A Chimeric UDP-Glucose Pyrophosphorylase Produced by Protein Engineering Exhibits Sensitivity to Allosteric Regulators

Abstract
:1. Introduction
2. Results and Discussion
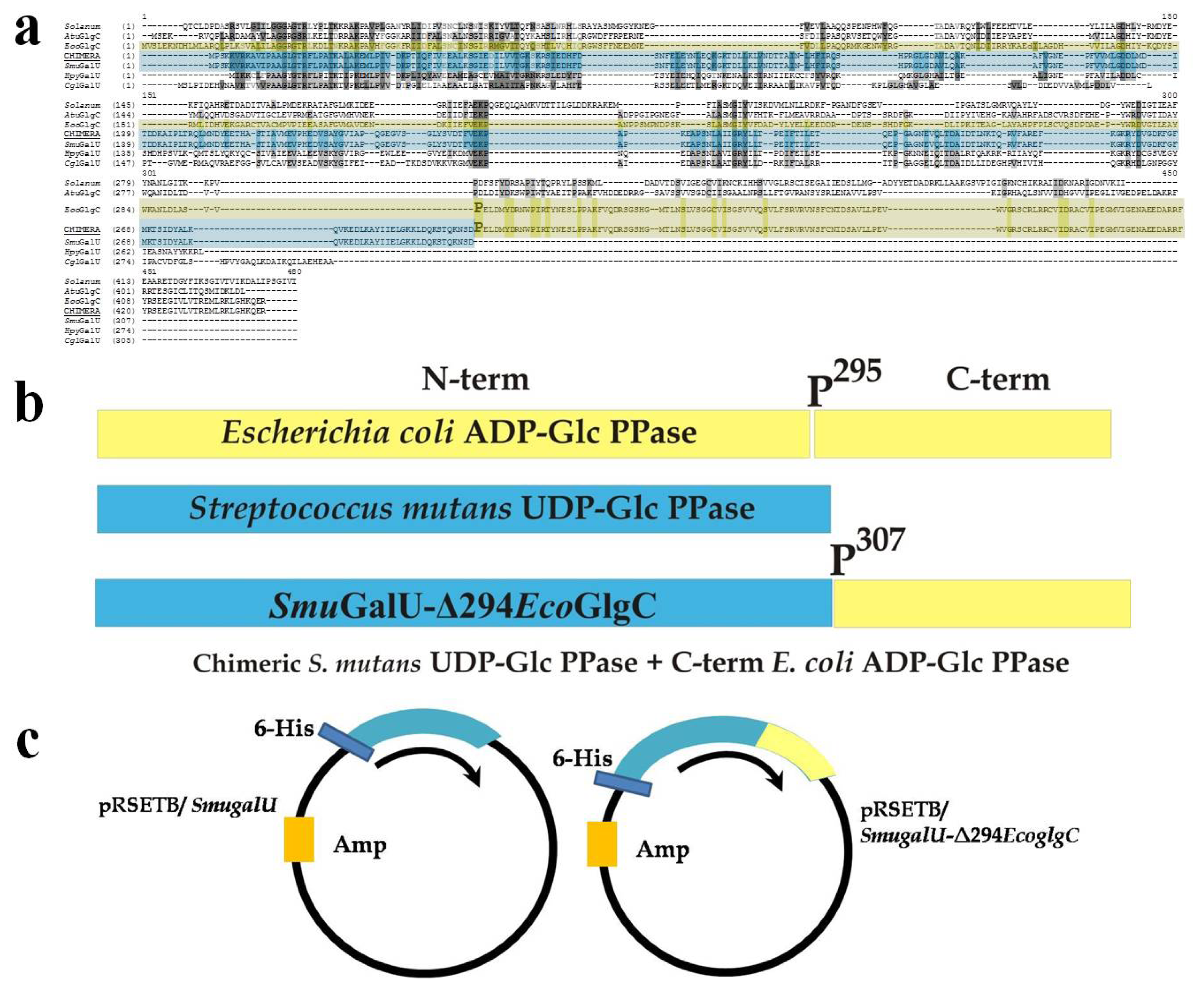
2.1. Isolation and Analysis of the Gene Coding for UDP-Glc PPase in S. mutans and Construction of the Chimeric Protein
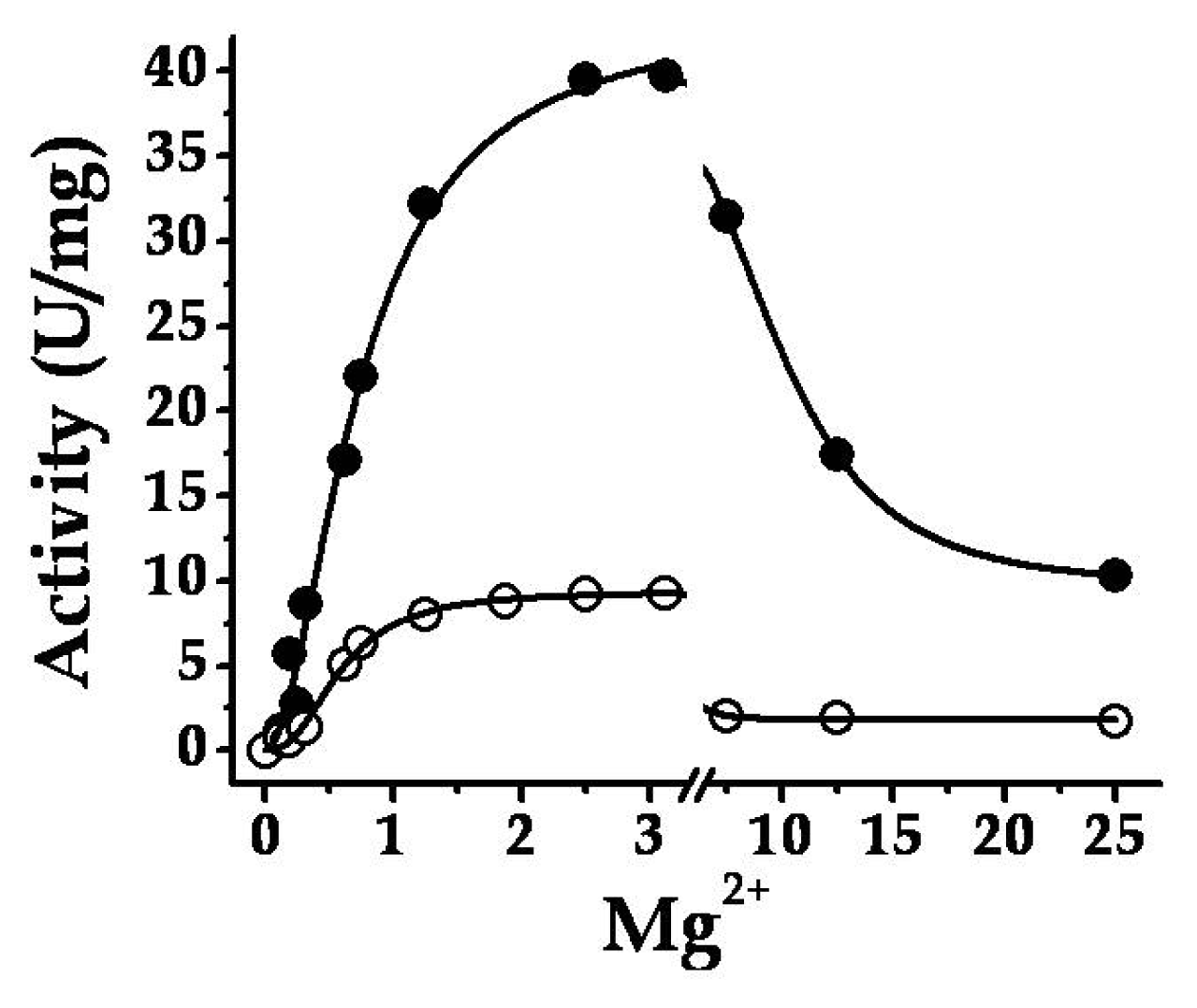
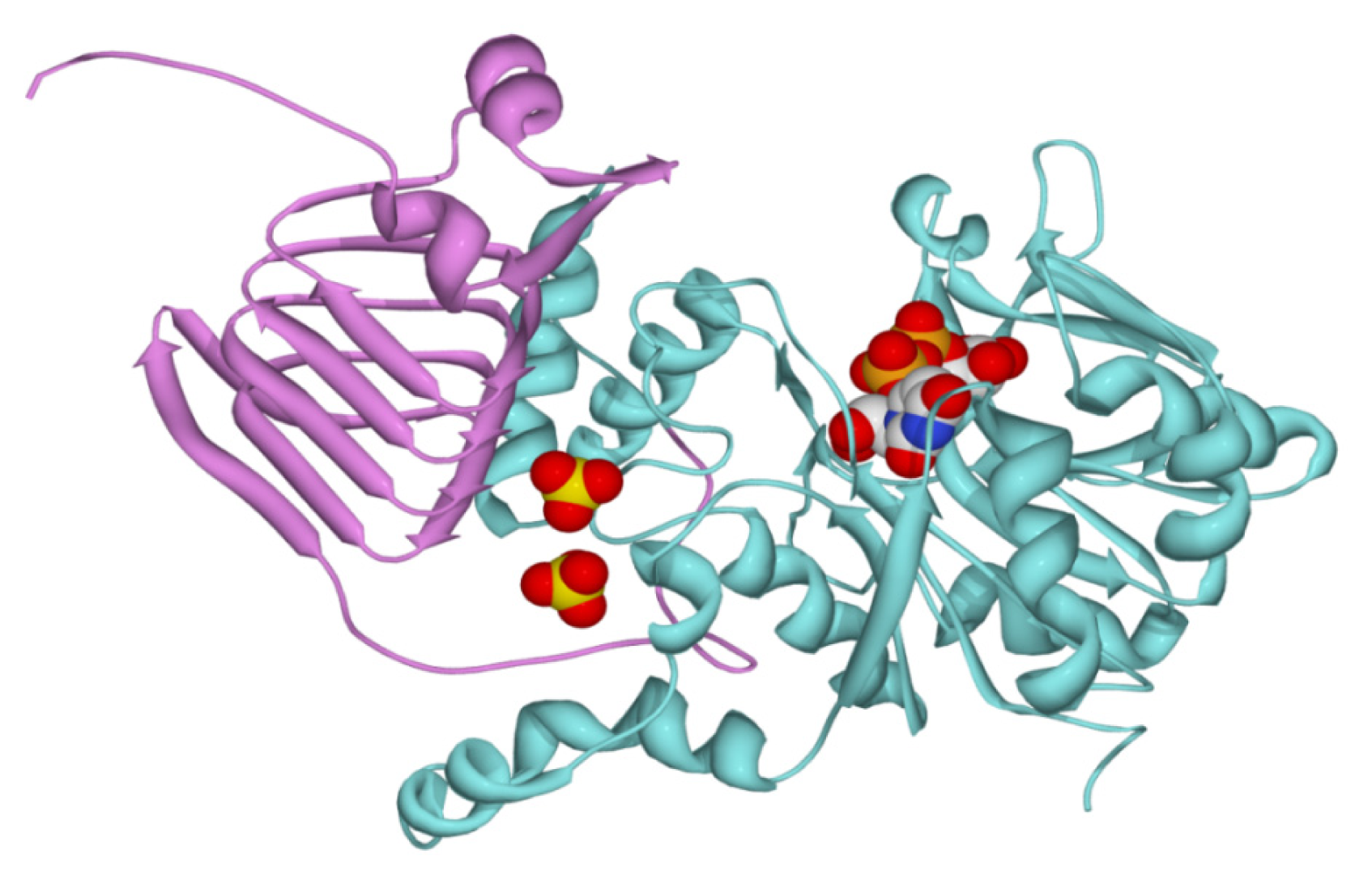
2.2. Structural and Kinetic Characterization of SmuGalU and Chimeric SmuGalU-Δ294EcoGlgC
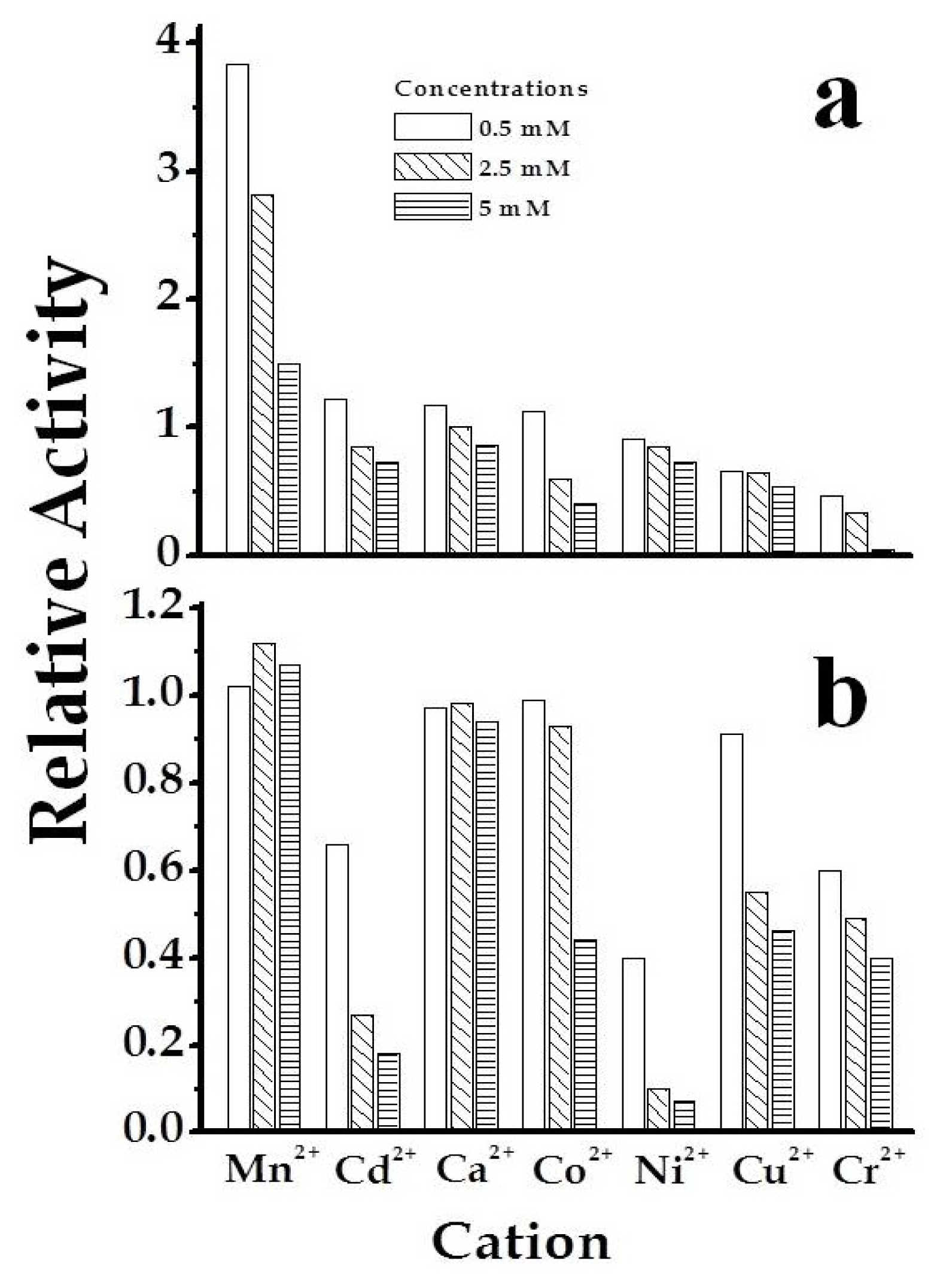
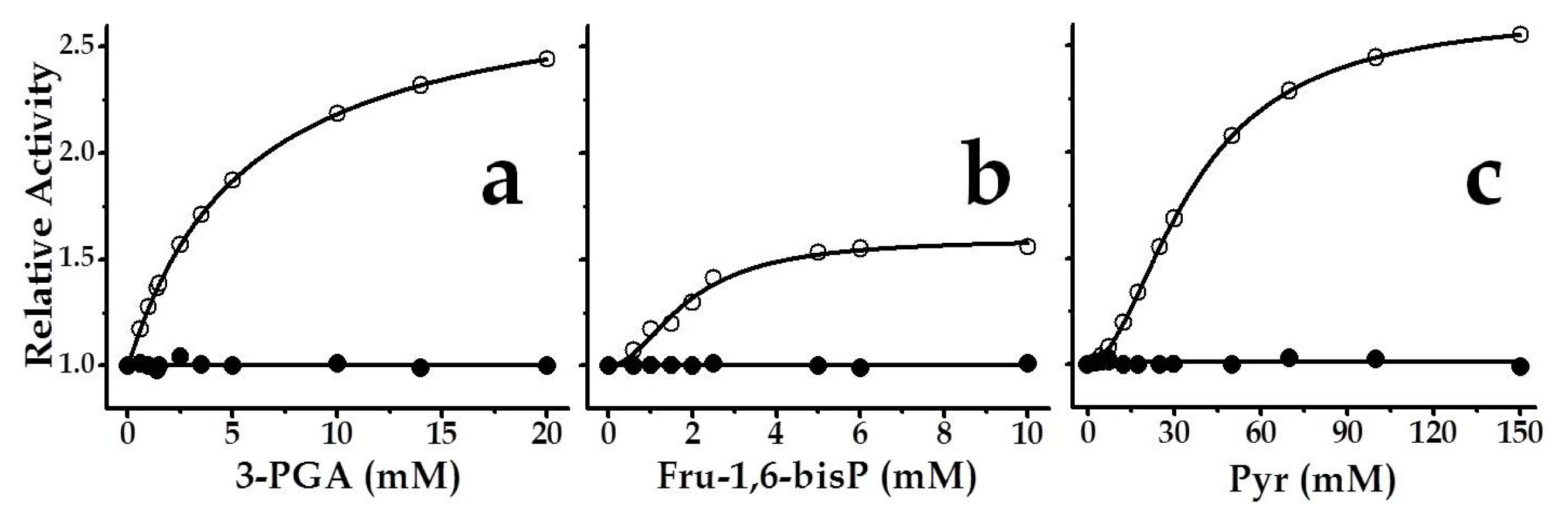
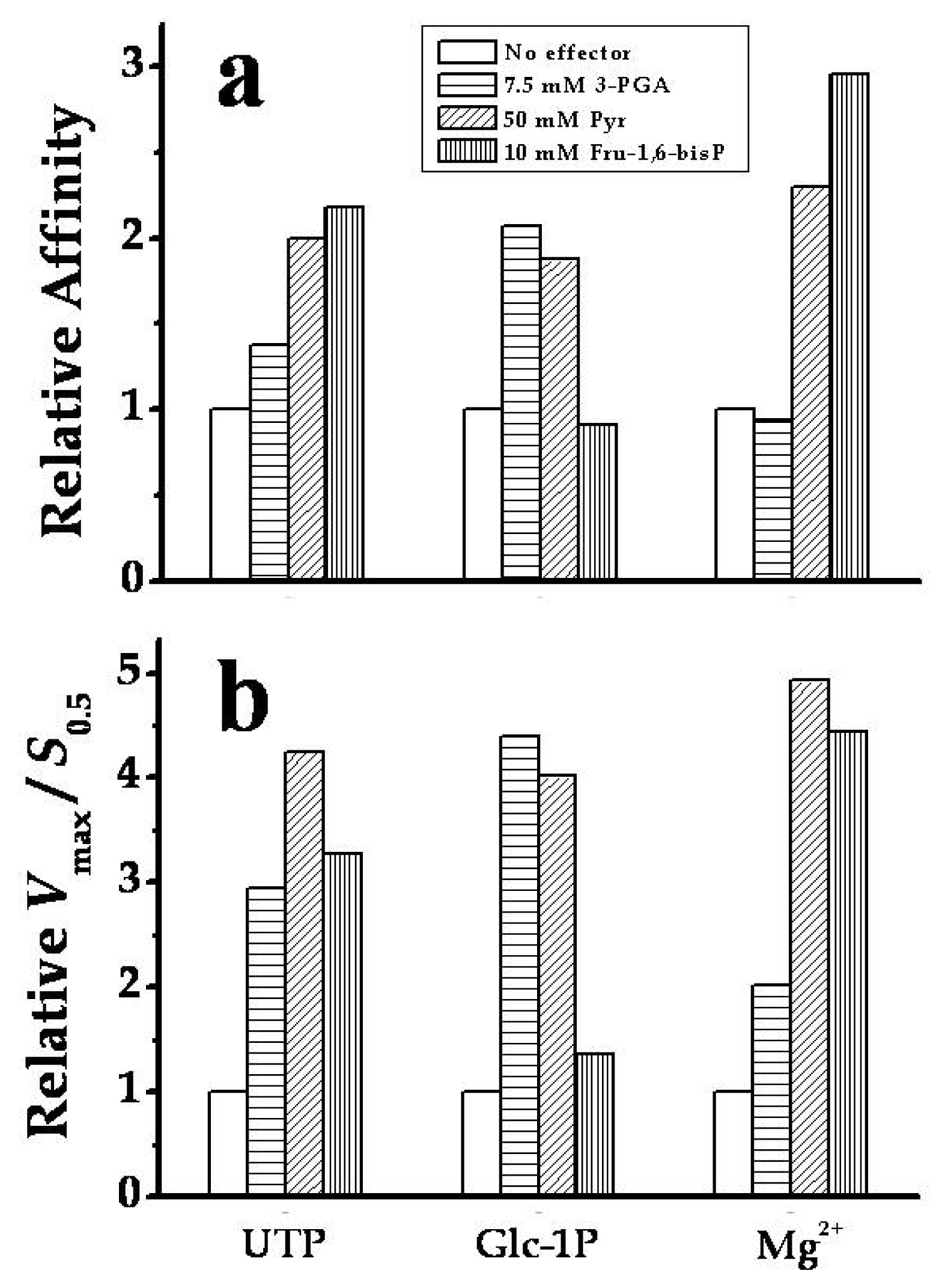
2.3. The Chimeric SmuGalU-Δ294EcoGlgC Protein Exhibits Allosteric Properties
3. Experimental Section
3.1. Chemicals
3.2. Bacteria and Plasmids
3.3. Amplification of galU Gene from S. mutans and Construction of Chimeric galU
3.4. Cloning of SmugalU and Chimeric SmugalU-Δ294EcoglgC Genes
3.5. Enzymes Expressions and Purifications
3.6. Protein Methods
3.7. Determination of Activity Optimal pH
3.8. Molecular Mass Determination
3.9. Enzyme Assays
3.10. Calculation of Kinetic Constants
3.11. Homology Modeling
4. Conclusions
Supplementary Information
ijms-14-09703-s001.pdfAcknowledgments
Conflict of Interest
References
- Ballicora, M.A.; Iglesias, A.A.; Preiss, J. ADP-glucose pyrophosphorylase, a regulatory enzyme for bacterial glycogen synthesis. Microbiol. Mol. Biol. Rev 2003, 67, 213–225. [Google Scholar]
- Ballicora, M.A.; Iglesias, A.A.; Preiss, J. ADP-glucose pyrophosphorylase: A regulatory enzyme for plant starch synthesis. Photosynth. Res 2004, 79, 1–24. [Google Scholar]
- Liu, H.W.; Thorson, J.S. Pathways and mechanisms in the biogenesis of novel deoxysugars by bacteria. Annu. Rev. Microbiol 1994, 48, 223–256. [Google Scholar]
