Melatonin and Pancreatic Islets: Interrelationships between Melatonin, Insulin and Glucagon

Abstract
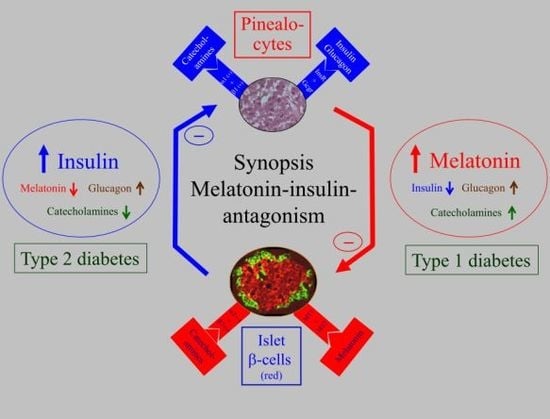
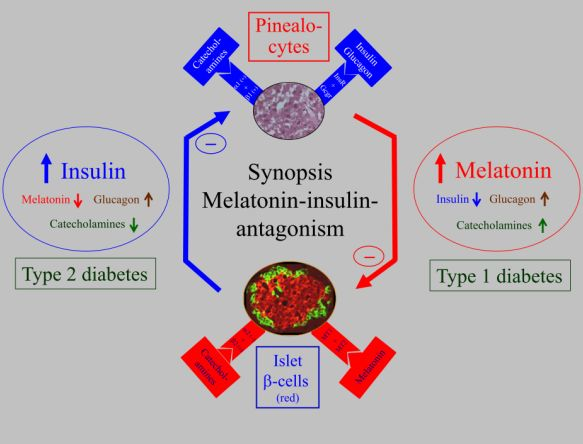
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction, Historical Aspects
2. Membrane Melatonin Receptors of the Pancreatic β-Cell
2.1. Function and Signaling of Mammalian Melatonin Membrane Receptors
2.2. Receptor Heterodimerization
2.3. Receptor-Associated Proteins
2.4. Sensitization and Desensitization
2.5. Function and Impact of Melatonin Receptors of the Pancreatic β-Cell on Insulin Secretion and Its Role within the Framework of Peripheral Circadian Clocks
2.6. Implications of Genetic Association Studies
3. Recent Investigations of Melatonin on Insulin Secretion
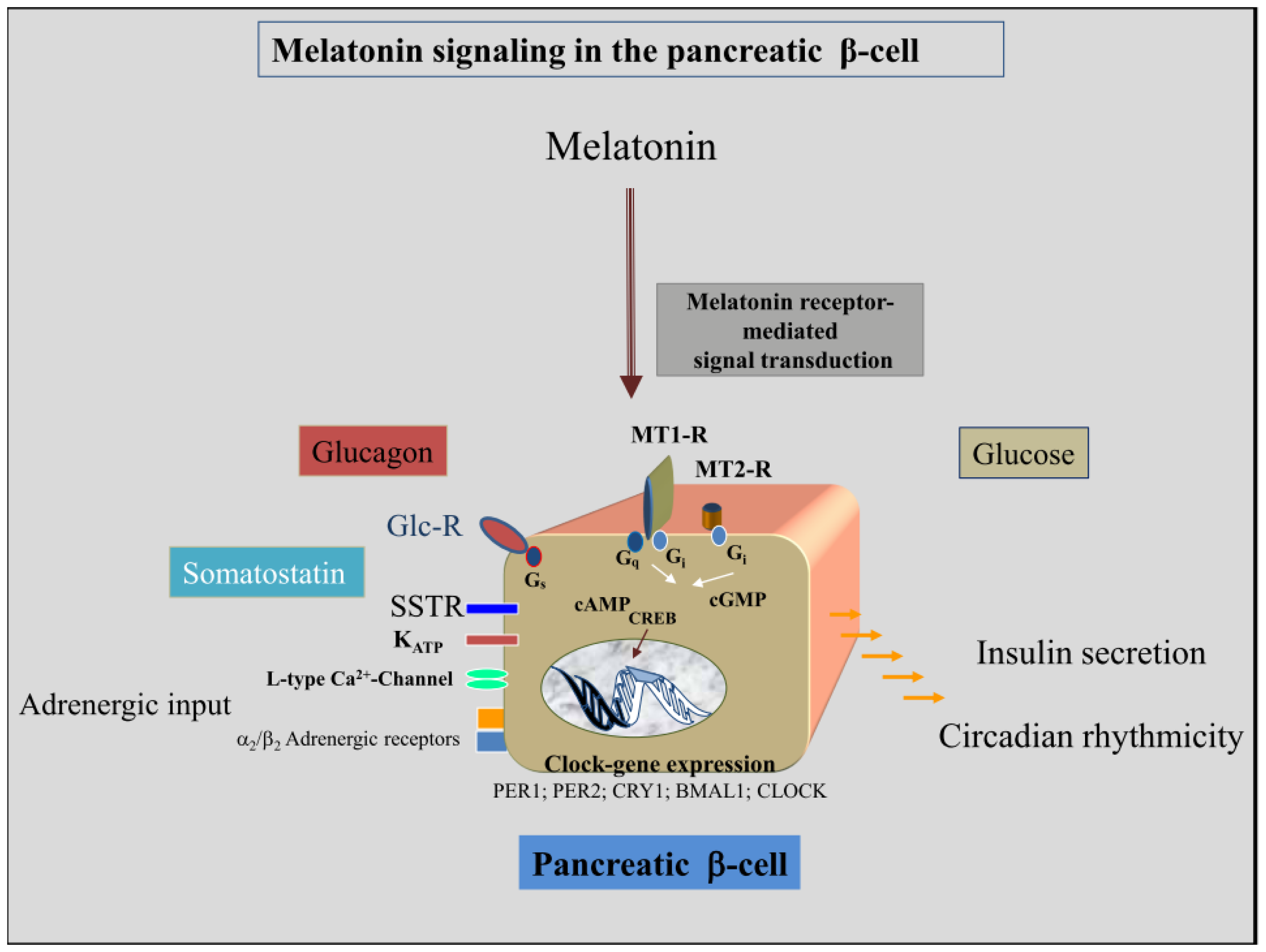
4. Melatonin-Receptor-Mediated Signal Transduction Pathways of the Insulin-Producing β-Cell
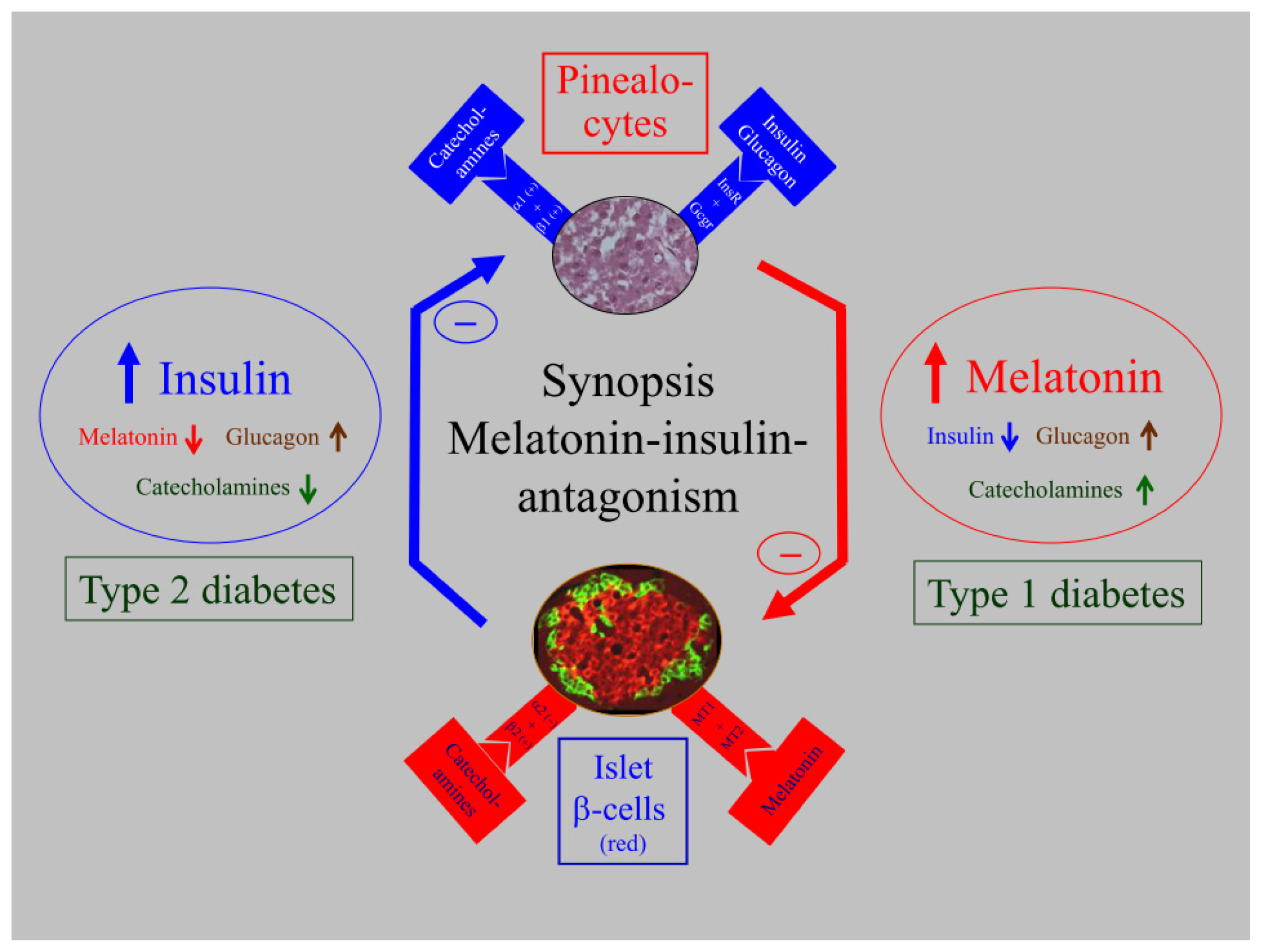
5. Biological Relevance of Melatonin-Insulin Antagonisms in Metabolic Disturbances, as well as in Type 1 and Type 2 Diabetes
6. Recent Investigations of Melatonin-Glucagon Interrelationships
7. Conclusions
Acknowledgements
References
- Parhon, C.I. Congrès d’Endocrinologie de Bucarest 1939, I, 187.
