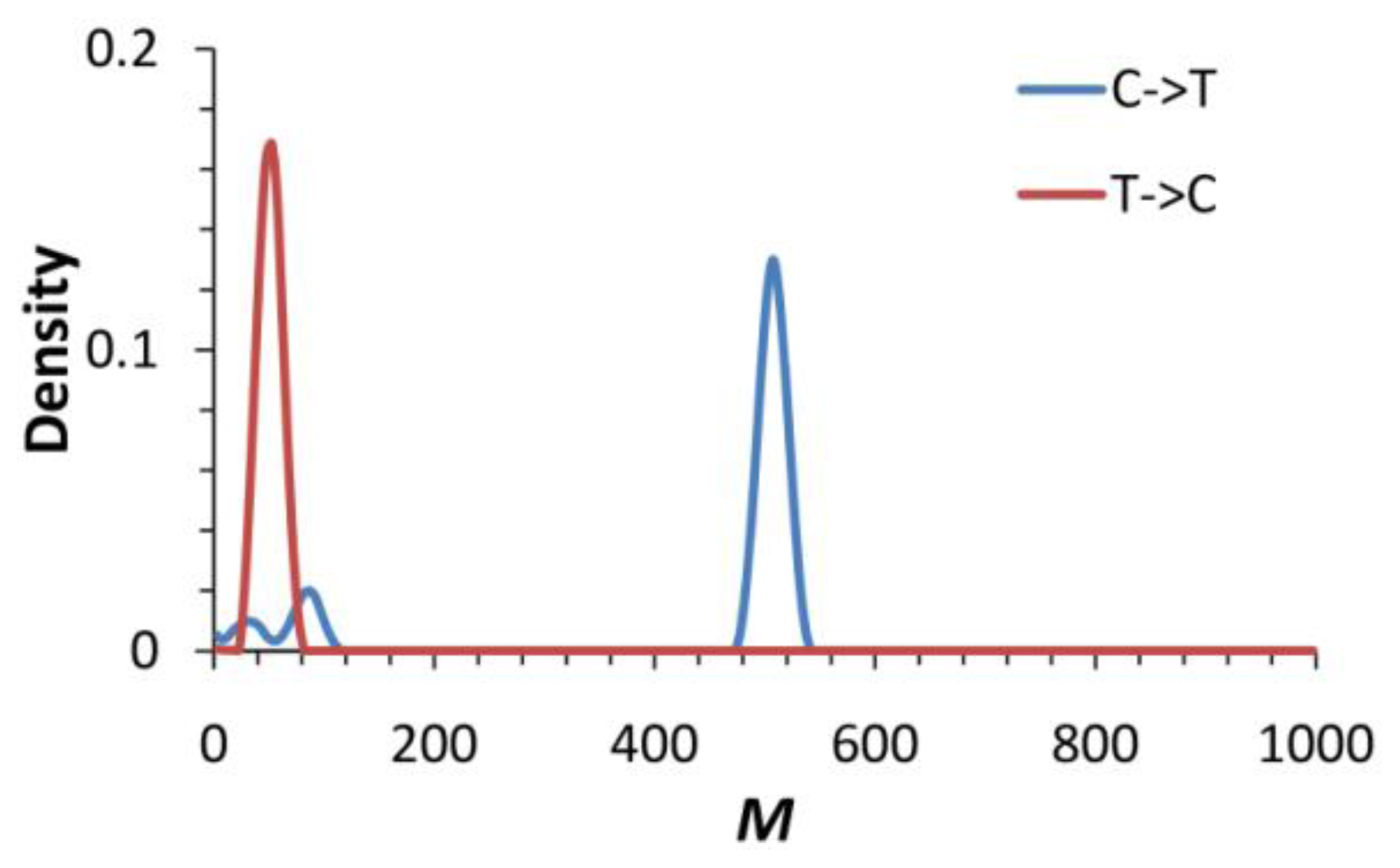
2.5. Historical and Contemporary Gene Flow
3. Discussion
4. Experimental Methods
4.1. Sample Preparation
4.2. Molecular Techniques
4.3. Data Analyses
4.3.1. Genetic Diversity and Neutrality Tests
4.3.2. Phylogenetic Analyses of Chloroplast DNA
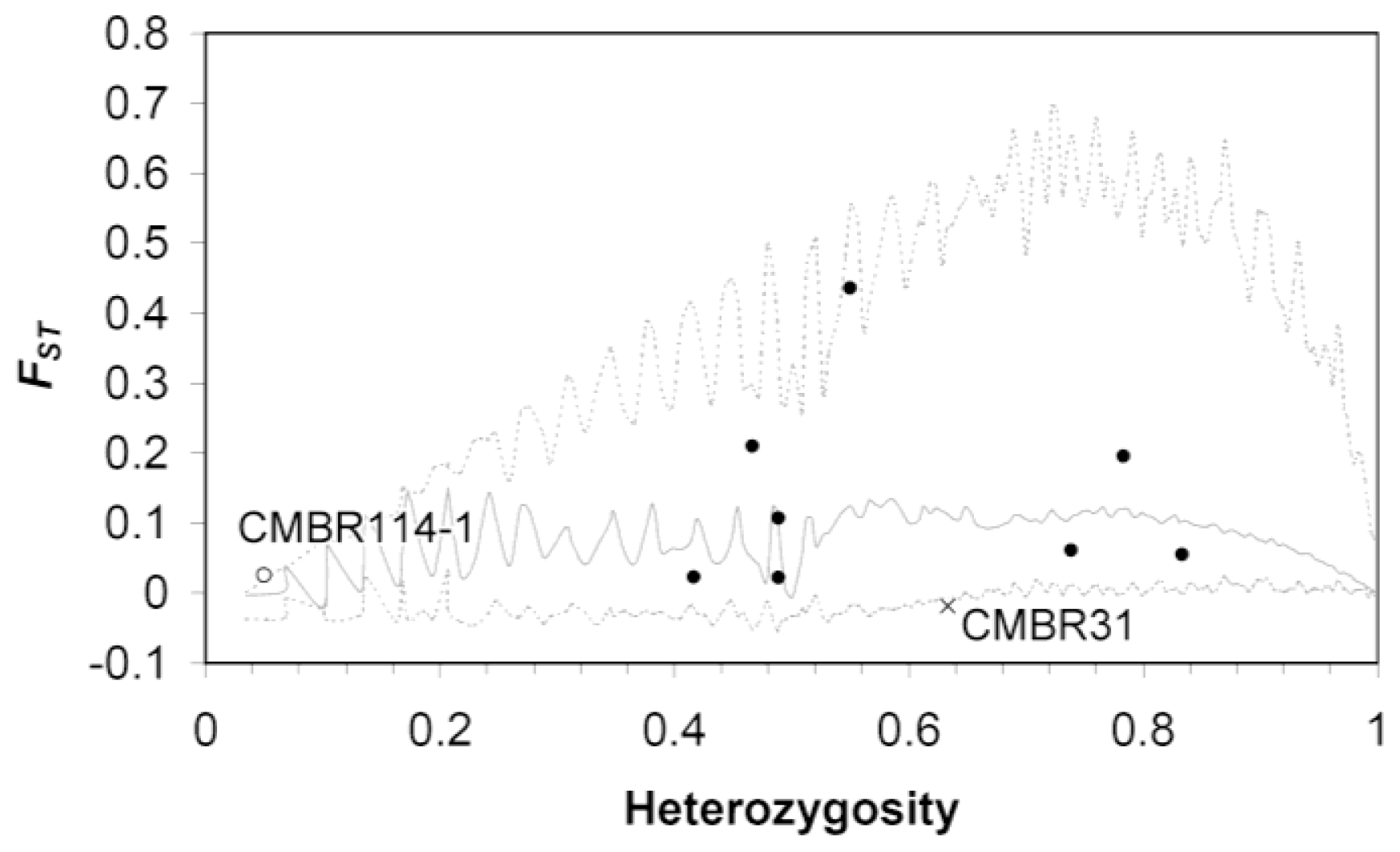
4.3.3. Neutrality Test by Detecting Outlier Loci for Microsatellite DNA
4.3.4. Genetic Diversity and Genetic Structure
4.3.5. Isolation with Migration (IM) Model Test
4.3.6. Inferring the Past and Recent Introgression
5. Conclusions
Acknowledgments
References
- Marr, K.L.; Mei, X.Y.; Bhattarai, N.K. Allozyme, morphological and nutritional analysis bearing on the domestication of Momordica charantia L. (Cucurbitaceae). Econ. Bot 2004, 58, 435–455. [Google Scholar]
- Lu, A.; Jeffrey, C. Momordica Linnaeus. Flora China 2011, 19, 28–30. [Google Scholar]
- Janick, J.; Paris, H.S. The cucurbit images (1515–1518) of the Villa Farnesina, Rome. Ann. Bot 2006, 97, 165–176. [Google Scholar]
- Joseph, J.K.; Antony, V.T. Ethnobotanical investigations in the genus Momordica L. in the southern Western Ghats of India. Genet. Resour. Crop Evol 2008, 55, 713–721. [Google Scholar]
- Wang, L.L.; Zhang, H.; Song, L.S.; Guo, X.M. Loss of allele diversity in introduced populations of the hermaphroditic bay scallop Argopecten irradians. Aquaculture 2007, 271, 252–259. [Google Scholar]
- Whitt, S.R.; Wilson, L.M.; Tenaillon, M.I.; Gaut, B.S.; Buckler, E.S. Genetic diversity and selection in the maize starch pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 12959–12962. [Google Scholar]
- Tripathi, U.N.; Chandra, D. Anti-hyperglycemic and anti-oxidative effect of aqueous extract of Momordica charantia pulp and Trigonella foenum graecum seed in alloxan-induced diabetic rats. Indian J. Biochem. Biophys 2010, 47, 227–233. [Google Scholar]
- Wu, S.J.; Ng, L.T. Antioxidant and free radical scavenging activities of wild bitter melon (Momordica charantia Linn. var. abbreviata Ser.) in Taiwan. LWT Food Sci Technol 2008, 41, 323–330. [Google Scholar]
- Horax, R.; Hettiarachchy, N.; Chen, P.Y. Extraction, quantification, and antioxidant activities of phenolics from pericarp and seeds of bitter melons (Momordica charantia) harvested at three maturity stages (immature, mature, and ripe). J. Agric. Food Chem 2010, 58, 4428–4433. [Google Scholar]
- Kobori, M.; Nakayama, H.; Fukushima, K.; Ohnishi-Kameyama, M.; Ono, H.; Fukushima, T.; Akimoto, Y.; Masumoto, S.; Yukizaki, C.; Hoshi, Y.; et al. Bitter gourd suppresses lipopolysaccharide-induced inflammatory responses. J. Agric. Food Chem 2008, 56, 4004–4011. [Google Scholar]