- Moretti, R.; Thorson, J.S. Enhancing the latent nucleotide triphosphate flexibility of the glucose-1-phosphate thymidylyltransferase RmlA. J. Biol. Chem 2007, 282, 16942–16947. [Google Scholar]
- Kleczkowski, L.A.; Geisler, M.; Ciereszko, I.; Johansson, H. UDP-glucose pyrophosphorylase. An old protein with new tricks. Plant Physiol 2004, 134, 912–918. [Google Scholar]
- Mollerach, M.; Garcia, E. The galU gene of Streptococcus pneumoniae that codes for a UDP-glucose pyrophosphorylase is highly polymorphic and suitable for molecular typing and phylogenetic studies. Gene 2000, 260, 77–86. [Google Scholar]
- Martinez, L.I.; Piattoni, C.V.; Garay, S.A.; Rodrigues, D.E.; Guerrero, S.A.; Iglesias, A.A. Redox regulation of UDP-glucose pyrophosphorylase from Entamoeba histolytica. Biochimie 2011, 93, 260–268. [Google Scholar]
- Asencion Diez, M.D.; Peiru, S.; Demonte, A.M.; Gramajo, H.; Iglesias, A.A. Characterization of recombinant UDP- and ADP-glucose pyrophosphorylases and glycogen synthase to elucidate glucose-1-phosphate partitioning into oligo- and polysaccharides in Streptomyces coelicolor. J. Bacteriol 2012, 194, 1485–1493. [Google Scholar]
- Mollerach, M.; Lopez, R.; Garcia, E. Characterization of the galU gene of Streptococcus pneumoniae encoding a uridine diphosphoglucose pyrophosphorylase: A gene essential for capsular polysaccharide biosynthesis. J. Exp. Med 1998, 188, 2047–2056. [Google Scholar]
- Bonofiglio, L.; Garcia, E.; Mollerach, M. Biochemical characterization of the pneumococcal glucose 1-phosphate uridylyltransferase (GalU) essential for capsule biosynthesis. Curr. Microbiol 2005, 51, 217–221. [Google Scholar]
- Bosco, M.B.; Machtey, M.; Iglesias, A.A.; Aleanzi, M. UDPglucose pyrophosphorylase from Xanthomonas spp. Characterization of the enzyme kinetics, structure and inactivation related to oligomeric dissociation. Biochimie 2009, 91, 204–213. [Google Scholar]