- Parhon, C.I.; Potop, I.; Felix, E.; Boeru, V. Influenta epifisectomiei si a administrarii de extract epifisar asupra unor data metabolice privind mineralele (Ca, K, P, Si, Mg), lipidele, protidele si glucidele, la sobolanul alb adult. Stud. Cercet. Endocr 1952, 3, 321–329. [Google Scholar]
- Milcou, I.; Nanu, L.; Marcean, R. De l’existence d’une hormone hypoglycémiante épiphysaire synergique de l’insuline. Ann. Endocrinol. (Paris) 1957, 18, 612–620. [Google Scholar]
- Milcu, S.M. Role de l’épiphyse dans le metabolisme glucidique. J. Annu. Diabetol. Dieu 1968, 9, 163–180. [Google Scholar]
- Milcu, S.M.; Milcu, I. Über ein hypoglykämisch wirkendes Hormon in der Zirbeldrüse. Medizinische 1958, 17, 711–715. [Google Scholar]
- Milcu, I.; Nanu, L.; Marcean, R.; Sitaru, S. L’action de l’extrait pinéal et de la pinéalectomie sur le glycogène hépatique et musculaire après infusion prolongée de glucose. Stud. Cercet Endocr 1963, 14, 651–655. [Google Scholar]
- Milcou, S.M.; Milcou, I.; Nanu, L. Le role de la glande pinéale dans le métabolisme des glucides. Ann. Endocrinol. (Paris) 1963, 24, 233–254. [Google Scholar]
- Lerner, A.B.; Case, J.D.; Takahashi, Y.; Lee, T.H.; Mori, W. Isolation of melatonin, the pineal gland factor that lightens melanocytes. J. Am. Chem. Soc 1958, 80, 2587. [Google Scholar]
- Lerner, A.B.; Case, J.D.; Heinzelman, R.V. Structure of melatonin. J. Am. Chem. Soc 1959, 81, 6084–6092. [Google Scholar]
- Milcu, I.; Nanu, L.; Marcean, R.; Sitaru, S. Research on the diabetogenic effects of epiphysectomy in the lamb. Stud. Cercet. Endocr 1964, 15, 507–513. [Google Scholar]
- Milcu, S.M.; Nanu-Ionescu, L.; Milcu, I. The Effect of Pinealectomy on Plasma Insulin in Rats. In The Pineal Gland; Wolstenholme, G.E.W., Knight, J., Eds.; Churchill Livingstone: Edinburgh-London, UK, 1971; pp. 345–357. [Google Scholar]
- Diaz, B.; Blazquez, E. Effect of pinealectomy on plasma glucose, insulin and glucagon levels in the rat. Horm. Metab. Res 1986, 18, 225–229. [Google Scholar]
- Mellado, M.C.; Rodriguez, M.V.; Diaz, B.; de Diego, J.G.; Alvarez, E.; Blázquez, E. Role of the Pineal Gland in the Normal Maintenance of Circulating Levels and of Liver Receptor Concentrations of Insulin and Glucagon in the Rat. Proceedings of the Workshop on the Pineal Gland, Salamanca, Spain, 26–28 May 1986; pp. 52–56.
- Rodriguez, V.; Mellado, C.; Alvarez, E.; de Diego, J.G.; Blazquez, E. Effect of pinealectomy on liver insulin and glucagon receptor concentrations in the rat. J. Pineal Res 1989, 6, 77–88. [Google Scholar]
- Munoz Barragán, L.; López Gil, J.A.; Toranzo, D.; Blázquez, J.L.; Pizarro, M.D.L.; Pastor, F.E.; Mosqueira, M.I. The Pineal Gland: A Neuroendocrine “Crossroad”. Proceedings of the Workshop on the Pineal Gland, Salamanca, Spain, 26–28 May 1986; pp. 57–61.
- Munoz Barragán, L.; Toranzo, D.; Blázquez, E.; Ghiglione, M.; Pastor, F.E. Role of the pineal gland on insulin and glucagon release of control and diabetic rats. Diabetes 1983, 32, 141A. [Google Scholar]
- Munoz Barragán, L.; Toranzo, D.; Blázquez, E.; Pastor, F.E.; Mosqueira, M.I.; López, J.A.; Blázquez, J.J. A radio-immunoanalytical and immunocytochemical study on A and B insular cells in response to pinealectomy or pineal denervation. Diabetologia 1984, 27, 313A. [Google Scholar]
- De Lima, L.M.; dos Reis, L.C.; de Lima, M.A. Influence of the pineal gland on the physiology, morphometry and morphology of pancreatic islets in rats. Braz. J. Biol 2001, 61, 333–340. [Google Scholar]
- Shima, T.; Chun, S.J.; Niijima, A.; Bizot-Espiard, J.G.; Guardiola-Lemaitre, B.; Hosokawa, M.; Nagai, K. Melatonin suppresses hyperglycemia caused by intracerebroventricular injection of 2-deoxy-D-glucose in rats. Neurosci. Lett 1997, 226, 119–122. [Google Scholar]
- Conti, A.; Maestroni, G.J. Melatonin rhythms in mice: Role in autoimmune and lymphoproliferative diseases. Ann. N. Y. Acad. Sci 1998, 840, 395–410. [Google Scholar]
- Conti, A.; Maestroni, G.J. Role of the pineal gland and melatonin in the development of autoimmune diabetes in non-obese diabetic mice. J. Pineal Res 1996, 20, 164–172. [Google Scholar]
- Benson, B.; Miller, C.W.; Sorrentino, S. Effects of blinding on blood glucose and serum insulin-like activity in rats. Texas Rep. Biol. Med 1971, 29, 513–525. [Google Scholar]
- Burns, J.K. Serum sodium and potassium and blood glucose levels in cynamolgus monkeys after administration of melatonin. J. Physiol. 1973, 232, 84P–85P. [Google Scholar]
- Csaba, G.; Barath, P. Are Langerhan’s islets influenced by the pineal body? Experientia 1971, 27, 962. [Google Scholar]
- Csaba, G.; Nagy, S.U. The regulatory role of the pineal gland on the thyroid gland, adrenal medulla and islets of Langerhans. Acta. Biol. Med. Ger 1973, 31, 617–619. [Google Scholar]
- Nanu-Ionescu, L.; Ionescu, V. La glande pinéale et le métabolisme de l’insuline. I. Le niveau sanguin de làctivité insulinique totale et de l’insuline “libre” et “liée” dans làpinéalisme experimental. Stud. Cercet. Endocr 1969, 20, 237–243. [Google Scholar]
- Nanu-Ionescu, L.; Marcean, R. La glande pinéale et le métabolisme de l’insuline. II. Les facteurs plasmatique anti-insuliniques dans lápinéalisme expérimental. Rev. Roum. Endocr 1970, 2, 55–61. [Google Scholar]
- Gorray, K.C.; Quay, W.B. Effects of pinealectomy and of sham-pinealectomy on blood glucose levels in the alloxan-diabetic rat. Horm. Metab. Res 1978, 10, 389–392. [Google Scholar]
- Quay, W.B.; Gorray, K.C. Pineal effects on metabolism and glucose homeostasis: Evidence for lines of humoral mediation of pineal influences on tumor growth. J. Neural Transm 1980, 47, 107–120. [Google Scholar]
- Rasmussen, D.D.; Boldt, B.M.; Wilkinson, C.W.; Yellon, S.M.; Matsumoto, A.M. Daily melatonin administration at middle age suppresses male rat visceral fat, plasma leptin, and plasma insulin to youthful levels. Endocrinology 1999, 140, 1009–1012. [Google Scholar]
- Wolden-Hanson, T.; Mitton, D.R.; McCants, R.L.; Yellon, S.M.; Wilkinson, C.W.; Matsumoto, A.M.; Rasmussen, D.D. Daily melatonin administration to middle-aged male rats suppresses body weight, intraabdominal adiposity, and plasma leptin and insulin independent of food intake and total body fat. Endocrinology 2000, 141, 487–497. [Google Scholar]
- Rasmussen, D.D.; Mitton, D.R.; Larsen, S.A.; Yellon, S.M. Aging-dependent changes in the effect of daily melatonin supplementation on rat metabolic and behavioral responses. J. Pineal Res 2001, 31, 89–94. [Google Scholar]
- Dhar, M.; Dayal, S.S.; Ramesh Babu, C.S.; Arora, S.R. Effect of melatonin on glucose tolerance and blood glucose circadian rhythm in rabbits. Indian J. Physiol. Pharmacol 1983, 27, 109–117. [Google Scholar]
- Cagnacci, A.; Arangino, S.; Renzi, A.; Paoletti, A.M.; Melis, G.B.; Cagnacci, P.; Volpe, A. Influence of melatonin administration on glucose tolerance and insulin sensitivity of postmenopausal women. Clin. Endocrinol. (Oxf. ) 2001, 54, 339–346. [Google Scholar]
- Champney, T.H.; Steger, R.W.; Christie, D.S.; Reiter, R.J. Alterations in components of the pineal melatonin synthetic pathway by acute insulin stress in the rat and Syrian hamster. Brain Res 1985, 338, 25–32. [Google Scholar]
- Champney, T.H.; Brainard, G.C.; Richardson, B.A.; Reiter, R.J. Experimentally-induced diabetes reduces nocturnal pineal melatonin content in the Syrian hamster. Comp. Biochem. Physiol. A 1983, 76, 199–201. [Google Scholar]