- Lii, C.K.; Chen, H.W.; Yun, W.T.; Liu, K.L. Suppressive effects of wild bitter gourd (Momordica charantia Linn. var. abbreviata Ser.) fruit extracts on inflammatory responses in RAW 264.7 macrophages. J. Ethnopharmacol 2009, 122, 227–233. [Google Scholar]
- Fang, Q.M.; Zhang, H.; Cao, Y.; Wang, C. Anti-inflammatory and free radical scavenging activities of ethanol extracts of three seeds used as “Bolengguazi”. J. Ethnopharmacol 2007, 114, 61–65. [Google Scholar]
- Kobori, M.; Amemiya, J.; Sakai, M.; Shiraki, M.; Sugishita, H.; Sakaue, N.; Hoshi, Y.; Yukizaki, C. Bitter gourd induces apoptosis in HL60 human leukemia cells and suppresses the production of inflammatory cytokine in RAW264.7 macrophage like cells. J. Jpn. Soc. Food Sci. Technol 2006, 53, 408–415. [Google Scholar]
- Abdollahi, M.; Zuki, A.B.Z.; Goh, Y.M.; Rezaeizadeh, A.; Noordin, M.M. The effects of Momordica charantia on the liver in streptozotocin-induced diabetes in neonatal rats. Afr. J. Biotechnol 2010, 9, 5004–5012. [Google Scholar]
- Yuan, X.Q.; Gu, X.H.; Tang, J. Optimization of the production of Momordica charantia L. var. abbreviata Ser. protein hydrolysates with hypoglycemic effect using Alcalase. Food Chem 2008, 111, 340–344. [Google Scholar]
- Schaefer, H.; Heibl, C.; Renner, S.S. Gourds afloat: A dated phylogeny reveals an Asian origin of the gourd family (Cucurbitaceae) and numerous oversea dispersal events. Proc. R. Soc. B 2009, 276, 843–851. [Google Scholar]
- Schaefer, H.; Renner, S.S. A three-genome phylogeny of Momordica (Cucurbitaceae) suggests seven returns from dioecy to monoecy and recent long-distance dispersal to Asia. Mol. Phylogenet. Evol 2010, 54, 553–560. [Google Scholar]
- Zhai, C.; Lin, F.; Dong, Z.; He, X.; Yuan, B.; Zeng, X.; Wang, L.; Pan, Q. The isolation and characterization of Pik, a rice blast resistance gene which emerged after rice domestication. New Phytol 2011, 189, 321–334. [Google Scholar]
- Ramsay, L.; Comadran, J.; Druka, A.; Marshall, D.F.; Thomas, W.T.; Macaulay, M.; Mackenzie, K.; Simpson, C.; Fuller, J.; Bonar, N.; et al. INTERMEDIUM-C, a modifier of lateral spikelet fertility in barley, is an ortholog of the maize domestication gene TEOSINTE BRANCHED 1. Nat. Genet 2011, 43, 169–172. [Google Scholar]
- Wright, S.I.; Bi, I.V.; Schroeder, S.G.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S. The effects of artificial selection on the maize genome. Science 2005, 308, 1310–1314. [Google Scholar]