- Marques, A.R.; Ferreira, P.B.; Sa-Correia, I.; Fialho, A.M. Characterization of the ugpG gene encoding a UDP-glucose pyrophosphorylase from the gellan gum producer Sphingomonas paucimobilis ATCC 31461. Mol. Genet. Genomics 2003, 268, 816–824. [Google Scholar]
- Hossain, S.A.; Tanizawa, K.; Kazuta, Y.; Fukui, T. Overproduction and characterization of recombinant UDP-glucose pyrophosphorylase from Escherichia coli K-12. J. Biochem 1994, 115, 965–972. [Google Scholar]
- Thoden, J.B.; Holden, H.M. The molecular architecture of glucose-1-phosphate uridylyltransferase. Protein Sci 2007, 16, 432–440. [Google Scholar]
- Aragao, D.; Fialho, A.M.; Marques, A.R.; Mitchell, E.P.; Sa-Correia, I.; Frazao, C. The complex of Sphingomonas elodea ATCC 31461 glucose-1-phosphate uridylyltransferase with glucose-1-phosphate reveals a novel quaternary structure, unique among nucleoside diphosphate-sugar pyrophosphorylase members. J. Bacteriol 2007, 189, 4520–4528. [Google Scholar]
- Thoden, J.B.; Holden, H.M. Active site geometry of glucose-1-phosphate uridylyltransferase. Protein Sci 2007, 16, 1379–1388. [Google Scholar]
- Cupp-Vickery, J.R.; Igarashi, R.Y.; Perez, M.; Poland, M.; Meyer, C.R. Structural analysis of ADP-glucose pyrophosphorylase from the bacterium Agrobacterium tumefaciens. Biochemistry 2008, 47, 4439–4451. [Google Scholar]
- Jin, X.; Ballicora, M.A.; Preiss, J.; Geiger, J.H. Crystal structure of potato tuber ADP-glucose pyrophosphorylase. Embo. J 2005, 24, 694–704. [Google Scholar]
- Ballicora, M.A.; Sesma, J.I.; Iglesias, A.A.; Preiss, J. Characterization of chimeric ADPglucose pyrophosphorylases of Escherichia coli and Agrobacterium tumefaciens. Importance of the C-terminus on the selectivity for allosteric regulators. Biochemistry 2002, 41, 9431–9437. [Google Scholar]
