3.1. Geometrical Structures
The optimized geometries of tetrahydroquinoline dyes C1-1, C1-2, C1-3, C1-4, C1-5, C2-1, C2-2, C2-3, C2-4, and C3-1 are shown in
Figures 1 and
2. The selected geometrical parameters, including bond lengths, bond angles, and dihedrals are listed in Tables S1–S3 in supporting information (SI). The calculated geometrical data indicate that the geometrical parameters of same group (tetrahydroquinoline, vinylene, thienyl and dithieno[3,2-b;2′,3′-d]thienyl,
etc.) in different dyes are very similar. This can be understood from the localized interaction of chemical bonds. The cyanoacrylic acid groups in the dyes are coplanar with the groups in conjugate bridge. The torsion angles among the thiophene-rings in bithiophene and dithieno-[3,2-b;2′,3′-d]thiophene groups are quite small. For instance, the torsion angle between thiophene-rings in bithiophene in C1-2 is about 1.7°. The conjugated effects, which can be enhanced by the quasi-planar structure of bridge and the coplanar character between acceptor moieties and conjugate bridges, extend the delocalization of electron, and it is favorable for efficient CT from the chromophore to the carboxyl group. Similar geometric characters are also appeared in the organic dye sensitizers JK-1 and JK-2 [
63], JK-16 and JK-17 [
64,
65], and other organic dyes which contain cyanoacrylic acid group [
66–
69].
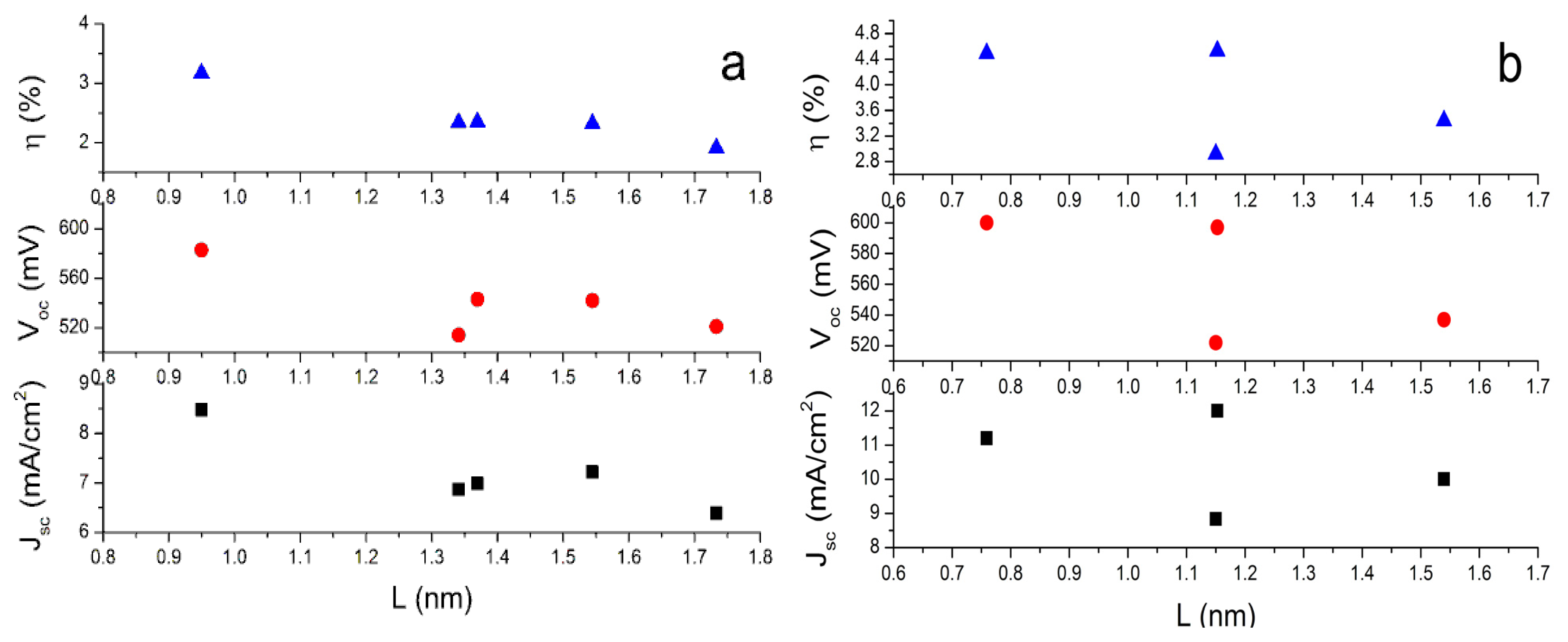
To investigate the role of conjugate bridge length in the performance of dyes in DSCs, we define “