- Boden, G.; Ruiz, J.; Urbain, J.L.; Chen, X. Evidence for a circadian rhythm of insulin secretion. Am. J. Physiol 1996, 271, E246–E252. [Google Scholar]
- Champney, T.H.; Holtorf, A.P.; Craft, C.M.; Reiter, R.J. Hormonal modulation of pineal melatonin synthesis in rats and Syrian hamsters: Effects of streptozotocin-induced diabetes and insulin injections. Comp. Biochem. Physiol. A 1986, 83, 391–395. [Google Scholar]
- O’Brien, I.A.; Lewin, I.G.; O’Hare, J.P.; Arendt, J.; Corrall, R.J. Abnormal circadian rhythm of melatonin in diabetic autonomic neuropathy. Clin. Endocrinol. (Oxf. ) 1986, 24, 359–364. [Google Scholar]
- John, T.M.; Viswanathan, M.; George, J.C.; Scanes, C.G. Influence of chronic melatonin implantation on circulating levels of catecholamines, growth hormone, thyroid hormones, glucose, and free fatty acids in the pigeon. Gen. Comp. Endocrinol 1990, 79, 226–232. [Google Scholar]
- Lynch, H.J.; Hsuan, M.; Wurtman, R.J. Sympathetic neural control of indoleamine metabolism in the rat pineal gland. Adv. Exp. Med. Biol 1975, 54, 93–114. [Google Scholar]
- Tannenbaum, M.G.; Reiter, R.J.; Vaughan, M.K.; Troiani, M.E.; Gonzalez-Brito, A. Adrenalectomy prevents changes in rat pineal melatonin content and N-acetyltransferase activity induced by acute insulin stress. J. Pineal Res 1987, 4, 395–402. [Google Scholar]
- Mahata, S.K.; Mandal, A.; Ghosh, A. Influence of age and splanchnic nerve on the action of melatonin in the adrenomedullary catecholamine content and blood glucose level in the avian group. J. Comp. Physiol. B 1988, 158, 601–607. [Google Scholar]
- Mahata, S.K. Effect of insulin on serotonin, 5-hydroxyindole-acetic acid, norepinephrine, epinephrine and corticosterone contents in chick. Neurosci. Lett 1991, 121, 115–118. [Google Scholar]
- Maitra, S.K.; Dey, M.; Dutta, S.; Bhattacharya, S.; Dey, R.; Sengupta, A. Influences of graded dose of melatonin on the levels of blood glucose and adrenal catecholamines in male roseringed parakeets (Psittacula krameri) under different photoperiods. Arch. Physiol. Biochem 2000, 108, 444–450. [Google Scholar]
- Maitra, S.K.; Dey, M.; Dey, R.; Bhattacharya, S.; Sengupta, A. Influence of photoperiods on glycemic and adrenal catecholaminergic responses to melatonin administrations in adult male roseringed parakeets, Psittacula krameri Neumann. Indian J. Exp. Biol 2000, 38, 1111–1116. [Google Scholar]
- Feldman, J.M.; Lebovitz, H.E. Structural determinants of indole amine action on in vitro insulin release. Endocrinology 1972, 91, 809–816. [Google Scholar]
- Frankel, B.J.; Strandberg, M.J. Insulin release from isolated mouse islets in vitro: No effect of physiological levels of melatonin or arginine vasotocin. J. Pineal Res 1991, 11, 145–148. [Google Scholar]
- Bizot-Espiard, J.G.; Double, A.; Cousin, B.; Lesieur, D.; Guardiola-Lemaitre, B.; Delagrange, P.; Ktorza, A.; Penicaud, L. Lack of melatonin effects on insulin action in normal rats. Horm. Metab. Res 1998, 30, 711–716. [Google Scholar]
- Niijima, A.; Chun, S.J.; Shima, T.; Bizot-Espiard, J.G.; Guardiola-Lemaitre, B.; Nagai, K. Effect of intravenous administration of melatonin on the efferent activity of the adrenal nerve. J. Auton. Nerv. Syst 1998, 71, 134–138. [Google Scholar]
- Peschke, E.; Fauteck, J.D.; Musshoff, U.; Schmidt, F.; Beckmann, A.; Peschke, D. Evidence for a melatonin receptor within pancreatic islets of neonate rats: Functional, autoradiographic, and molecular investigations. J. Pineal Res 2000, 28, 156–164. [Google Scholar]
- Kemp, D.M.; Ubeda, M.; Habener, J.F. Identification and functional characterization of melatonin Mel 1a receptors in pancreatic beta cells: Potential role in incretin-mediated cell function by sensitization of cAMP signaling. Mol. Cell Endocrinol 2002, 191, 157–166. [Google Scholar]
- Reppert, S.M.; Weaver, D.R.; Ebisawa, T. Cloning and characterization of a mammalian melatonin receptor that mediates reproductive and circadian responses. Neuron 1994, 13, 1177–85. [Google Scholar]
- Reppert, S.M.; Godson, C.; Mahle, C.D.; Weaver, D.R.; Slaugenhaupt, S.A.; Gusella, J.F. Molecular characterization of a second melatonin receptor expressed in human retina and brain: The Mel1b melatonin receptor. Proc. Natl. Acad. Sci. USA 1995, 92, 8734–8738. [Google Scholar]
- Reppert, S.M.; Weaver, D.R.; Ebisawa, T.; Mahle, C.D.; Kolakowski, L.F., Jr. Cloning of a melatonin-related receptor from human pituitary. FEBS Lett. 1996, 386, 219–224. [Google Scholar]
- Dufourny, L.; Migaud, M.; Thiery, J.C.; Malpaux, B. Development of an in vivo adeno-associated virus-mediated siRNA approach to knockdown tyrosine hydroxylase in the lateral retrochiasmatic area of the ovine brain. J. Neurosci. Methods 2008, 170, 56–66. [Google Scholar]
- Brydon, L.; Barrett, P.; Morgan, P.J.; Strosberg, A.D.; Jockers, R. Investigation of the human Mel 1a melatonin receptor using anti-receptor antibodies. Adv. Exp. Med. Biol 1999, 460, 215–220. [Google Scholar]
- Roka, F.; Brydon, L.; Waldhoer, M.; Strosberg, A.D.; Freissmuth, M.; Jockers, R.; Nanoff, C. Tight association of the human Mel(1a)-melatonin receptor and G(i): Precoupling and constitutive activity. Mol. Pharmacol 1999, 56, 1014–1024. [Google Scholar]
- Browning, C.; Beresford, I.; Fraser, N.; Giles, H. Pharmacological characterization of human recombinant melatonin mt(1) and MT(2) receptors. Br. J. Pharmacol 2000, 129, 877–886. [Google Scholar]
- Godson, C.; Reppert, S.M. The Mel1a melatonin receptor is coupled to parallel signal transduction pathways. Endocrinology 1997, 138, 397–404. [Google Scholar]
- Brydon, L.; Roka, F.; Petit, L.; de Coppet, P.; Tissot, M.; Barrett, P.; Morgan, P.J.; Nanoff, C.; Strosberg, A.D.; Jockers, R. Dual signaling of human Mel1a melatonin receptors via G(i2), G(i3), and G(q/11) proteins. Mol. Endocrinol 1999, 13, 2025–2038. [Google Scholar]
- Blumenau, C.; Berger, E.; Fauteck, J.D.; Madeja, M.; Wittkowski, W.; Speckmann, E.J.; Musshoff, U. Expression and functional characterization of the mt1 melatonin receptor from rat brain in Xenopus oocytes: Evidence for coupling to the phosphoinositol pathway. J. Pineal Res 2001, 30, 139–146. [Google Scholar]
- Ross, A.W.; Webster, C.A.; Thompson, M.; Barrett, P.; Morgan, P.J. A novel interaction between inhibitory melatonin receptors and protein kinase C-dependent signal transduction in ovine pars tuberalis cells. Endocrinology 1998, 139, 1723–1730. [Google Scholar]
- Jarzynka, M.J.; Passey, D.K.; Ignatius, P.F.; Melan, M.A.; Radio, N.M.; Jockers, R.; Rasenick, M.M.; Brydon, L.; Witt-Enderby, P.A. Modulation of melatonin receptors and G-protein function by microtubules. J. Pineal Res 2006, 41, 324–336. [Google Scholar]
- Brydon, L.; Petit, L.; de Coppet, P.; Barrett, P.; Morgan, P.J.; Strosberg, A.D.; Jockers, R. Polymorphism and signalling of melatonin receptors. Reprod. Nutr. Dev 1999, 39, 315–324. [Google Scholar]
- MacKenzie, R.S.; Melan, M.A.; Passey, D.K.; Witt-Enderby, P.A. Dual coupling of MT(1) and MT(2) melatonin receptors to cyclic AMP and phosphoinositide signal transduction cascades and their regulation following melatonin exposure. Biochem. Pharmacol 2002, 63, 587–595. [Google Scholar]