- Ballicora, M.A.; Erben, E.D.; Yazaki, T.; Bertolo, A.L.; Demonte, A.M.; Schmidt, J.R.; Aleanzi, M.; Bejar, C.M.; Figueroa, C.M.; Fusari, C.M.; et al. Identification of regions critically affecting kinetics and allosteric regulation of the Escherichia coli ADP-glucose pyrophosphorylase by modeling and pentapeptide-scanning mutagenesis. J. Bacteriol 2007, 189, 5325–5333. [Google Scholar]
- Gomez-Casati, D.F.; Igarashi, R.Y.; Berger, C.N.; Brandt, M.E.; Iglesias, A.A.; Meyer, C.R. Identification of functionally important amino-terminal arginines of Agrobacterium tumefaciens ADP-glucose pyrophosphorylase by alanine scanning mutagenesis. Biochemistry 2001, 40, 10169–10178. [Google Scholar]
- Bejar, C.M.; Ballicora, M.A.; Iglesias, A.A.; Preiss, J. ADPglucose pyrophosphorylase’s N-terminus: Structural role in allosteric regulation. Biochem. Biophys. Res. Commun 2006, 343, 216–221. [Google Scholar]
- Meyer, C.R.; Bork, J.A.; Nadler, S.; Yirsa, J.; Preiss, J. Site-directed mutagenesis of a regulatory site of Escherichia coli ADP-glucose pyrophosphorylase: The role of residue 336 in allosteric behavior. Arch. Biochem. Biophys 1998, 353, 152–159. [Google Scholar]
- Meyer, C.R.; Yirsa, J.; Gott, B.; Preiss, J. A kinetic study of site-directed mutants of Escherichia coli ADP-glucose pyrophosphorylase: The role of residue 295 in allosteric regulation. Arch. Biochem. Biophys 1998, 352, 247–254. [Google Scholar]
- Bejar, C.M.; Ballicora, M.A.; Gomez-Casati, D.F.; Iglesias, A.A.; Preiss, J. The ADP-glucose pyrophosphorylase from Escherichia coli comprises two tightly bound distinct domains. FEBS Lett 2004, 573, 99–104. [Google Scholar]