L” as the distance between two special C atoms. One is the C atom in the carboxyl group. The other one is the C atom in tetrahydroquinoline moiety where the conjugate bridge connects. When the dyes adsorbed on the surface of semiconductor nanoparticles, the photo-induced CT occurs at the interface between dyes and semiconductor nanoparticles. Thus, the
L can describe the CT distance and it can describe conjugate length to some extent. The Table S4 in SI lists the calculated
L. The data indicate that the lengths of conjugate bridge with bithiophene and dithieno-[3,2-b;2′,3′-d]thiophene groups are very similar. On the basis of the calculated
L and the reported photovoltaic experimental data [
47], the dependence of open-circuit photovoltage (
Voc), short-circuit photocurrent density (
Jsc), and solar-to-electrical energy conversion efficiencies (η) on the length of conjugate bridges are plotted in
Figure 3. It can be found that the longest
L with terthiophene is unfavorable to improve dye performance in DSCs, while the shorter
L with thiophene (C2-1) or thienylvinyl (C1-1) have better performance. It is understandable because the longer
L may increase the possibility that the excited electrons decay to the electrolyte [
70].
3.2. Electronic Structures
In order to analyze the charge distribution and the electron-transfer mechanism of D-π-A dyes, the Natural Bond Orbital (NBO) analysis has been performed, based upon the optimized structure of the ground state. The calculated NBO results are listed in