- Frary, A.; Doganlar, S.; Daunay, M.C.; Tanksley, S.D. QTL analysis of morphological traits in eggplant and implications for conservation of gene function during evolution of solanaceous species. Theor. Appl. Genet 2003, 107, 359–370. [Google Scholar]
- Doganlar, S.; Frary, A.; Daunay, M.C.; Lester, R.N.; Tanksley, S.D. Conservation of gene function in the solanaceae as revealed by comparative mapping of domestication traits in eggplant. Genetics 2002, 161, 1713–1726. [Google Scholar]
- Hancock, J.F. Contributions of domesticated plant studies to our understanding of plant evolution. Ann. Bot 2005, 96, 953–963. [Google Scholar]
- Haygood, R.; Ives, A.R.; Andow, D.A. Consequences of recurrent gene flow from crops to wild relatives. Proc. R. Soc. Lond. B Biol. Sci 2003, 270, 1879–1886. [Google Scholar]
- Teng, L.S. Geotectonic evolution of late Cenozoic arc-continent collision in Taiwan. Tectonophysics 1990, 183, 57–76. [Google Scholar]
- Lu, C.Y.; Hsu, K.J. Tectonic evolution of the Taiwan mountain belt. Petrol. Geol. Taiwan 1992, 27, 21–46. [Google Scholar]
- Wei, K.-Y. Environmental changes during the Late Quaternary in Taiwan and adjacent seas: An overview of recent results of the past decade (1990–2000). West Pac. Earth Sci 2002, 2, 149–160. [Google Scholar]
- Chiang, T.Y.; Schaal, B.A. Phylogeography of plants in Taiwan and the Ryukyu archipelago. Taxon 2006, 55, 31–41. [Google Scholar]
- Edwards, S.V.; Beerli, P. Perspective: Gene divergence, population divergence, and the variance in coalescence time in phylogeographic studies. Evolution 2000, 54, 1839–1854. [Google Scholar]
- Ritschel, P.S.; Lins, T.C.; Tristan, R.L.; Buso, G.S.; Buso, J.A.; Ferreira, M.E. Development of microsatellite markers from an enriched genomic library for genetic analysis of melon (Cucumis melo L.). BMC Plant Biol 2004, 4. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Hey, J.; Nielsen, R. Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics 2004, 167, 747–760. [Google Scholar]
- Hey, J.; Nielsen, R. Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc. Natl. Acad. Sci. USA 2007, 104, 2785–2790. [Google Scholar]
- Beerli, P.; Palczewski, M. Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics 2010, 185, 313–326. [Google Scholar]
- Rannala, B.; Wilson, G.A. Bayesian inference of recent migration rates using multilocus genotypes. Genetics 2003, 163, 1177–1191. [Google Scholar]
- Stevison, L.S.; Kohn, M.H. Divergence population genetic analysis of hybridization between rhesus and cynomolgus macaques. Mol. Ecol 2009, 18, 2457–2475. [Google Scholar]