- Witt-Enderby, P.A.; Masana, M.I.; Dubocovich, M.L. Physiological exposure to melatonin supersensitizes the cyclic adenosine 3′,5′-monophosphate-dependent signal transduction cascade in Chinese hamster ovary cells expressing the human mt1 melatonin receptor. Endocrinology 1998, 139, 3064–3071. [Google Scholar]
- Shiu, S.Y.; Pang, B.; Tam, C.W.; Yao, K.M. Signal transduction of receptor-mediated antiproliferative action of melatonin on human prostate epithelial cells involves dual activation of Galpha(s) and Galpha(q) proteins. J. Pineal Res 2010, 49, 301–311. [Google Scholar]
- Brydon, L.; Petit, L.; Delagrange, P.; Strosberg, A.D.; Jockers, R. Functional expression of MT2 (Mel1b) melatonin receptors in human PAZ6 adipocytes. Endocrinology 2001, 142, 4264–4271. [Google Scholar]
- Picinato, M.C.; Hirata, A.E.; Cipolla-Neto, J.; Curi, R.; Carvalho, C.R.; Anhe, G.F.; Carpinelli, A.R. Activation of insulin and IGF-1 signaling pathways by melatonin through MT1 receptor in isolated rat pancreatic islets. J. Pineal Res 2008, 44, 88–94. [Google Scholar]
- Anhe, G.F.; Caperuto, L.C.; Pereira-Da-Silva, M.; Souza, L.C.; Hirata, A.E.; Velloso, L.A.; Cipolla-Neto, J.; Carvalho, C.R. In vivo activation of insulin receptor tyrosine kinase by melatonin in the rat hypothalamus. J. Neurochem 2004, 90, 559–566. [Google Scholar]
- Von Gall, C.; Weaver, D.R.; Moek, J.; Jilg, A.; Stehle, J.H.; Korf, H.W. Melatonin plays a crucial role in the regulation of rhythmic clock gene expression in the mouse pars tuberalis. Ann. N. Y. Acad. Sci 2005, 1040, 508–511. [Google Scholar]
- Hunt, A.E.; Al-Ghoul, W.M.; Gillette, M.U.; Dubocovich, M.L. Activation of MT(2) melatonin receptors in rat suprachiasmatic nucleus phase advances the circadian clock. Am. J. Physiol. Cell Physiol 2001, 280, C110–C118. [Google Scholar]
- Slominski, R.M.; Reiter, R.J.; Schlabritz-Loutsevitch, N.; Ostrom, R.S.; Slominski, A.T. Melatonin membrane receptors in peripheral tissues: Distribution and functions. Mol. Cell Endocrinol 2012, 351, 152–166. [Google Scholar]
- Albizu, L.; Moreno, J.L.; Gonzalez-Maeso, J.; Sealfon, S.C. Heteromerization of G protein-coupled receptors: Relevance to neurological disorders and neurotherapeutics. CNS Neurol. Disord. Drug Targets 2010, 9, 636–650. [Google Scholar]
- Ayoub, M.A.; Couturier, C.; Lucas-Meunier, E.; Angers, S.; Fossier, P.; Bouvier, M.; Jockers, R. Monitoring of ligand-independent dimerization and ligand-induced conformational changes of melatonin receptors in living cells by bioluminescence resonance energy transfer. J. Biol. Chem 2002, 277, 21522–21528. [Google Scholar]
- Ayoub, M.A.; Levoye, A.; Delagrange, P.; Jockers, R. Preferential formation of MT1/MT2 melatonin receptor heterodimers with distinct ligand interaction properties compared with MT2 homodimers. Mol. Pharmacol 2004, 66, 312–321. [Google Scholar]
- Levoye, A.; Dam, J.; Ayoub, M.A.; Guillaume, J.L.; Couturier, C.; Delagrange, P.; Jockers, R. The orphan GPR50 receptor specifically inhibits MT1 melatonin receptor function through heterodimerization. EMBO J 2006, 25, 3012–3023. [Google Scholar]
- Guillaume, J.L.; Daulat, A.M.; Maurice, P.; Levoye, A.; Migaud, M.; Brydon, L.; Malpaux, B.; Borg-Capra, C.; Jockers, R. The PDZ protein mupp1 promotes Gi coupling and signaling of the Mt1 melatonin receptor. J. Biol. Chem 2008, 283, 16762–16771. [Google Scholar]
- Daulat, A.M.; Maurice, P.; Froment, C.; Guillaume, J.L.; Broussard, C.; Monsarrat, B.; Delagrange, P.; Jockers, R. Purification and identification of G protein-coupled receptor protein complexes under native conditions. Mol. Cell Proteomics 2007, 6, 835–844. [Google Scholar]
- Daulat, A.M.; Maurice, P.; Jockers, R. Recent methodological advances in the discovery of GPCR-associated protein complexes. Trends Pharmacol. Sci 2009, 30, 72–78. [Google Scholar]
- Maurice, P.; Daulat, A.M.; Broussard, C.; Mozo, J.; Clary, G.; Hotellier, F.; Chafey, P.; Guillaume, J.L.; Ferry, G.; Boutin, J.A.; et al. A generic approach for the purification of signaling complexes that specifically interact with the carboxyl-terminal domain of G protein-coupled receptors. Mol. Cell Proteomics 2008, 7, 1556–1569. [Google Scholar]
- Maurice, P.; Daulat, A.M.; Turecek, R.; Ivankova-Susankova, K.; Zamponi, F.; Kamal, M.; Clement, N.; Guillaume, J.L.; Bettler, B.; Gales, C.; et al. Molecular organization and dynamics of the melatonin MT receptor/RGS20/G(i) protein complex reveal asymmetry of receptor dimers for RGS and G(i) coupling. EMBO J 2010, 29, 3646–3659. [Google Scholar]
- Devavry, S.; Legros, C.; Brasseur, C.; Cohen, W.; Guenin, S.P.; Delagrange, P.; Malpaux, B.; Ouvry, C.; Coge, F.; Nosjean, O.; et al. Molecular pharmacology of the mouse melatonin receptors MT(1) and MT(2). Eur. J. Pharmacol 2012, 677, 15–21. [Google Scholar]
- Dubocovich, M.L.; Delagrange, P.; Krause, D.N.; Sugden, D.; Cardinali, D.P.; Olcese, J. International Union of Basic and Clinical Pharmacology. LXXV. Nomenclature, classification, and pharmacology of G protein-coupled melatonin receptors. Pharmacol. Rev 2010, 62, 343–380. [Google Scholar]
- Hardeland, R. Investigational melatonin receptor agonists. Expert Opin. Investig. Drugs 2010, 19, 747–764. [Google Scholar]
- Barrett, P.; Choi, W.S.; Morris, M.; Morgan, P. A role for tyrosine phosphorylation in the regulation and sensitization of adenylate cyclase by melatonin. FASEB J 2000, 14, 1619–1628. [Google Scholar]
- Von Gall, C.; Garabette, M.L.; Kell, C.A.; Frenzel, S.; Dehghani, F.; Schumm-Draeger, P.M.; Weaver, D.R.; Korf, H.W.; Hastings, M.H.; Stehle, J.H. Rhythmic gene expression in pituitary depends on heterologous sensitization by the neurohormone melatonin. Nat. Neurosci 2002, 5, 234–238. [Google Scholar]
- Barrett, P.; Schuster, C.; Mercer, J.; Morgan, P.J. Sensitization: A mechanism for melatonin action in the pars tuberalis. J. Neuroendocrinol 2003, 15, 415–421. [Google Scholar]
- Gerdin, M.J.; Masana, M.I.; Rivera-Bermudez, M.A.; Hudson, R.L.; Earnest, D.J.; Gillette, M.U.; Dubocovich, M.L. Melatonin desensitizes endogenous MT2 melatonin receptors in the rat suprachiasmatic nucleus: relevance for defining the periods of sensitivity of the mammalian circadian clock to melatonin. FASEB J 2004, 18, 1646–1656. [Google Scholar]
- Bondi, C.D.; McKeon, R.M.; Bennett, J.M.; Ignatius, P.F.; Brydon, L.; Jockers, R.; Melan, M.A.; Witt-Enderby, P.A. MT1 melatonin receptor internalization underlies melatonin-induced morphologic changes in Chinese hamster ovary cells and these processes are dependent on Gi proteins, MEK 1/2 and microtubule modulation. J. Pineal Res 2008, 44, 288–298. [Google Scholar]
- Kokkola, T.; Vaittinen, M.; Laitinen, J.T. Inverse agonist exposure enhances ligand binding and G protein activation of the human MT1 melatonin receptor, but leads to receptor down-regulation. J. Pineal Res 2007, 43, 255–262. [Google Scholar]
- Maury, E.; Ramsey, K.M.; Bass, J. Circadian rhythms and metabolic syndrome: From experimental genetics to human disease. Circ. Res 2010, 106, 447–462. [Google Scholar]
- Welsh, D.K.; Yoo, S.H.; Liu, A.C.; Takahashi, J.S.; Kay, S.A. Bioluminescence imaging of individual fibroblasts reveals persistent, independently phased circadian rhythms of clock gene expression. Curr. Biol 2004, 14, 2289–2295. [Google Scholar]
- Damiola, F.; Le Minh, N.; Preitner, N.; Kornmann, B.; Fleury-Olela, F.; Schibler, U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev 2000, 14, 2950–2961. [Google Scholar]