Table 3. The positive charges of tetrahydroquinoline moieties represent the acts as an effective electron-donor unit. Contrarily, the negative NBO charges of cyanoacrylic acid reveal that the electrons trap in the electron-acceptor unit. The data also suggest the charges of similar acceptors in different dyes are very similar, but the charges of donors and conjugate bridges are more flexible. This means that the natural charges in dye sensitizers are dominated by acceptor moieties.
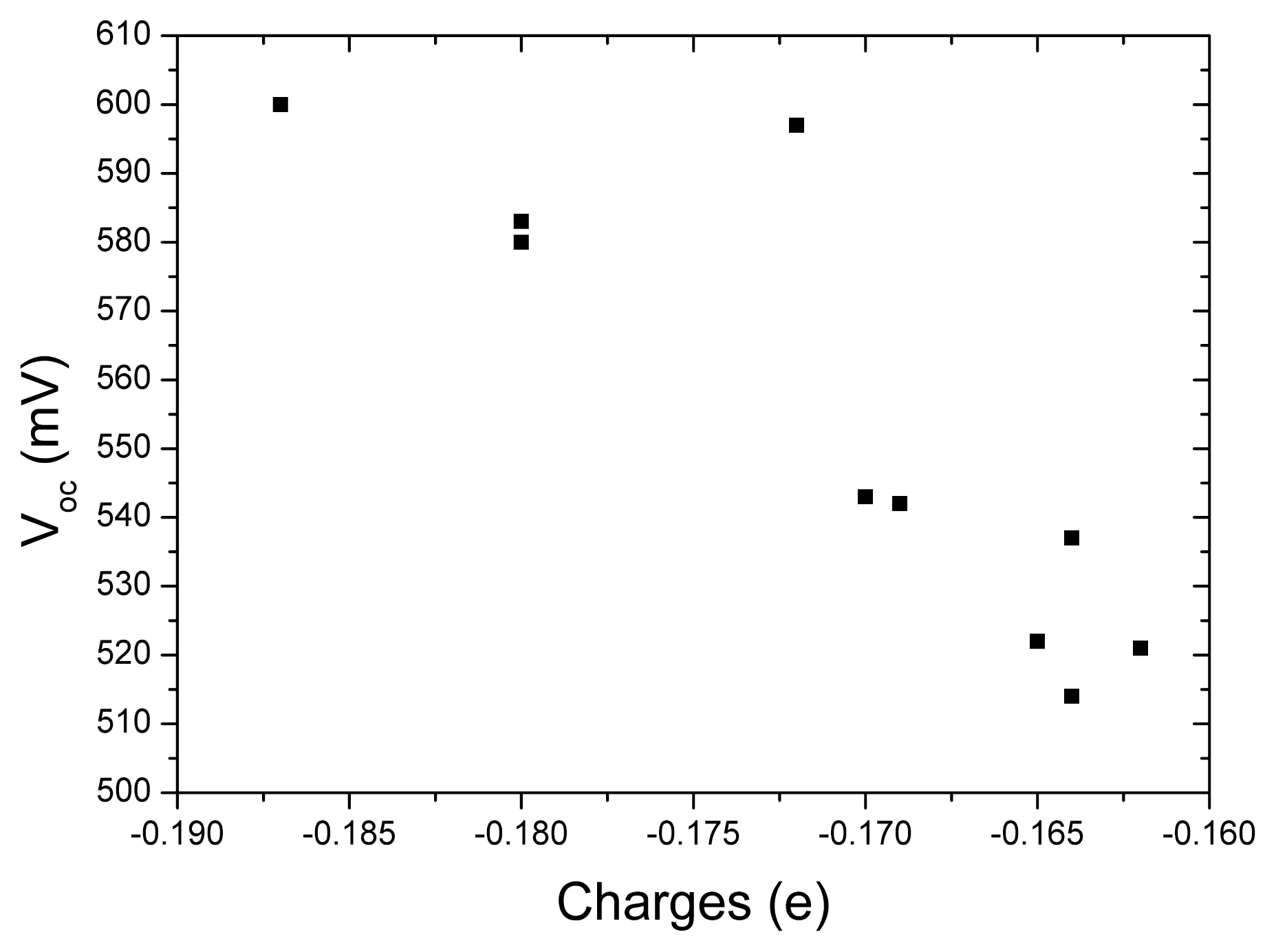
The open-circuit voltage
Vocversus the charges of acceptor groups in tetrahydroquinoline dyes is presented in
Figure 4 in order to investigate the relationship between these quantities. It can be found that more charges populated in acceptor groups correspond to larger
Voc. This means more charges populated in acceptor are favorable to increase
Voc. So, introducing strong electron-withdrawing group in acceptor of dye sensitizer is a possible way to increase
Voc of DSC. This tendency is similar to our previous work for the dyes of coumarin NKX derivatives [
70].
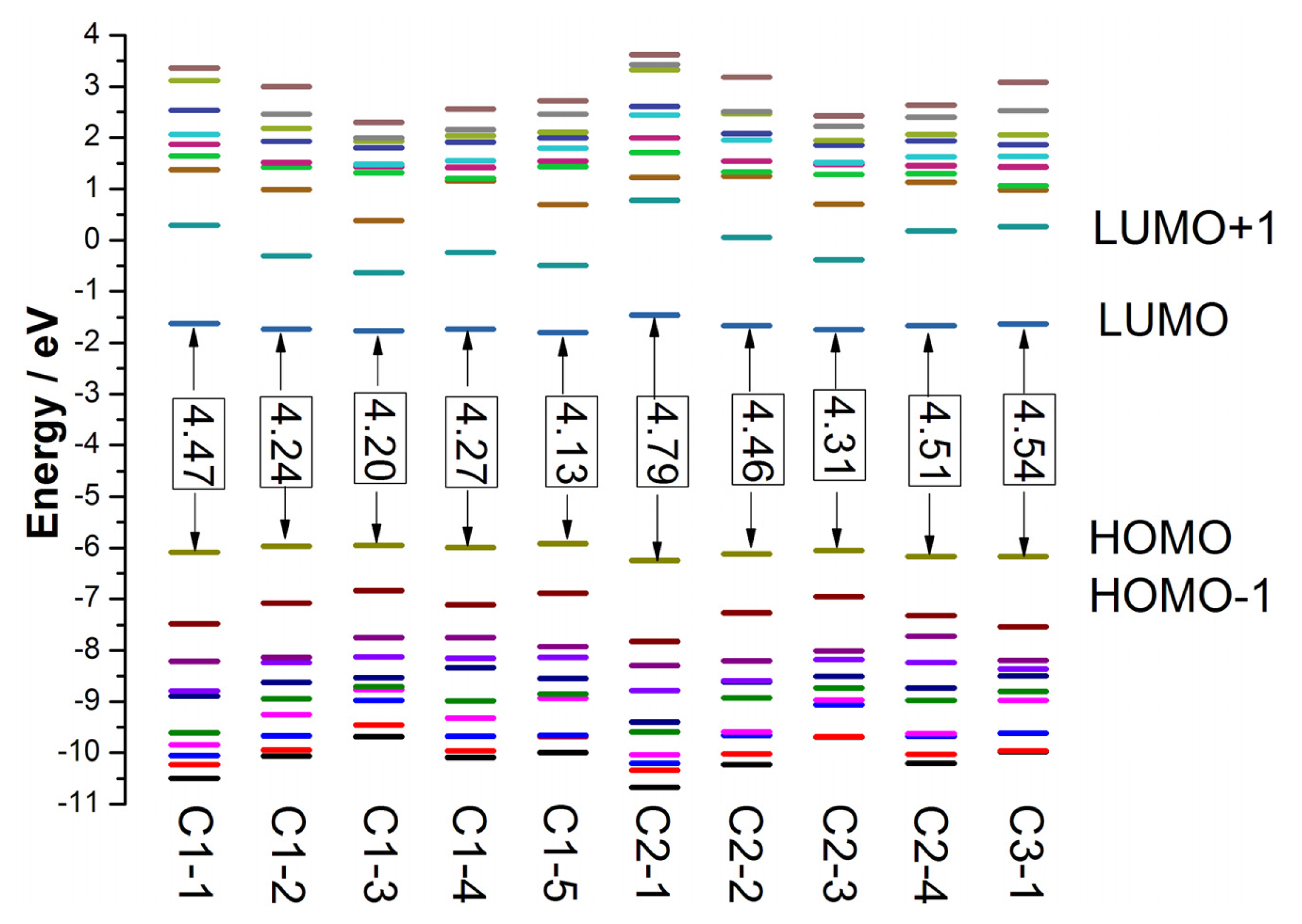
The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energies of better dye sensitizers are required to locate suitable values for matching the conduction band edge of semiconductor and redox potential of electrolyte in DSCs. The HOMO level corresponds to the oxidation potential of dye sensitizer [
71], and the larger oxidation potential increase the driving force for the reduction of oxidized dye [
72]. The HOMO and LUMO energies, as well as the HOMO-LUMO gaps (
Eg) of tetrahydroquinoline dyes in solvent are presented in