- Lin, S.M.; Chen, C.A.; Lue, K.Y. Molecular phylogeny and biogeography of the grass lizards genus Takydromus (Reptilia : Lacertidae) of East Asia. Mol. Phylogenet. Evol 2002, 22, 276–288. [Google Scholar]
- Chang, C.H.; Chen, J.H. Three new species of octothecate pheretimoid earthworms from Taiwan, with discussion on the biogeography of related species. J. Nat. Hist 2005, 39, 1469–1482. [Google Scholar]
- Long, Y.; Wan, H.; Yan, F.M.; Xu, C.R.; Lei, G.C.; Li, S.W.; Wang, R.J. Glacial effects on sequence divergence of mitochondrial COII of Polyura eudamippus (Lepidoptera: Nymphalidae) in China. Biochem. Genet 2006, 44, 361–377. [Google Scholar]
- Yin, W.; Fu, C.Z.; Guo, L.; He, Q.X.; Li, J.; Jin, B.S.; Wu, Q.H.; Li, B. Species delimitation and historical biogeography in the genus Helice (Brachyura: Varunidae) in the Northwestern Pacific. Zool. Sci 2009, 26, 467–475. [Google Scholar]
- Wu, C.I. The genic view of the process of speciation. J. Evol. Biol 2001, 14, 851–865. [Google Scholar]
- Eyre-Walker, A.; Gaut, R.L.; Hilton, H.; Feldman, D.L.; Gaut, B.S. Investigation of the bottleneck leading to the domestication of maize. Proc. Natl. Acad. Sci. USA 1998, 95, 4441–4446. [Google Scholar]
- Lawton-Rauh, A.; Burgos, N. Cultivated and weedy rice interactions and the domestication process. Mol. Ecol 2010, 19, 3243–3245. [Google Scholar]
- Glemin, S.; Bataillon, T. A comparative view of the evolution of grasses under domestication. New Phytol 2009, 183, 273–290. [Google Scholar]
- Strasburg, J.L.; Rieseberg, L.H. Interpreting the estimated timing of migration events between hybridizing species. Mol. Ecol 2011, 20, 2353–2366. [Google Scholar]
- Sousa, V.C.; Grelaud, A.; Hey, J. On the nonidentifiability of migration time estimates in isolation with migration models. Mol. Ecol 2011, 20, 3956–3962. [Google Scholar]
- Chiang, T.Y.; Schaal, B.A.; Peng, C.I. Universal primers for amplification and sequencing a noncoding spacer between the atpB and rbcL genes of chloroplast DNA. Bot. Bull. Acad. Sin 1998, 39, 245–250. [Google Scholar]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 1997, 25, 4876–4882. [Google Scholar]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser 1999, 41, 95–98. [Google Scholar]
- Rozas, J.; Sanchez-DelBarrio, J.C.; Messeguer, X.; Rozas, R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 2003, 19, 2496–2497. [Google Scholar]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol 2011, 28, 2731–2739. [Google Scholar]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol 1993, 10, 512–526. [Google Scholar]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol 2007, 7. [Google Scholar] [CrossRef]