- Peschke, E.; Peschke, D. Evidence for a circadian rhythm of insulin release from perifused rat pancreatic islets. Diabetologia 1998, 41, 1085–1092. [Google Scholar]
- Delattre, E.; Cipolla Neto, J.; Boschero, A. Diurnal variations in insulin secretion and K+ permeability in isolated rat islets. Clin. Exp. Pharmacol. Physiol 1999, 26, 505–510. [Google Scholar]
- Picinato, M.C.; Haber, E.P.; Carpinelli, A.R.; Cipolla-Neto, J. Daily rhythm of glucose-induced insulin secretion by isolated islets from intact and pinealectomized rat. J. Pineal Res 2002, 33, 172–177. [Google Scholar]
- Boden, G.; Chen, X.; Polansky, M. Disruption of circadian insulin secretion is associated with reduced glucose uptake in first-degree relatives of patients with type 2 diabetes. Diabetes 1999, 48, 2182–2188. [Google Scholar]
- Kalsbeek, A.; Buijs, R.M. Output pathways of the mammalian suprachiasmatic nucleus: Coding circadian time by transmitter selection and specific targeting. Cell Tissue Res 2002, 309, 109–118. [Google Scholar]
- Kalsbeek, A.; Foppen, E.; Schalij, I.; Van Heijningen, C.; van der Vliet, J.; Fliers, E.; Buijs, R.M. Circadian control of the daily plasma glucose rhythm: An interplay of GABA and glutamate. PLoS One 2008, 3, e3194. [Google Scholar]
- Muhlbauer, E.; Wolgast, S.; Finckh, U.; Peschke, D.; Peschke, E. Indication of circadian oscillations in the rat pancreas. FEBS Lett 2004, 564, 91–96. [Google Scholar]
- Stamenkovic, J.A.; Olsson, A.H.; Nagorny, C.L.; Malmgren, S.; Dekker-Nitert, M.; Ling, C.; Mulder, H. Regulation of core clock genes in human islets. Metabolism 2012, 61, 978–985. [Google Scholar]
- Marcheva, B.; Ramsey, K.M.; Buhr, E.D.; Kobayashi, Y.; Su, H.; Ko, C.H.; Ivanova, G.; Omura, C.; Mo, S.; Vitaterna, M.H.; et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature 2010, 466, 627–631. [Google Scholar]
- Peschke, E.; Muhlbauer, E.; Musshoff, U.; Csernus, V.J.; Chankiewitz, E.; Peschke, D. Receptor (MT(1)) mediated influence of melatonin on cAMP concentration and insulin secretion of rat insulinoma cells INS-1. J. Pineal Res 2002, 33, 63–71. [Google Scholar]
- Peschke, E.; Stumpf, I.; Bazwinsky, I.; Litvak, L.; Dralle, H.; Muhlbauer, E. Melatonin and type 2 diabetes—A possible link? J. Pineal Res 2007, 42, 350–358. [Google Scholar]
- Muhlbauer, E.; Peschke, E. Evidence for the expression of both the MT1- and in addition, the MT2-melatonin receptor, in the rat pancreas, islet and beta-cell. J. Pineal Res 2007, 42, 105–106. [Google Scholar]
- Lyssenko, V.; Nagorny, C.L.; Erdos, M.R.; Wierup, N.; Jonsson, A.; Spegel, P.; Bugliani, M.; Saxena, R.; Fex, M.; Pulizzi, N.; et al. Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nat. Genet 2009, 41, 82–88. [Google Scholar]
- Prokopenko, I.; Langenberg, C.; Florez, J.C.; Saxena, R.; Soranzo, N.; Thorleifsson, G.; Loos, R.J.; Manning, A.K.; Jackson, A.U.; Aulchenko, Y.; et al. Variants in MTNR1B influence fasting glucose levels. Nat. Genet 2009, 41, 77–81. [Google Scholar]
- Nagorny, C.L.F.; Sathanoori, R.; Voss, U.; Mulder, H.; Wierup, N. Distribution of melatonin receptors in murine pancreatic islets. J. Pineal Res 2011, 50, 412–417. [Google Scholar]
- Bouatia-Naji, N.; Bonnefond, A.; Cavalcanti-Proenca, C.; Sparso, T.; Holmkvist, J.; Marchand, M.; Delplanque, J.; Lobbens, S.; Rocheleau, G.; Durand, E.; et al. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat. Genet 2009, 41, 89–94. [Google Scholar]
- Ramracheya, R.D.; Muller, D.S.; Squires, P.E.; Brereton, H.; Sugden, D.; Huang, G.C.; Amiel, S.A.; Jones, P.M.; Persaud, S.J. Function and expression of melatonin receptors on human pancreatic islets. J. Pineal Res 2008, 44, 273–279. [Google Scholar]
- Liu, C.; Weaver, D.R.; Jin, X.; Shearman, L.P.; Pieschl, R.L.; Gribkoff, V.K.; Reppert, S.M. Molecular dissection of two distinct actions of melatonin on the suprachiasmatic circadian clock. Neuron 1997, 19, 91–102. [Google Scholar]
- Jin, X.; von Gall, C.; Pieschl, R.L.; Gribkoff, V.K.; Stehle, J.H.; Reppert, S.M.; Weaver, D.R. Targeted disruption of the mouse Mel(1b) melatonin receptor. Mol. Cell Biol 2003, 23, 1054–1060. [Google Scholar]
- Picinato, M.C.; Haber, E.P.; Cipolla-Neto, J.; Curi, R.; de Oliveira Carvalho, C.R.; Carpinelli, A.R. Melatonin inhibits insulin secretion and decreases PKA levels without interfering with glucose metabolism in rat pancreatic islets. J. Pineal Res 2002, 33, 156–160. [Google Scholar]
- Tasali, E.; Leproult, R.; Ehrmann, D.A.; Van Cauter, E. Slow-wave sleep and the risk of type 2 diabetes in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 1044–1049. [Google Scholar]
- Van Cauter, E.; Blackman, J.D.; Roland, D.; Spire, J.P.; Refetoff, S.; Polonsky, K.S. Modulation of glucose regulation and insulin secretion by circadian rhythmicity and sleep. J. Clin. Invest 1991, 88, 934–942. [Google Scholar]
- Braun, M.; Ramracheya, R.; Johnson, P.R.; Rorsman, P. Exocytotic properties of human pancreatic β-cells. Ann. N. Y. Acad. Sci 2009, 1152, 187–193. [Google Scholar]
- Tengholm, A.; Gylfe, E. Oscillatory control of insulin secretion. Mol. Cell Endocrinol 2009, 297, 58–72. [Google Scholar]
- Mears, D. Regulation of insulin secretion in islets of Langerhans by Ca2+ channels. J. Membr. Biol 2004, 200, 57–66. [Google Scholar]
- Muhlbauer, E.; Albrecht, E.; Bazwinsky-Wutschke, I.; Peschke, E. Melatonin influences insulin secretion primarily via MT(1) receptors in rat insulinoma cells (INS-1) and mouse pancreatic islets. J. Pineal Res 2012, 52, 446–459. [Google Scholar]
- Stumpf, I.; Muhlbauer, E.; Peschke, E. Involvement of the cGMP pathway in mediating the insulin-inhibitory effect of melatonin in pancreatic beta-cells. J. Pineal Res 2008, 45, 318–327. [Google Scholar]
- Stumpf, I.; Bazwinsky, I.; Peschke, E. Modulation of the cGMP signaling pathway by melatonin in pancreatic beta-cells. J. Pineal Res 2009, 46, 140–147. [Google Scholar]
- Mühlbauer, E.; Albrecht, E.; Hofmann, K.; Bazwinsky-Wutschke, I.; Peschke, E. Melatonin inhibits insulin secretion in rat insulinoma beta-cells (INS-1) heterologously expressing the human melatonin receptor isoform MT2. J. Pineal Res 2011, 51, 361–372. [Google Scholar]
- Bazwinsky-Wutschke, I.; Wolgast, S.; Muhlbauer, E.; Albrecht, E.; Peschke, E. Phosphorylation of cyclic AMP-response element-binding protein (CREB) is influenced by melatonin treatment in pancreatic rat insulinoma beta-cells (INS-1). J. Pineal Res 2012, 53, 344–357. [Google Scholar]
- Furstenau, U.; Schwaninger, M.; Blume, R.; Jendrusch, E.M.; Knepel, W. Characterization of a novel calcium response element in the glucagon gene. J. Biol. Chem 1999, 274, 5851–5860. [Google Scholar]
- Oetjen, E.; Grapentin, D.; Blume, R.; Seeger, M.; Krause, D.; Eggers, A.; Knepel, W. Regulation of human insulin gene transcription by the immunosuppressive drugs cyclosporin A and tacrolimus at concentrations that inhibit calcineurin activity and involving the transcription factor CREB. Naunyn. Schmiedebergs. Arch. Pharmacol 2003, 367, 227–236. [Google Scholar]
- Sander, M.; German, M.S. The beta cell transcription factors and development of the pancreas. J. Mol. Med. (Berl.) 1997, 75, 327–340. [Google Scholar]