Figure 5, and the data are listed in Table S5 in SI. The ranges of HOMO, LUMO, and
Eg for tetrahydroquinoline dyes in gas phase are about −5.85 to −6.26 eV, −1.32 to −1.77 eV and 4.09 to 4.94 eV, respectively, and the corresponding data in solvent are about −5.92 to −6.25 eV, −1.46 to −1.79 eV and 4.13 to 4.79 eV, respectively. For C1-
n and C2-
n (
n = 1–3) dyes, the HOMO energies are increased and the LUMO energies are decreased in turn since the size of the thiophene-containing increases. Therefore, the E
g of the dyes are reduced in turn. Compared the C1-
n and C2-
n (
n = 1–4) dyes with giving
n, the corresponding π-conjugated linker of C1-
n dyes have one vinylene group more than that of C2-
n dyes, though they have same electron donor and acceptor groups, and then the HOMO energies of C1-
n are more positive, the LUMO energies of C1-
n are more negative, and the
Eg of C1-
n is smaller. The comparison of the dyes with bithiophene and terthiophene in conjugate bridges suggests that introducing rigid planar dithieno-[3,2-b;2′,3′-d] thiophene in conjugate bridges generate lower HOMO energies and broader E
g. From the data of C1-2 and C1-5, it can be found that inserting vinylene group in the middle of bithiophene elevate HOMO about 0.09 eV in gas phase (0.05 eV in solvent), decline LUMO about 0.09 eV in gas phase (0.06 eV in solvent), and thus reduce
Eg about 0.17 eV in gas phase (0.11 eV in solvent). The comparison of C2-1 and C3-1 indicates that substitution of hydrogen with phenyl increase HOMO about 0.08 eV in solvent, and the reduced
Eg by about 0.25 eV.