- Wolfe, K.H.; Li, W.-H.; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar]
- Beaumont, M.A.; Nichols, R.A. Evaluating loci for use in the genetic analysis of population structure. Proc. R. Soc. Lond. B Biol. Sci 1996, 263, 1619–1626. [Google Scholar]
- Antao, T.; Lopes, A.; Lopes, R.J.; Beja-Pereira, A.; Luikart, G. LOSITAN: A workbench to detect molecular adaptation based on a FST-outlier method. BMC Bioinform 2008, 9. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evol. Bioinform. Online 2005, 1, 47–50. [Google Scholar]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar]
- Weir, B.C.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar]
- JMP, version 7; SAS Institute Inc: Cary, NC, USA, 2007.
- Ellstrand, N.C. Current knowledge of gene flow in plants: Implications for transgene flow. Philos. Trans. R. Soc. Lond. B Biol. Sci 2003, 358, 1163–1170. [Google Scholar]
- Tang, H.B.; Sezen, U.; Paterson, A.H. Domestication and plant genomes. Curr. Opin. Plant Biol 2010, 13, 160–166. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | N a | H b | S c | θ d | π e | Tajima’s D f |
|---|---|---|---|---|---|---|
| The cultivar accession | 20 | 3 | 2 | 0.0005 | 0.0002 | −1.2414 |
| The Taiwan accession | 7 | 2 | 1 | 0.0004 | 0.0003 | −1.0062 |
| Hybrid | 2 | 1 | 0 | 0 | 0 | −1.0488 |
| Total | 29 | 5 | 4 | 0.001 | 0.0011 | 0.1977 |
| Locus | Primer set | Tm | Type * | cultivars | wild population (Taiwan) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Hexp | θ | Na | Ne | Hexp | θ | Na | Ne | ||||
| CMBR21 | F: 5′-AGATTCTGGTTGTTGGGCAG-3′ R: 5′-CAGCGATGATCAACAGAAACA-3′ | 59 °C | P | 0.528 | 1.506 | 3 | 2.062 | 0.264 | 2.075 | 2 | 1.324 |
| CMBR22 | F: 5′-CCAAAACGACCAAATGTTCC-3′ R: 5′-ATACAGACACGCCTTCCACC-3′ | 56 °C | P | 0.554 | 1.523 | 5 | 2.174 | 0.615 | 1.613 | 3 | 2.333 |
| CMBR30 | F: 5′-CACTGCATACACACACATCCA-3′ R: 5′-AAAAGAAGGAGGAGGAGGG-3′ | 58 °C | P | 0.097 | 5.186 | 2 | 1.105 | 0.440 | 1.530 | 2 | 1.690 |
| CMBR47 | F: 5′-ATCCCAACCCATCACTCTCA-3′ R: 5′-TGGGGACAGGTGAGAATATTAGA-3′ | 59 °C | P | 0.738 | 2.089 | 6 | 3.571 | 0.791 | 2.527 | 5 | 3.769 |
| CMBR57 | F: 5′-GCTCTGAAGAGTGGAATGAGAGA-3′ R: 5′-CCATTTGGGAAGTAGGCATC-3′ | 59 °C | P | 0.708 | 1.917 | 6 | 3.226 | 0.659 | 1.726 | 4 | 2.579 |
| CMBR66 | F: 5′-TCAAGCAAAAACCATAATCAGAA-3′ R: 5′-TCCCTTTTCATCATTTCTCTTCA-3′ | 52 °C | P | 0.272 | 2.026 | 3 | 1.361 | 0.659 | 1.726 | 3 | 2.579 |
| CMBR82 | F: 5′-ACGACTCTTGGAAATCGGTC-3′ R: 5′-TTTAGAAAAGAATCACGAAGAGAGC-3′ | 54 °C | P | 0.344 | 1.717 | 3 | 1.504 | 0.527 | 1.506 | 2 | 1.960 |