- Staiger, H.; Machicao, F.; Schafer, S.A.; Kirchhoff, K.; Kantartzis, K.; Guthoff, M.; Silbernagel, G.; Stefan, N.; Haring, H.U.; Fritsche, A. Polymorphisms within the novel type 2 diabetes risk locus MTNR1B determine beta-cell function. PLoS One 2008, 3, e3962. [Google Scholar]
- Simonis-Bik, A.M.; Nijpels, G.; van Haeften, T.W.; Houwing-Duistermaat, J.J.; Boomsma, D.I.; Reiling, E.; van Hove, E.C.; Diamant, M.; Kramer, M.H.; Heine, R.J.; et al. Gene variants in the novel type 2 diabetes loci CDC123/CAMK1D, THADA, ADAMTS9, BCL11A, and MTNR1B affect different aspects of pancreatic beta-cell function. Diabetes 2010, 59, 293–301. [Google Scholar]
- Chambers, J.C.; Zhang, W.; Zabaneh, D.; Sehmi, J.; Jain, P.; McCarthy, M.I.; Froguel, P.; Ruokonen, A.; Balding, D.; Jarvelin, M.R.; et al. Common genetic variation near melatonin receptor MTNR1B contributes to raised plasma glucose and increased risk of type 2 diabetes among Indian Asians and European Caucasians. Diabetes 2009, 58, 2703–2708. [Google Scholar]
- Olsson, L.; Pettersen, E.; Ahlbom, A.; Carlsson, S.; Midthjell, K.; Grill, V. No effect by the common gene variant rs10830963 of the melatonin receptor 1B on the association between sleep disturbances and type 2 diabetes: Results from the nord-trondelag health study. Diabetologia 2011, 54, 1375–1378. [Google Scholar]
- Andersson, E.A.; Holst, B.; Sparso, T.; Grarup, N.; Banasik, K.; Holmkvist, J.; Jorgensen, T.; Borch-Johnsen, K.; Egerod, K.L.; Lauritzen, T.; et al. MTNR1B G24E variant associates with BMI and fasting plasma glucose in the general population in studies of 22,142 Europeans. Diabetes 2010, 59, 1539–1548. [Google Scholar]
- Bonnefond, A.; Clement, N.; Fawcett, K.; Yengo, L.; Vaillant, E.; Guillaume, J.L.; Dechaume, A.; Payne, F.; Roussel, R.; Czernichow, S.; et al. Rare MTNR1B variants impairing melatonin receptor 1B function contribute to type 2 diabetes. Nat. Genet 2012, 44, 297–301. [Google Scholar]
- Peschke, E.; Frese, T.; Chankiewitz, E.; Peschke, D.; Preiss, U.; Schneyer, U.; Spessert, R.; Muhlbauer, E. Diabetic Goto Kakizaki rats as well as type 2 diabetic patients show a decreased diurnal serum melatonin level and an increased pancreatic melatonin-receptor status. J. Pineal Res 2006, 40, 135–143. [Google Scholar]
- Bizot-Espiard, J.G.; Double, A.; Guardiola-Lemaitre, B.; Delagrange, P.; Ktorza, A.; Penicaud, L. Diurnal rhythms in plasma glucose, insulin, growth hormone and melatonin levels in fasted and hyperglycaemic rats. Diabetes Metab 1998, 24, 235–240. [Google Scholar]
- la Fleur, S.E.; Kalsbeek, A.; Wortel, J.; van der Vliet, J.; Buijs, R.M. Role for the pineal and melatonin in glucose homeostasis: pinealectomy increases night-time glucose concentrations. J. Neuroendocrinol 2001, 13, 1025–1032. [Google Scholar]
- Peschke, E.; Peschke, D.; Hammer, T.; Csernus, V. Influence of melatonin and serotonin on glucose-stimulated insulin release from perifused rat pancreatic islets in vitro. J. Pineal Res 1997, 23, 156–163. [Google Scholar]
- Reppert, S.M.; Weaver, D.R.; Cassone, V.M.; Godson, C.; Kolakowski, L.F., Jr. Melatonin receptors are for the birds: Molecular analysis of two receptor subtypes differentially expressed in chick brain. Neuron 1995, 15, 1003–1015. [Google Scholar]
- Vanecek, J. Cellular mechanisms of melatonin action. Physiol. Rev 1998, 78, 687–721. [Google Scholar]
- Simonneaux, V.; Ribelayga, C. Generation of the melatonin endocrine message in mammals: A review of the complex regulation of melatonin synthesis by norepinephrine, peptides, and other pineal transmitters. Pharmacol Rev 2003, 55, 325–395. [Google Scholar]
- Peschke, E. Zum Einfluss von Melatonin auf Insulinsekretion, Signaltransduktion und Sekretionsrhythmus pankreatischer B-Zellen in vitro. Abh. Sächs. Akad. Wiss. Math.-nat. Kl 2003, 60, 89–119. [Google Scholar]
- Peschke, E. Über den phylogenetischen Funktionswandel des Pinealorgans und seine Bedeutung für die Insulinsekretion bei Mammalia. Sitzungsber. Sächs. Akad. Wiss. Math.-nat. Kl 2004, 129, 1–34. [Google Scholar]
- Popova, J.S.; Dubocovich, M.L. Melatonin receptor-mediated stimulation of phosphoinositide breakdown in chick brain slices. J. Neurochem 1995, 64, 130–138. [Google Scholar]
- Zemkova, H.; Vanecek, J. Differences in gonadotropin-releasing hormone-induced calcium signaling between melatonin-sensitive and melatonin-insensitive neonatal rat gonadotrophs. Endocrinology 2000, 141, 1017–1026. [Google Scholar]
- Balik, A.; Kretschmannova, K.; Mazna, P.; Svobodova, I.; Zemkova, H. Melatonin action in neonatal gonadotrophs. Physiol. Res 2004, 53, S153–S166. [Google Scholar]
- Girouard, H.; de Champlain, J. Inhibitory effect of melatonin on alpha1-adrenergic-induced vasoconstriction in mesenteric beds of spontaneously hypertensive rats. Am. J. Hypertens 2004, 17, 339–346. [Google Scholar]
- Bach, A.G.; Wolgast, S.; Muhlbauer, E.; Peschke, E. Melatonin stimulates inositol-1,4,5-trisphosphate and Ca2+ release from INS1 insulinoma cells. J. Pineal Res 2005, 39, 316–323. [Google Scholar]
- Bach, A.G.; Peschke, E. Melatonin and Type 2 Diabetes. In Melatonin in the Promotion of Health, 2nd ed; Watson, R.R., Ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2012; pp. 147–165. [Google Scholar]
- Lai, F.P.; Mody, S.M.; Yung, L.Y.; Pang, C.S.; Pang, S.F.; Wong, Y.H. Chimeric Galphaq subunits can distinguish the long form of the Xenopus Mel1c melatonin receptor from the mammalian mt1 and MT2 melatonin receptors. J. Pineal Res 2001, 30, 171–179. [Google Scholar]
- Lai, F.P.; Mody, S.M.; Yung, L.Y.; Kam, J.Y.; Pang, C.S.; Pang, S.F.; Wong, Y.H. Molecular determinants for the differential coupling of Galpha(16) to the melatonin MT1, MT2 and Xenopus Mel1c receptors. J. Neurochem 2002, 80, 736–745. [Google Scholar]
- Peschke, E. Melatonin, endocrine pancreas and diabetes. J. Pineal Res 2008, 44, 26–40. [Google Scholar]
- Mulder, H.; Nagorny, C.L.; Lyssenko, V.; Groop, L. Melatonin receptors in pancreatic islets: Good morning to a novel type 2 diabetes gene. Diabetologia 2009, 52, 1240–1249. [Google Scholar]
- Radziuk, J.; Pye, S. Diurnal rhythm in endogenous glucose production is a major contributor to fasting hyperglycaemia in type 2 diabetes. Suprachiasmatic deficit or limit cycle behaviour? Diabetologia 2006, 49, 1619–1628. [Google Scholar]
- Van Cauter, E. Putative roles of melatonin in glucose regulation. Therapie 1998, 53, 467–472. [Google Scholar]
- Peschke, D.; Peschke, E.; Mess, B. Circannual rhythm and increase of body weight and food intake in the young Wistar-rat following pinealectomy and ganglionectomy. Neuroendocrinol. Lett 1987, 9, 321–327. [Google Scholar]
- Murialdo, G.; Costelli, P.; Fonzi, S.; Parodi, C.; Torre, F.; Cenacchi, T.; Polleri, A. Circadian secretion of melatonin and thyrotropin in hospitalized aged patients. Aging (Milano) 1993, 5, 39–46. [Google Scholar]
- Reiter, R.J. Oxidative damage in the central nervous system: Protection by melatonin. Prog. Neurobiol 1998, 56, 359–384. [Google Scholar]
- Tarquini, B.; Cornelissen, G.; Perfetto, F.; Tarquini, R.; Halberg, F. Chronome assessment of circulating melatonin in humans. In Vivo 1997, 11, 473–484. [Google Scholar]
- Morgan, L.; Hampton, S.; Gibbs, M.; Arendt, J. Circadian aspects of postprandial metabolism. Chronobiol. Int 2003, 20, 795–808. [Google Scholar]
- Qin, L.Q.; Li, J.; Wang, Y.; Wang, J.; Xu, J.Y.; Kaneko, T. The effects of nocturnal life on endocrine circadian patterns in healthy adults. Life Sci 2003, 73, 2467–2475. [Google Scholar]