3.3. Absorption and Emission Spectra: UV-Vis and Fluorescence Spectra
The electronic absorption spectra of tetrahydroquinoline dyes C1-1, C1-2, C1-3, C1-5, C2-1, C2-2, and C3-1 were measured in ethanol solution, and the absorption spectra of the dyes C1-4, C2-3, and C2-4 were measured in DMF solution. The experimental and calculated electronic absorption λ
max (nm/eV), as well as the λ
max errors (nm/eV) between the experiment and the calculation are listed in
Table 4. It can be found that the calculated results agree well with that of experiment. So, the results of CAM-B3LYP functional combined with PCM method are reliable for analyzing excited state properties of tetrahydroquinoline dyes. Apparently, with the elongating conjugate bridge by thiophene units, the corresponding λ
max has a red-shift, which is favorable for matching the solar radiation spectra. The corresponding oscillator strength also increases in the order because the longer conjugate bridge extends the overlap of HOMO and LUMO.
To obtain the microscopic information about the electronic transitions, we check the corresponding MO properties. The absorptions in visible and near-UV region are the most important regions for photo to current conversion, so only the singlet→singlet transitions of the absorption bands with the wavelength longer than 300 nm and the oscillator strength larger than 0.1 are listed in
Table 5. The isodensity plots of the frontier MOs that related to the absorptions in visible and near-UV region are presented in
Table 6. The MOs indicate that the HOMO-1 of the dyes are delocalized in the molecules, and the HOMOs are mainly contributed from the framework of quinoline moiety to conjugate bridge groups, whereas the LUMOs and LUMO + 1 mainly locate on the acceptor cyanoacetic acid moieties and conjugate bridge groups. In addition, the molecular orbital overlaps of HOMO and LUMO are highly coupled to the π-conjugated linker. For these dyes, the maximum absorptions in UV/vis spectra are dominated by HOMO→LUMO π→π* transitions. Furthermore, the overlap between HOMO and LUMO of the dyes suggest the maximum absorptions have some local excited transitions in conjugate bridge. While the relocations of the HOMOs and LUMOs in the dyes support that the transitions at maximum absorptions have intramolecular charge transfer (IMCT) character. The further MO analysis indicates that the quinoline groups are effective chromophores in IMCT, and then they play an important role in the sensitization of DSC.
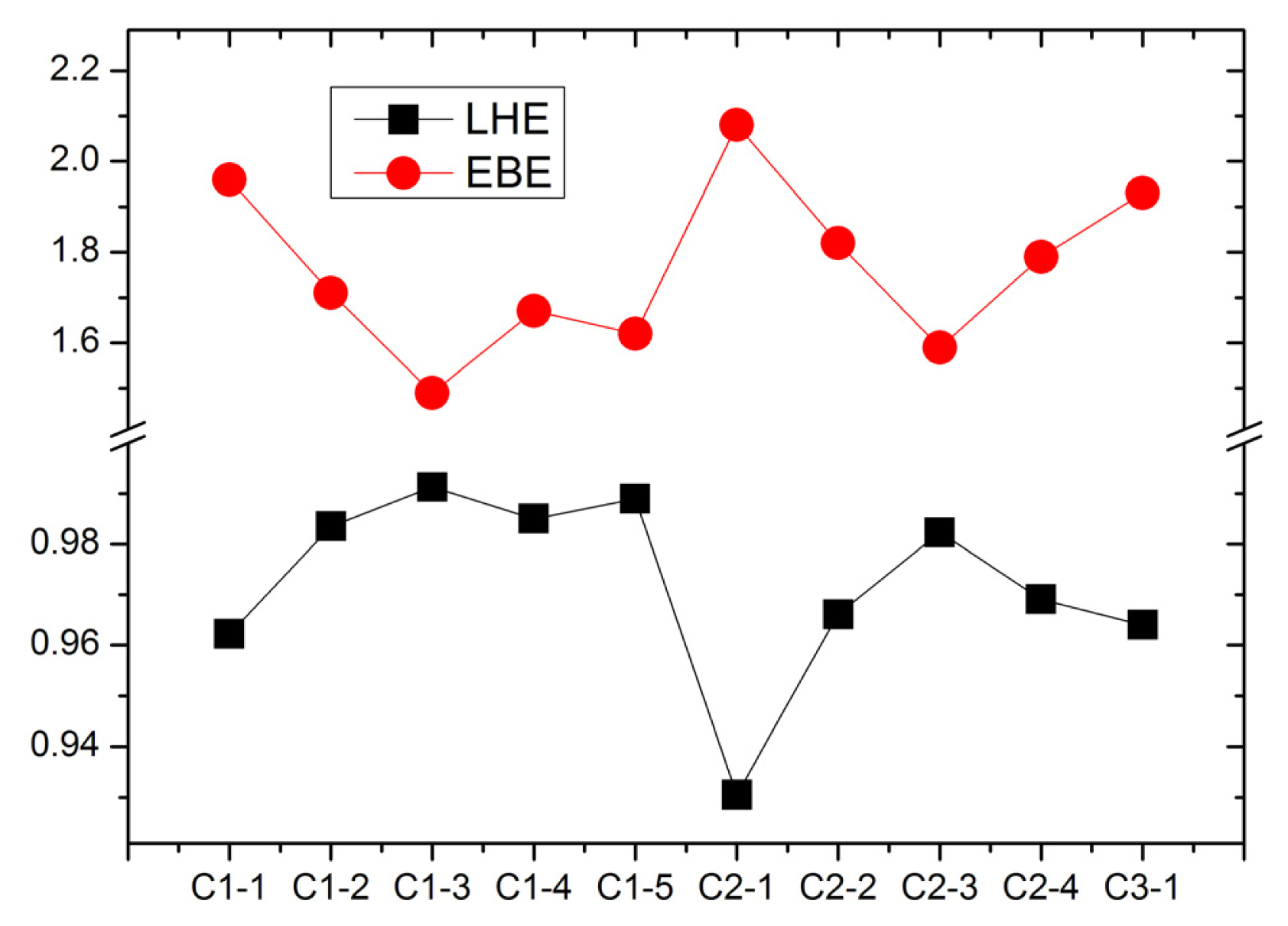
For the dye sensitizers with good performance in DSCs, the LHE are expected to be as high as possible to increase the photocurrent if the excited processes have CT character. LHE can be calculated as [
73]:
where
A is the absorption coefficient and
f is oscillator strength of the excited state associated to the