| CMBR152 | F: 5′-CCCACATTGGTCTCAACAAG-3′ R: 5′-AAAAAATTTGGCATTAGCTATAAAAA-3′ | 54 °C | P | 0.590 | 1.567 | 4 | 2.353 | 0.440 | 1.530 | 2 | 1.690 |
| CMBR31 | F: 5′-AAACAAACCAAACCAAACCG-3′ R: 5′-AAAAAGAAGCGGGAGTAATGA-3′ | 58 °C | BS | 0.610 | 1.602 | 4 | 2.469 | 0.703 | 1.896 | 3 | 2.882 |
| CMBR114-1 | F: 5′-TGCTTTGCCTTAACCGTCTT-3′ R: 5′-TGAGTGCCCAAGATGTTGTC-3′ | 52 °C | PS | 0.097 | 5.186 | 2 | 1.105 | 0 | 0 | 1 | 1.000 |
| CMBR114-2 | F: 5′-TGCTTTGCCTTAACCGTCTT-3′ R: 5′-TGAGTGCCCAAGATGTTGTC-3′ | 52 °C | M | - | - | - | - | - | - | - | - |
| CMBR145 | F: 5′-TGTGACAATGTGCAACCAG-3′ R: 5′-AAAAATGGTGTTAAACGACATGG-3′ | 56 °C | M | - | - | - | - | - | - | - | - |
| Source of variation | d.f. | Sum of Squares | Variance Components | Percentage of Variance | RST | P |
|---|---|---|---|---|---|---|
| Between accessions | 1 | 104.955 | 0.528 | 0.56 | 0.0056 | 0.615 |
| Within accessions | 52 | 4887.786 | 93.996 | 99.44 | ||
| Total | 53 | 4992.741 | 94.524 | 100 |
| Model | T | θC | θT | θA | mC→T | mT→C | df | 2LLR † | P |
|---|---|---|---|---|---|---|---|---|---|
| Full model | |||||||||
| mode | 0.135 | 0.3314 | 0.1767 | 3.3572 | 9.95 | 0.01 | |||
| 95% CI Low | 0.065 | 0.1736 | 0.0589 | 1.708 | 1.61 | 0.05 | |||
| 95% CI High | 0.545 | 1.9409 | 0.6086 | 29.5471 | 19.37 | 18.85 | |||
| Null models | |||||||||
| m1 = m2 | 0.1167 | 0.5634 | 0.1469 | 2.1057 | 3.2863 | 3.2863 | 1 | 4.3693 | 0.0366 |
| m2 = 0 | 0.0625 | 0.2105 | 0.2230 | 2.6982 | 19.9976 | 0.0001 | 1 | 0.0056 | 0.4702 |
| m1 = 0 | 0.0995 | 0.4138 | 0.2023 | 2.5454 | 0.0001 | 9.4322 | 1 | 14.8471 | 5.829 × 10−5 |
| m1 = m2 = 0 | 0.1785 | 0.7870 | 0.4202 | 1.8314 | 0.0001 | 0.0001 | 2 | 718.0318 | 6.03 × 10−157 |
| θC = θT | 0.0625 | 0.2163 | 0.2163 | 2.7202 | 19.9992 | 0.0002 | 1 | 0.3680 | 0.5441 |
| θC = θT = θA | 0.2213 | 0.6101 | 0.6101 | 0.6101 | 7.8461 | 2.8492 | 2 | 16.3993 | 2.747 × 10−4 |
| θC = θT, m1 = m2 | 0.0902 | 0.1906 | 0.1906 | 2.1566 | 19.5826 | 19.5826 | 2 | 9.8940 | 0.0071 |
| θC = θT, m1 = m2 = 0 | 0.1785 | 0.7026 | 0.7026 | 1.8250 | 0.0001 | 0.0001 | 3 | 746.7677 | 7.58 × 10−162 |
| θC = θT = θA, m1 = m2 | 0.2213 | 0.6107 | 0.6107 | 0.6107 | 5.4662 | 5.4662 | 3 | 51.8731 | 3.19 × 10−11 |
| θC = θT = θA, m1 = m2 = 0 | 0.1785 | 0.8188 | 0.8188 | 0.8188 | 0.0001 | 0.0001 | 4 | 803.2170 | 7.72 × 10−173 |
| θC = θA | 0.2537 | 1.0702 | 0.4156 | 1.0702 | 7.3973 | 0.0478 | 1 | 5.9515 | 0.0147 |
| θC = θA, m1 = m2 | 0.2980 | 1.5204 | 0.3881 | 1.5204 | 1.4217 | 1.4217 | 2 | 25.8596 | 2.42 × 10−6 |
| θC = θA, m1 = m2 = 0 | 0.1785 | 0.9223 | 0.4204 | 0.9223 | 0.0001 | 0.0001 | 3 | 759.3913 | 1.39 × 10−164 |