- Ajdic, D.; McShan, W.M.; McLaughlin, R.E.; Savic, G.; Chang, J.; Carson, M.B.; Primeaux, C.; Tian, R.; Kenton, S.; Jia, H.; et al. Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proc. Natl. Acad. Sci. USA 2002, 99, 14434–14439. [Google Scholar]
- Bonofiglio, L.; Garcia, E.; Mollerach, M. The galU gene expression in Streptococcus pneumoniae. FEMS Microbiol. Lett 2012, 332, 47–53. [Google Scholar]
- Varon, D.; Boylan, S.A.; Okamoto, K.; Price, C.W. Bacillus subtilis gtaB encodes UDP-glucose pyrophosphorylase and is controlled by stationary-phase transcription factor sigma B. J. Bacteriol 1993, 175, 3964–3971. [Google Scholar]
- Kim, H.; Choi, J.; Kim, T.; Lokanath, N.K.; Ha, S.C.; Suh, S.W.; Hwang, H.Y.; Kim, K.K. Structural basis for the reaction mechanism of UDP-glucose pyrophosphorylase. Mol. Cells 2010, 29, 397–405. [Google Scholar]
- Iglesias, A.A.; Ballicora, M.A.; Sesma, J.I.; Preiss, J. Domain swapping between a cyanobacterial and a plant subunit ADP-glucose pyrophosphorylase. Plant Cell Physiol 2006, 47, 523–530. [Google Scholar]
- Silva, E.; Marques, A.R.; Fialho, A.M.; Granja, A.T.; Sa-Correia, I. Proteins encoded by Sphingomonas elodea ATCC 31461 rmlA and ugpG genes, involved in gellan gum biosynthesis, exhibit both dTDP- and UDP-glucose pyrophosphorylase activities. App. Environ. Microbiol 2005, 71, 4703–4712. [Google Scholar]
- Weissborn, A.C.; Liu, Q.; Rumley, M.K.; Kennedy, E.P. UTP: alpha-d-glucose-1-phosphate uridylyltransferase of Escherichia coli: Isolation and DNA sequence of the galU gene and purification of the enzyme. J. Bacteriol 1994, 176, 2611–2618. [Google Scholar]
- Asencion Diez, M.D.; Demonte, A.; Giacomelli, J.; Garay, S.; Rodrigues, D.; Hofmann, B.; Hecht, H.J.; Guerrero, S.A.; Iglesias, A.A. Functional characterization of GDP-mannose pyrophosphorylase from Leptospira interrogans serovar Copenhageni. Arch. Microbiol 2010, 192, 103–114. [Google Scholar]