. The calculated LHE of tetrahydroquinoline dyes are presented in
Figure 6. It can be found that C1-3 and C2-3 have the largest LHE in C1-
n (
n = 1–5) and C2-
n (
n = 1–4) series, respectively. In addition, the elongating of conjugate bridge generates the larger oscillator strength and LHE because of the enhanced overlap between the ground states (mainly contributed by HOMO) and the first excited states (mainly contributed by LUMO). So, extending the conjugate bridge in D-π-A organic dye is an effective way to increase the harvest of solar light.
The EBE, as an important quantity for the efficiency of excitonic solar cells, determines the charge separation in solar cells [
42]. The EBE can be calculated as the difference between the electronic and optical band gap energies [
74]. The electronic band gap is calculated as the energy difference between the HOMO and LUMO levels, while the first excitation energy is adopted as the optical gap [
42,
75]. The calculated EBE (in eV) of tetrahydroquinoline dyes are shown in
Figure 6. It indicates that the elongating of conjugate bridge generates the decreasing of EBE, which is favorable for photo-to-current energy conversion via the dissociation of a bound exciton (hole/electron pair). Furthermore, the smallest EBE of C1-3 and C2-3 dyes suggest the exciton can dissociate more effectively than that in other dyes. More interestingly, the figure reveals that the EBE and the LHE are inversely correlated, implying that the dyes with lower EBE produce more efficient light harvesting. This agrees with the relationship between the EBE and quantum yield [
42].
Concerning fluorescence, the calculated and experimental emission maxima λ and Stokes shift (SS) for the tetrahydroquinoline dyes are listed in
Table 7. For most of the dyes, except C1-1 and C2-1, the computed emission maxima are in good agreement with the experimental data because the discrepancy is lower than 0.3 eV. Particularly, for C2-3 and C1-4, a quantitatively agreement with the experimental results is obtained since the errors are about 0.03 and 0.08 eV, respectively. While in the case of C1-1 and C2-1, the errors (0.34 and 0.45 eV, respectively) are larger than that of others. This is probably to be ascribed to the neglect of direct solute-solvent interactions [
76]. After state specific correction, the calculated emission maxima of C1-1 and C2-1 dyes are about 2.14 and 2.41 eV, respectively, and therefore the errors of emission maxima are reduced to 0.22 and 0.32 eV, respectively. Compared with emission maxima, the SS has similar errors. It can be found that, except C1-3, elongating conjugate bridge generates red shift of emission maxima, larger oscillator strength, and increasing of SS due to the enhanced flexibility by the longer conjugate linker, allowing for a larger structural relaxation from the excited state S
1 to the ground state S
0.