| θT = θA | 0.2213 | 0.6338 | 0.5800 | 0.5800 | 7.8300 | 2.8436 | 1 | 15.4141 | 8.63 × 10−5 |
| θT = θA, m1 = m2 | 0.2213 | 0.6347 | 0.5806 | 0.5806 | 5.4683 | 5.4683 | 2 | 50.8823 | 8.93 × 10−12 |
| θT = θA, m1 = m2 = 0 | 0.1785 | 0.7864 | 0.8914 | 0.8914 | 0.0001 | 0.0001 | 3 | 801.3394 | 1.11 × 10−173 |
| IMa a | MIGRATE-n a | BayesAss a | ||||
|---|---|---|---|---|---|---|
| Source | Cultivar accession | Taiwan accession | Cultivar accession | Taiwan accession | Cultivar accession | Taiwan accession |
| Cultivar accession | - | 2.655 | - | 507.5 | 0.985 | 0.071 |
| Taiwan accession | 0.195 | - | 52.5 | - | 0.015 | 0.929 |
| Species | Strain | Accessions | Sample size | Resource a, b | Abbreviation |
|---|---|---|---|---|---|
| M. charantia (white cultivar) | Pai Bitter Gourd | TVI006693 | 6 | NPGRC, TARI, COA | BM1 |
| M. charantia (white cultivar) | Pin Tung He Tzu Ku Kua No. 8 | TVI007217 | 6 | NPGRC, TARI, COA | BM2 |
| M. charantia (white cultivar) | Pai Pi Bitter Gourd | TVI007364 | 6 | NPGRC, TARI, COA | BM3 |
| M. charantia (white cultivar) | Chin Lien Bitter Gourd | TVI007723 | 6 | NPGRC, TARI, COA | BM4 |
| M. charantia (white cultivar) | Pin Tung He Tzu Bitter Gourd | TVI007895 | 6 | NPGRC, TARI, COA | BM5 |
| M. charantia (white cultivar) | Ming Hua | TVI006970 | 6 | NPGRC, TARI, COA | BM8 |
| M. charantia (white cultivar) | Lin Nei Tzu Liu Chung | TVI009016 | 5 | NPGRC, TARI, COA | BM12 |
| M. charantia (white cultivar) | Chiang Men Ta Ting Bitter Gourd | TVI010175 | 6 | NPGRC, TARI, COA | BM16 |
| M. charantia (white cultivar) | Ping Tung Li Kang Bitter Gourd | TVI009615 | 5 | NPGRC, TARI, COA | BM18 |
| M. charantia (white cultivar) | Small Bitter Melon | TVI009995 | 5 | NPGRC, TARI, COA | BM20 |
| M. charantia (white cultivar) | Chen Chu | TVI006900 | 6 | NPGRC, TARI, COA | BM24 |
| M. charantia (white cultivar) | Kuang Han Te Ta Chang Pai Bitter Gourd | TVI009892 | 5 | NPGRC, TARI, COA | BM27 |
| M. charantia (white cultivar) | Pai Pi Tsu Mi Bitter Gourd | TVI009534 | 5 | NPGRC, TARI, COA | BM31 |
| M. charantia (white cultivar) | Ping Tung Bitter Gourd | TVI007365 | 5 | NPGRC, TARI, COA | BM33 |
| M. charantia (white cultivar) | Chinese Gui-Nin No. 2 | SYSU-BM-1 | 5 | Exchange from South China Botanical Garden, China. | CBM2 |
| M. charantia (green cultivar) | Chin Pi Bitter Gourd | TVI008603 | 6 | NPGRC, TARI, COA | BM22 |
| M. charantia (green cultivar) | Chin Pi Bitter Gourd | SYSU-BM-2 | 6 | Exchange from Kunming Institute of Botany, China. | BBM |