- Ribeiro, D.C.; Hampton, S.M.; Morgan, L.; Deacon, S.; Arendt, J. Altered postprandial hormone and metabolic responses in a simulated shift work environment. J. Endocrinol 1998, 158, 305–310. [Google Scholar]
- Robeva, R.; Kirilov, G.; Tomova, A.; Kumanov, P. Low testosterone levels and unimpaired melatonin secretion in young males with metabolic syndrome. Andrologia 2006, 38, 216–220. [Google Scholar]
- Peschke, E.; Schucht, H.; Muhlbauer, E. Long-term enteral administration of melatonin reduces plasma insulin and increases expression of pineal insulin receptors in both Wistar and type 2-diabetic Goto-Kakizaki rats. J. Pineal Res 2010, 49, 373–381. [Google Scholar]
- Frese, T.; Bach, A.G.; Muhlbauer, E.; Ponicke, K.; Bromme, H.J.; Welp, A.; Peschke, E. Pineal melatonin synthesis is decreased in type 2 diabetic Goto-Kakizaki rats. Life Sci 2009, 85, 526–533. [Google Scholar]
- Peschke, E.; Bach, A.G.; Muhlbauer, E. Parallel signaling pathways of melatonin in the pancreatic beta-cell. J. Pineal Res 2006, 40, 184–191. [Google Scholar]
- Peschke, E.; Muhlbauer, E. New evidence for a role of melatonin in glucose regulation. Best Pract. Res. Clin. Endocrinol. Metab 2010, 24, 829–841. [Google Scholar]
- Bach, A.G.; Mühlbauer, E.; Peschke, E. Adrenoceptor expression and diurnal rhythms of melatonin and its precursors in the pineal gland of type 2 diabetic Goto-Kakizaki rats. Endocrinology 2010, 151, 2483–2493. [Google Scholar]
- Peschke, E.; Hofmann, K.; Ponicke, K.; Wedekind, D.; Muhlbauer, E. Catecholamines are the key for explaining the biological relevance of insulin-melatonin antagonisms in type 1 and type 2 diabetes. J. Pineal Res 2012, 52, 389–396. [Google Scholar]
- Peschke, E.; Wolgast, S.; Bazwinsky, I.; Ponicke, K.; Muhlbauer, E. Increased melatonin synthesis in pineal glands of rats in streptozotocin induced type 1 diabetes. J. Pineal Res 2008, 45, 439–448. [Google Scholar]
- Nishida, S.; Segawa, T.; Murai, I.; Nakagawa, S. Long-term melatonin administration reduces hyperinsulinemia and improves the altered fatty-acid compositions in type 2 diabetic rats via the restoration of Delta-5 desaturase activity. J. Pineal Res 2002, 32, 26–33. [Google Scholar]
- Nishida, S. Metabolic effects of melatonin on oxidative stress and diabetes mellitus. Endocrine 2005, 27, 131–136. [Google Scholar]
- Bojkova, B.; Orendas, P.; Friedmanova, L.; Kassayova, M.; Datelinka, I.; Ahlersova, E.; Ahlers, I. Prolonged melatonin administration in 6-month-old Sprague-Dawley rats: metabolic alterations. Acta Physiol. Hung 2008, 95, 65–76. [Google Scholar]
- Nishida, S.; Sato, R.; Murai, I.; Nakagawa, S. Effect of pinealectomy on plasma levels of insulin and leptin and on hepatic lipids in type 2 diabetic rats. J. Pineal Res 2003, 35, 251–256. [Google Scholar]
- Peschke, E.; Hofmann, K.; Bahr, I.; Streck, S.; Albrecht, E.; Wedekind, D.; Muhlbauer, E. The insulin-melatonin antagonism: studies in the LEW.1AR1-iddm rat (an animal model of human type 1 diabetes mellitus). Diabetologia 2011, 54, 1831–1840. [Google Scholar]
- Ahrén, B. Islet G protein-coupled receptors as potential tragets for treatment of type 2 diabetes. Nat. Rev. Drug Discov 2009, 8, 369–385. [Google Scholar]
- Ahrén, B. GLP-1 for type 2 diabetes. Exp. Cell Res 2011, 317, 1239–1245. [Google Scholar]
- Saunders, C.; Limbird, L.E. Localization and trafficking of alpha2-adrenergic receptor subtypes in cells and tissues. Pharmacol. Ther. 1999, 84, 193–205. [Google Scholar]
- Klein, D.C. Arylalkylamine N-acetyltransferase: “the Timezyme”. J. Biol. Chem 2007, 282, 4233–4237. [Google Scholar]
- Arendt, J. Importance and relevance of melatonin to human biological rhythms. J. Neuroendocrinol 2003, 15, 427–431. [Google Scholar]
- Korf, H.W.; Schomerus, C.; Stehle, J.H. The pineal organ, its hormone melatonin, and the photoneuroendocrine system. Adv. Anat. Embryol. Cell Biol 1998, 146, 1–100. [Google Scholar]
- Korf, H.W.; Stehle, J.H. The circadian system: Circuits-cells-clock genes. Cell Tissue Res 2002, 309, 1–2. [Google Scholar]
- Reiter, R.J.; Paredes, S.D.; Manchester, L.C.; Tan, D.X. Reducing oxidative/nitrosative stress: A newly-discovered genre for melatonin. Crit. Rev. Biochem. Mol. Biol 2009, 44, 175–200. [Google Scholar]
- Lenzen, S. Oxidative stress: The vulnerable beta cell. Biochem. Soc. Trans 2008, 36, 343–347. [Google Scholar]
- Unger, R.H. Role of glucagon in the pathogenesis of diabetes: The status of the controversy. Metabolism 1978, 27, 1691–1709. [Google Scholar]
- Reaven, G.M.; Chen, Y.D.; Golay, A.; Swislocki, A.L.; Jaspan, J.B. Documentation of hyperglucagonemia throughout the day in nonobese and obese patients with noninsulin-dependent diabetes mellitus. J. Clin. Endocrinol. Metab 1987, 64, 106–110. [Google Scholar]
- Larsson, H.; Ahren, B. Islet dysfunction in insulin resistance involves impaired insulin secretion and increased glucagon secretion in postmenopausal women with impaired glucose tolerance. Diabetes Care 2000, 23, 650–657. [Google Scholar]
- Burcelin, R.; Knauf, C.; Cani, P.D. Pancreatic alpha-cell dysfunction in diabetes. Diabetes Metab 2008, 34, S49–S55. [Google Scholar]
- Quesada, I.; Tuduri, E.; Ripoll, C.; Nadal, A. Physiology of the pancreatic alpha-cell and glucagon secretion: Role in glucose homeostasis and diabetes. J. Endocrinol 2008, 199, 5–19. [Google Scholar]
- Yamamoto, H.; Nagai, K.; Nakagawa, H. Role of SCN in daily rhythms of plasma glucose, FFA, insulin and glucagon. Chronobiol. Int 1987, 4, 483–491. [Google Scholar]
- Tasaka, Y.; Inoue, S.; Maruno, K.; Hirata, Y. Twenty-four-hour variations of plasma pancreatic polypeptide, insulin and glucagon in normal human subjects. Endocrinol. Jpn 1980, 27, 495–498. [Google Scholar]
- Gagliardino, J.J.; Pessacq, M.T.; Hernandez, R.E.; Rebolledo, O.R. Circadian variations in serum glucagon and hepatic glycogen and cyclic amp concentrations. J. Endocrinol 1978, 78, 297–298. [Google Scholar]
- Ruiter, M.; La Fleur, S.E.; van Heijningen, C.; van der Vliet, J.; Kalsbeek, A.; Buijs, R.M. The daily rhythm in plasma glucagon concentrations in the rat is modulated by the biological clock and by feeding behavior. Diabetes 2003, 52, 1709–1715. [Google Scholar]
- Kosa, E.; Maurel, D.; Siaud, P. Effects of pinealectomy on glucagon responsiveness to hypoglycaemia induced by insulin injections in fed rats. Exp. Physiol 2001, 86, 617–620. [Google Scholar]
- Schmid, S.M.; Hallschmid, M.; Jauch-Chara, K.; Bandorf, N.; Born, J.; Schultes, B. Sleep loss alters basal metabolic hormone secretion and modulates the dynamic counterregulatory response to hypoglycemia. J. Clin. Endocrinol. Metab 2007, 92, 3044–3051. [Google Scholar]
- Schmid, S.M.; Jauch-Chara, K.; Hallschmid, M.; Schultes, B. Mild sleep restriction acutely reduces plasma glucagon levels in healthy men. J. Clin. Endocrinol. Metab 2009, 94, 5169–5173. [Google Scholar]
- Ravier, M.A.; Rutter, G.A. Glucose or insulin, but not zinc ions, inhibit glucagon secretion from mouse pancreatic alpha-cells. Diabetes 2005, 54, 1789–1797. [Google Scholar]
- Bähr, I.; Mühlbauer, E.; Schucht, H.; Peschke, E. Melatonin stimulates glucagon secretion in vitro and in vivo. J. Pineal Res 2011, 50, 336–344. [Google Scholar]