- Ning, B.; Elbein, A.D. Purification and properties of mycobacterial GDP-mannose pyrophosphorylase. Arch. Biochem. Biophys 1999, 362, 339–345. [Google Scholar]
- Frueauf, J.B.; Ballicora, M.A.; Preiss, J. ADP-glucose pyrophosphorylase from potato tuber: Site-directed mutagenesis of homologous aspartic acid residues in the small and large subunits. Plant J 2003, 33, 503–511. [Google Scholar]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, New York, NY, USA, 2001. [Google Scholar]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Wheeler, D.L. GenBank. Nucleic Acids Res 2005, 33, D34–D38. [Google Scholar]
- Higuchi, R.; Krummel, B.; Saiki, R.K. A general method of in vitro preparation and specific mutagenesis of DNA fragments: Study of protein and DNA interactions. Nucleic Acids Res 1988, 16, 7351–7367. [Google Scholar]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem 1976, 72, 248–254. [Google Scholar]
- Fusari, C.; Demonte, A.M.; Figueroa, C.M.; Aleanzi, M.; Iglesias, A.A. A colorimetric method for the assay of ADP-glucose pyrophosphorylase. Anal. Biochem 2006, 352, 145–147. [Google Scholar]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol 1993, 234, 779–815. [Google Scholar]
- Luthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar]
- Decker, D.; Meng, M.; Gornicka, A.; Hofer, A.; Wilczynska, M.; Kleczkowski, L.A. Substrate kinetics and substrate effects on the quaternary structure of barley UDP-glucose pyrophosphorylase. Phytochemistry 2012, 79, 39–45. [Google Scholar]
- Kleczkowski, L.A.; Martz, F.; Wilczynska, M. Factors affecting oligomerization status of UDP-glucose pyrophosphorylase. Phytochemistry 2005, 66, 2815–2821. [Google Scholar]
- Martz, F.; Wilczynska, M.; Kleczkowski, L.A. Oligomerization status, with the monomer as active species, defines catalytic efficiency of UDP-glucose pyrophosphorylase. Biochem. J 2002, 367, 295–300. [Google Scholar]