| M. charantia (green cultivar) | Kao Mien Bitter Gourd | TVI009317 | 5 | NPGRC, TARI, COA | FBM4 |
| M. charantia (wild population from Korea) | Han Ch’eng K’u Kua | CN93MJF135 | 5 | NPGRC, TARI, COA | FBM1 |
| M. charantia (wild population from India) | Momordica charantia variety | TVI009464 | 5 | NPGRC, TARI, COA | CBM1 |
| M. charantia (wild population from Taiwan) | Ping Tung Chiu Chow Bitter Gourd | SYSU-BW-1 | 8 | N 22°39′14″, E 120°38′10″, 205 m altitude | BMW1 |
| M. charantia (wild population from Taiwan) | Nan Tou Bitter Gourd | SYSU-BW-2 | 8 | N 23°55′06″, E 120°53′04″, 700 m altitude | BMW2 |
| M. charantia (wild population from Taiwan) | Yeh Sheng Bitter Gourd | TVI009560 | 5 | NPGRC, TARI, COA | BMW5 |
| M. charantia (wild population from Taiwan) | Hua Lien Yeh Sheng Bitter Melon No. 1 | TVI009930 | 5 | NPGRC, TARI, COA | BMW9 |
| M. charantia (wild population from Taiwan) | Hua Lien Yeh Sheng Bitter Melon No. 5 | TVI009934 | 5 | NPGRC, TARI, COA | BMW10 |
| M. charantia (wild population from Taiwan) | Hua Lien Yeh Sheng Bitter Melon No. 10 | TVI009959 | 5 | NPGRC, TARI, COA | BMW11 |
| M. charantia (wild population from Taiwan) | Ping Tung Bitter Gourd | SYSU-BW-3 | 8 | N 22°24′57″, E 120°39′57″, 686 m altitude | BMW12 |
| Hybrid between wild population and cultivars (H) | Hua Lien Bitter Gourd No. 2 | HB002 | 5 | HDARES, COA | H2 |
| Hybrid between wild population and cultivars (H) | Hua Lien Bitter Gourd No. 15 | HB015 | 5 | HDARES, COA | H15 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liao, P.-C.; Tsai, C.-C.; Chou, C.-H.; Chiang, Y.-C. Introgression Between Cultivars and Wild Populations of Momordica charantia L. (Cucurbitaceae) in Taiwan. Int. J. Mol. Sci. 2012, 13, 6469-6491. https://doi.org/10.3390/ijms13056469
Liao P-C, Tsai C-C, Chou C-H, Chiang Y-C. Introgression Between Cultivars and Wild Populations of Momordica charantia L. (Cucurbitaceae) in Taiwan. International Journal of Molecular Sciences. 2012; 13(5):6469-6491. https://doi.org/10.3390/ijms13056469
Chicago/Turabian StyleLiao, Pei-Chun, Chi-Chu Tsai, Chang-Hung Chou, and Yu-Chung Chiang. 2012. "Introgression Between Cultivars and Wild Populations of Momordica charantia L. (Cucurbitaceae) in Taiwan" International Journal of Molecular Sciences 13, no. 5: 6469-6491. https://doi.org/10.3390/ijms13056469
APA StyleLiao, P. -C., Tsai, C. -C., Chou, C. -H., & Chiang, Y. -C. (2012). Introgression Between Cultivars and Wild Populations of Momordica charantia L. (Cucurbitaceae) in Taiwan. International Journal of Molecular Sciences, 13(5), 6469-6491. https://doi.org/10.3390/ijms13056469