- Bahr, I.; Muhlbauer, E.; Albrecht, E.; Peschke, E. Evidence of the receptor-mediated influence of melatonin on pancreatic glucagon secretion via the Galphaq protein-coupled and PI3K signaling pathways. J. Pineal Res 2012, 53, 390–398. [Google Scholar]
- Ward, W.K.; Bolgiano, D.C.; McKnight, B.; Halter, J.B.; Porte, D., Jr. Diminished B cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J. Clin. Invest. 1984, 74, 1318–1328. [Google Scholar]
- Cryer, P.E. Hypoglycaemia: The limiting factor in the glycaemic management of Type I and Type II diabetes. Diabetologia 2002, 45, 937–948. [Google Scholar]
- Suzuki, N.; Aizawa, T.; Asanuma, N.; Sato, Y.; Komatsu, M.; Hidaka, H.; Itoh, N.; Yamauchi, K.; Hashizume, K. An early insulin intervention accelerates pancreatic beta-cell dysfunction in young Goto-Kakizaki rats, a model of naturally occurring noninsulin-dependent diabetes. Endocrinology 1997, 138, 1106–1110. [Google Scholar]
- Seica, R.M.; Martins, M.J.; Pessa, P.B.; Santos, R.M.; Rosario, L.M.; Suzuki, K.I.; Martins, M.I. Morphological changes of islet of Langerhans in an animal model of type 2 diabetes. Acta Med. Port 2003, 16, 381–388. [Google Scholar]
- Li, C.; Shi, Y.; You, L.; Wang, L.; Chen, Z.J. Melatonin receptor 1A gene polymorphism associated with polycystic ovary syndrome. Gynecol. Obstet. Invest 2011, 72, 130–134. [Google Scholar]
- New, D.C.; Tsim, S.T.; Wong, Y.H. G protein-linked effector and second messenger systems involved in melatonin signal transduction. Neurosignals 2003, 12, 59–70. [Google Scholar]
- Lai, L.; Yuan, L.; Chen, Q.; Dong, C.; Mao, L.; Rowan, B.; Frasch, T.; Hill, S.M. The Galphai and Galphaq proteins mediate the effects of melatonin on steroid/thyroid hormone receptor transcriptional activity and breast cancer cell proliferation. J. Pineal Res 2008, 45, 476–488. [Google Scholar]
- Yibchok-anun, S.; Cheng, H.; Abu-Basha, E.A.; Ding, J.; Ioudina, M.; Hsu, W.H. Mechanisms of bradykinin-induced glucagon release in clonal alpha-cells In-R1-G9: Involvement of Ca2+-dependent and -independent pathways. Mol. Cell Endocrinol 2002, 192, 27–36. [Google Scholar]
- Yibchok-Anun, S.; Cheng, H.; Chen, T.H.; Hsu, W.H. Mechanisms of AVP-induced glucagon release in clonal alpha-cells in-R1-G9: Involvement of Ca2+-dependent and -independent pathways. Br. J. Pharmacol 2000, 129, 257–264. [Google Scholar]
- Bode, H.P.; Yule, D.I.; Fehmann, H.C.; Goke, B.; Williams, J.A. Spontaneous calcium oscillations in clonal endocrine pancreatic glucagon-secreting cells. Biochem. Biophys. Res. Commun 1994, 205, 435–440. [Google Scholar]
- Gromada, J.; Bokvist, K.; Ding, W.G.; Barg, S.; Buschard, K.; Renstrom, E.; Rorsman, P. Adrenaline stimulates glucagon secretion in pancreatic A-cells by increasing the Ca2+ current and the number of granules close to the L-type Ca2+ channels. J. Gen. Physiol 1997, 110, 217–228. [Google Scholar]
- Kong, P.J.; Byun, J.S.; Lim, S.Y.; Lee, J.J.; Hong, S.J.; Kwon, K.J.; Kim, S.S. Melatonin induces Akt phosphorylation through melatonin receptor- and PI3K-dependent pathways in primary astrocytes. Korean J. Physiol. Pharmacol 2008, 12, 37–41. [Google Scholar]
- Cui, P.; Yu, M.; Luo, Z.; Dai, M.; Han, J.; Xiu, R.; Yang, Z. Intracellular signaling pathways involved in cell growth inhibition of human umbilical vein endothelial cells by melatonin. J. Pineal Res 2008, 44, 107–114. [Google Scholar]
- Batty, I.H.; Hickinson, D.M.; Downes, C.P. Cross-talk between phospholipase C and phosphoinositide 3-kinase signalling pathways. Biochem. Soc. Trans 1997, 25, 1132–1137. [Google Scholar]
- Schwaninger, M.; Lux, G.; Blume, R.; Oetjen, E.; Hidaka, H.; Knepel, W. Membrane depolarization and calcium influx induce glucagon gene transcription in pancreatic islet cells through the cyclic AMP-responsive element. J. Biol. Chem 1993, 268, 5168–5177. [Google Scholar]
- Ishikawa, K.; Shimazu, T. Daily rhythms of glycogen synthetase and phosphorylase activities in rat liver: influence of food and light. Life Sci 1976, 19, 1873–1878. [Google Scholar]
- Kida, K.; Nishio, T.; Yokozawa, T.; Nagai, K.; Matsuda, H.; Nakagawa, H. The circadian change of gluconeogenesis in the liver in vivo in fed rats. J. Biochem 1980, 88, 1009–1013. [Google Scholar]
- Roesler, W.J.; Khandelwal, R.L. Diurnal variations in the activities of the glycogen metabolizing enzymes in mouse liver. Int. J. Biochem 1985, 17, 81–85. [Google Scholar]
- Stokkan, K.A.; Yamazaki, S.; Tei, H.; Sakaki, Y.; Menaker, M. Entrainment of the circadian clock in the liver by feeding. Science 2001, 291, 490–493. [Google Scholar]
- Rudic, R.D.; McNamara, P.; Curtis, A.M.; Boston, R.C.; Panda, S.; Hogenesch, J.B.; Fitzgerald, G.A. BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol 2004, 2, e377. [Google Scholar]
- Muhlbauer, E.; Gross, E.; Labucay, K.; Wolgast, S.; Peschke, E. Loss of melatonin signalling and its impact on circadian rhythms in mouse organs regulating blood glucose. Eur. J. Pharmacol 2009, 606, 61–71. [Google Scholar]
- Poon, A.M.; Choy, E.H.; Pang, S.F. Modulation of blood glucose by melatonin: A direct action on melatonin receptors in mouse hepatocytes. Biol. Signals Recept 2001, 10, 367–379. [Google Scholar]
- Mazepa, R.C.; Cuevas, M.J.; Collado, P.S.; Gonzalez-Gallego, J. Melatonin increases muscle and liver glycogen content in nonexercised and exercised rats. Life Sci 2000, 66, 153–160. [Google Scholar]
- Shieh, J.M.; Wu, H.T.; Cheng, K.C.; Cheng, J.T. Melatonin ameliorates high fat diet-induced diabetes and stimulates glycogen synthesis via a PKCzeta-Akt-GSK3beta pathway in hepatic cells. J. Pineal Res 2009, 47, 339–344. [Google Scholar]
- Gottlieb, D.J.; Punjabi, N.M.; Newman, A.B.; Resnick, H.E.; Redline, S.; Baldwin, C.M.; Nieto, F.J. Association of sleep time with diabetes mellitus and impaired glucose tolerance. Arch. Intern. Med 2005, 165, 863–867. [Google Scholar]
- Yaggi, H.K.; Araujo, A.B.; McKinlay, J.B. Sleep duration as a risk factor for the development of type 2 diabetes. Diabetes Care 2006, 29, 657–661. [Google Scholar]
- Donga, E.; van Dijk, M.; van Dijk, J.G.; Biermasz, N.R.; Lammers, G.J.; van Kralingen, K.W.; Corssmit, E.P.; Romijn, J.A. A single night of partial sleep deprivation induces insulin resistance in multiple metabolic pathways in healthy subjects. J. Clin. Endocrinol. Metab 2010, 95, 2963–2968. [Google Scholar]


© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Peschke, E.; Bähr, I.; Mühlbauer, E. Melatonin and Pancreatic Islets: Interrelationships between Melatonin, Insulin and Glucagon. Int. J. Mol. Sci. 2013, 14, 6981-7015. https://doi.org/10.3390/ijms14046981
Peschke E, Bähr I, Mühlbauer E. Melatonin and Pancreatic Islets: Interrelationships between Melatonin, Insulin and Glucagon. International Journal of Molecular Sciences. 2013; 14(4):6981-7015. https://doi.org/10.3390/ijms14046981
Chicago/Turabian StylePeschke, Elmar, Ina Bähr, and Eckhard Mühlbauer. 2013. "Melatonin and Pancreatic Islets: Interrelationships between Melatonin, Insulin and Glucagon" International Journal of Molecular Sciences 14, no. 4: 6981-7015. https://doi.org/10.3390/ijms14046981
APA StylePeschke, E., Bähr, I., & Mühlbauer, E. (2013). Melatonin and Pancreatic Islets: Interrelationships between Melatonin, Insulin and Glucagon. International Journal of Molecular Sciences, 14(4), 6981-7015. https://doi.org/10.3390/ijms14046981