- Gardiol, A.; Preiss, J. Escherichia coli E-39 ADPglucose synthetase has different activation kinetics from the wild-type allosteric enzyme. Arch. Biochem. Biophys 1990, 280, 175–180. [Google Scholar]
- Parsons, T.F.; Preiss, J. Biosynthesis of bacterial glycogen. Isolation and characterization of the pyridoxal-P allosteric activator site and the ADP-glucose-protected pyridoxal-P binding site of Escherichia coli B ADP-glucose synthase. J. Biol. Chem 1978, 253, 7638–7645. [Google Scholar]
- Parsons, T.F.; Preiss, J. Biosynthesis of bacterial glycogen. Incorporation of pyridoxal phosphate into the allosteric activator site and an ADP-glucose-protected pyridoxal phosphate binding site of Escherichia coli B ADP-glucose synthase. J. Biol. Chem 1978, 253, 6197–6202. [Google Scholar]
- Wu, M.X.; Preiss, J. The N-terminal region is important for the allosteric activation and inhibition of the Escherichia coli ADP-glucose pyrophosphorylase. Arch. Biochem. Biophys 1998, 358, 182–188. [Google Scholar]
- Wu, M.X.; Preiss, J. Truncated forms of the recombinant Escherichia coli ADP-glucose pyrophosphorylase: The importance of the N-terminal region for allosteric activation and inhibition. Arch. Biochem. Biophys 2001, 389, 159–165. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Kinetic parameter | SmuGalU | SmuGalU-Δ294EcoGlgC |
|---|---|---|---|
| Vmax (U/mg) | 62.1 ± 2.2 | 15.4 ± 0.5 | |
| UTP | S0.5 (mM) | 0.68 ± 0.06 | 0.24 ± 0.02 |
| nH | 1.5 ± 0.2 | 1.2 ± 0.1 | |
| Vmax/S0.5 (U/mg mM) | 91.3 | 64.2 | |
| Glc-1P | S0.5 (mM) | 0.090 ± 0.005 | 0.060 ± 0.005 |
| nH | 1.3 ± 0.1 | 1.4 ± 0.1 | |
| Vmax/S0.5 (U/mg mM) | 690 | 257 | |
| Mg2+ | S0.5 (mM) | 0.81 ± 0.06 | 0.62 ± 0.05 |