Radiative lifetimes for spontaneous emission from S
1 were computed from fluorescene energies (
Efluo) and oscillator strength, which represents the transition probability (
f in a.u.) as [
76]:
where, me is the electron mass, ɛ0 is the vacuum permittivity, h̄ is the reduced Plank constant, c is the speed of light, and e is the elementary charge. To investigate the role of conjugate bridge in radiative lifetime, rather than accurate reproduce the radiative lifetime, we define relative radiative lifetime (τrel) of the dye molecule by assuming C1-1 radiative lifetime (ιC1–1) as reference value:
where
EfluoC1–1,
Efluoi,
fC1–1, and
fi are fluorescene energies and oscillator strength of C1-1 and the selected dye molecule, respectively. The calculated relative radiative lifetime of the tetrahydroquinoline dyes are listed in
Table 6. Similar to oscillator strength, the relative radiative lifetime is increasing with the elongating conjugate bridge by thiophene unit. The shortest lifetime of C1-4 and C2-4 may be ascribed to their planar conjugation (by dithienothiophene group), favoring charge recombination.
3.4. The Driving Force of Electron Injection and Dye Regeneration
The electron injection from the excited dyes to semiconductor conduction band and the dye regeneration processes can be described as CT reaction. The driving force, which is defined as the potential difference between the oxidized dyes and the electrolyte, determines the yield of electron collection and dye regeneration. Furthermore, the oxidation potential of the dyes must be more positive than that of electrolyte, thus ensuring enough driving force for a fast and efficient regeneration of the dye cation radical and avoiding the geminate charge recombination between oxidized dye sensitizers and the nanocrystalline TiO
2 film where the photo-excited electrons are injected. In terms of the Marcus theory for electron transfer [
77], the CT rate constants can be affected by free energy change related to the reaction. The free energy change for electron injection ( Δ
Ginject ) affects the electron injection rate and therefore the
Jsc in DSCs. Δ
Ginject can be viewed as the electron injection driving force [
44]. According to Preat’s method [
73], assuming the electron injection occurs from the unrelaxed excited state of the dye, the Δ
Ginject can be calculated by the following Equation:
where
is the oxidation potential of the dye in the excited state and
is the reduction potential of the conduction band of the semiconductor. The reported
for TiO
2[
78] was adopted in this work. The
can be calculated as following [
79]:
in which
is the redox potential of the ground state and λmax is the absorption maximum with IMCT character. Whereas the free energy change of dye regeneration (
) can affect on the rate constant of redox process between the oxidized dyes and electrolyte. The
can be calculated as
where
is the redox potential of electrolyte. The
of commonly used redox couple iodide/triiodide is about 4.85 eV (0.35 V
vs. NHE) [
80]. The calculated
, Δ
Ginject, and
for tetrahydroquinoline dyes are listed in
Table 8.
In terms of the calculated results, the negative ΔGinject of the tetrahydroquinoline dyes indicates that the excited state with IMCT character lies above the TiO2 conduction band edge. For C1-n (n = 1–5) dyes, the absolute value of ΔGinject for C1-3 is larger than that of other dyes, and the absolute value of ΔGinject increases with the elongating of conjugate bridges with thiophene units. The ΔGinject of C1-4 which has dithienothiophene in conjugate linker is almost as same as that of C1-3. In addition, the data of C1-5 means that inserting a vinyl unit between bithiophene in conjugate bridges induces a slight decreasing of ΔGinject (about 0.02 eV). Furthermore, with elongating conjugate bridges, the
of the dyes decrease in the corresponding order. So, introducing longer conjugate bridges in dyes can suppress electron recombination. For C2-n (n = 1–4) dyes, the variation of ΔGinject is only about 0.05 eV (from -0.59 to -0.64 eV). The C2-3 and C2-4 have largest absolute value of ΔGinject, and the dependence of
on the number of thiophene unit in conjugate bridge is similar to that of C1-n (n = 1–4) dyes. Comparing the data of C1-n and C2-n (n = 1–4), it is found that inserting vinyl units between quinoline and thiophene generate a reduction of absolute value ΔGinject of and
.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


















