| nH | 2.4 ± 0.6 | 2.7 ± 0.6 | |
| Vmax/S0.5 (U/mg mM) | 76.7 | 24.8 | |
| Primer | Sequence | Restriction site |
|---|---|---|
| Smu1 | 5′-GGATCCCATGCCAAGTAAAAAAGTCAG-3′ | BamHI |
| Smu2 | 5′-GAATTCCTTAATCCGAGTTCTTTTGAG-3′ | EcoRI |
| Qmr1 | 5′-CTCGGACCCGGAACTGGATATGTACGATC-3′ | -- |
| Qmr2 | 5′-GTTCCGGGTCCGAGTTCTTTTGAGTCG-3′ | -- |
| Qmr3 | 5′-CTCGAGTTATCGCTCCTGTTTATGCCC-3′ | XhoI |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Asención Diez, M.D.; Ebrecht, A.C.; Martínez, L.I.; Aleanzi, M.C.; Guerrero, S.A.; Ballícora, M.A.; Iglesias, A.A. A Chimeric UDP-Glucose Pyrophosphorylase Produced by Protein Engineering Exhibits Sensitivity to Allosteric Regulators. Int. J. Mol. Sci. 2013, 14, 9703-9721. https://doi.org/10.3390/ijms14059703
Asención Diez MD, Ebrecht AC, Martínez LI, Aleanzi MC, Guerrero SA, Ballícora MA, Iglesias AA. A Chimeric UDP-Glucose Pyrophosphorylase Produced by Protein Engineering Exhibits Sensitivity to Allosteric Regulators. International Journal of Molecular Sciences. 2013; 14(5):9703-9721. https://doi.org/10.3390/ijms14059703
Chicago/Turabian StyleAsención Diez, Matías D., Ana C. Ebrecht, Lucila I. Martínez, Mabel C. Aleanzi, Sergio A. Guerrero, Miguel A. Ballícora, and Alberto A. Iglesias. 2013. "A Chimeric UDP-Glucose Pyrophosphorylase Produced by Protein Engineering Exhibits Sensitivity to Allosteric Regulators" International Journal of Molecular Sciences 14, no. 5: 9703-9721. https://doi.org/10.3390/ijms14059703
APA StyleAsención Diez, M. D., Ebrecht, A. C., Martínez, L. I., Aleanzi, M. C., Guerrero, S. A., Ballícora, M. A., & Iglesias, A. A. (2013). A Chimeric UDP-Glucose Pyrophosphorylase Produced by Protein Engineering Exhibits Sensitivity to Allosteric Regulators. International Journal of Molecular Sciences, 14(5), 9703-9721. https://doi.org/10.3390/ijms14059703