Sod1 Loss Induces Intrinsic Superoxide Accumulation Leading to p53-Mediated Growth Arrest and Apoptosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
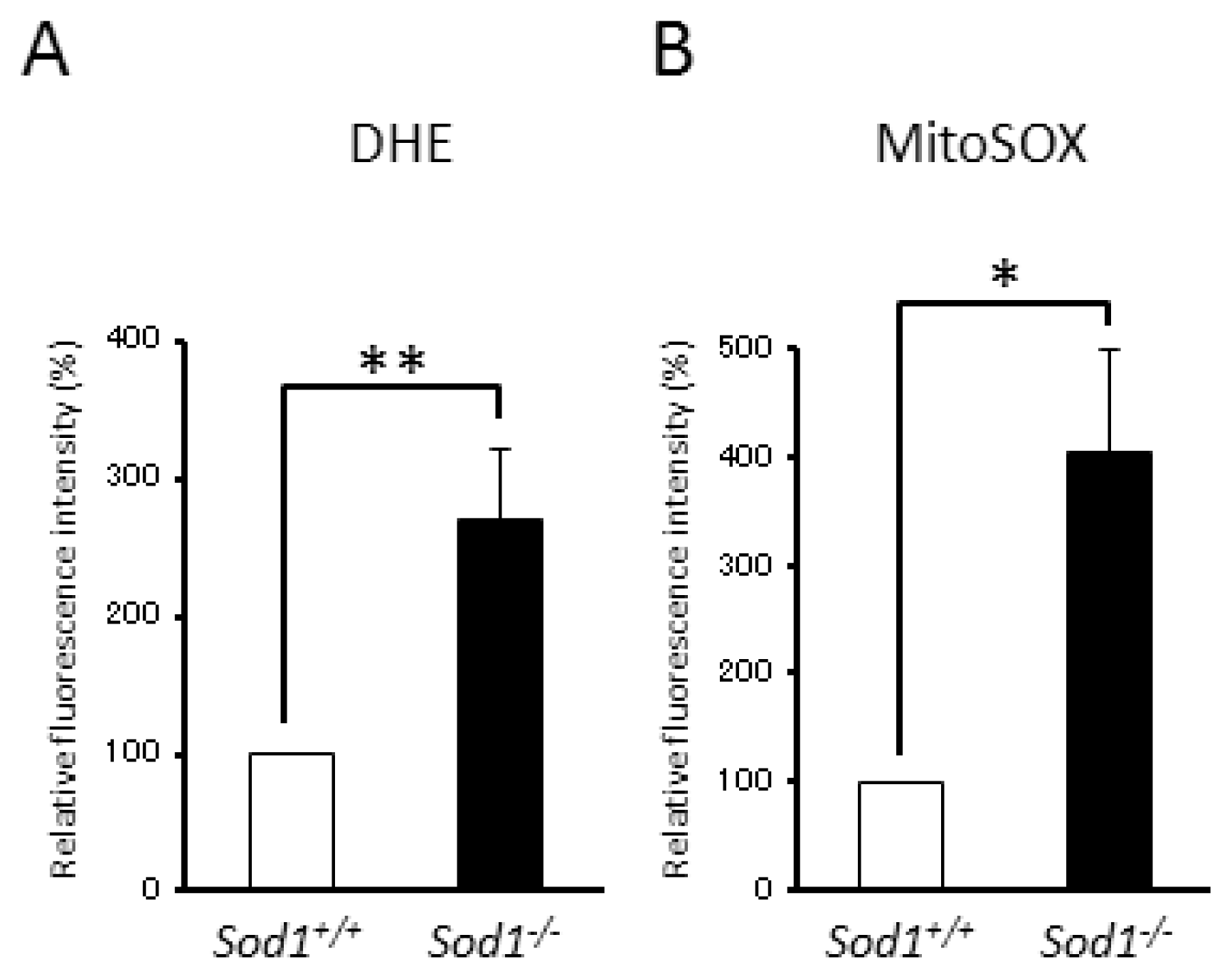
2.1. Sod1 Deficiency Induced Apoptotic Cell Death with Increased Superoxide Accumulation in Fibroblasts
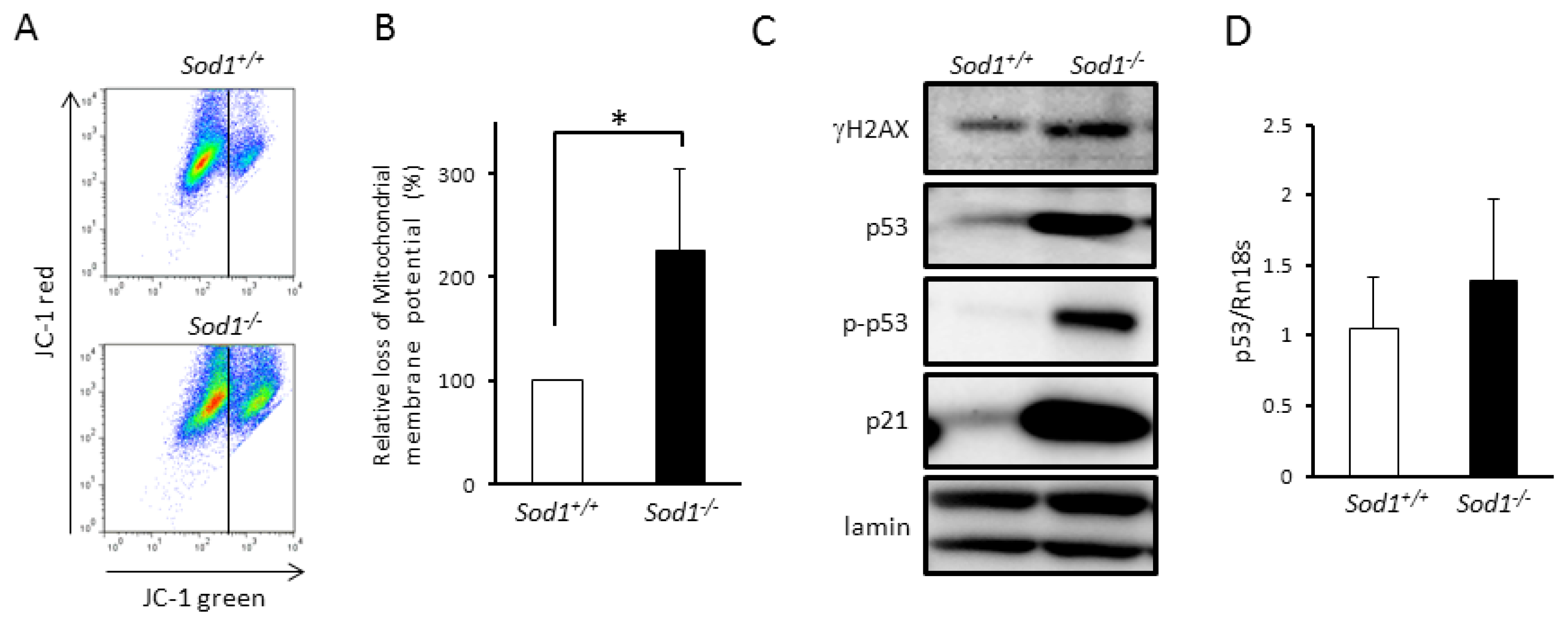
2.2. Sod1 Loss Caused p53 Upregulation Associated with Mitochondrial Dysfunction in Fibroblasts
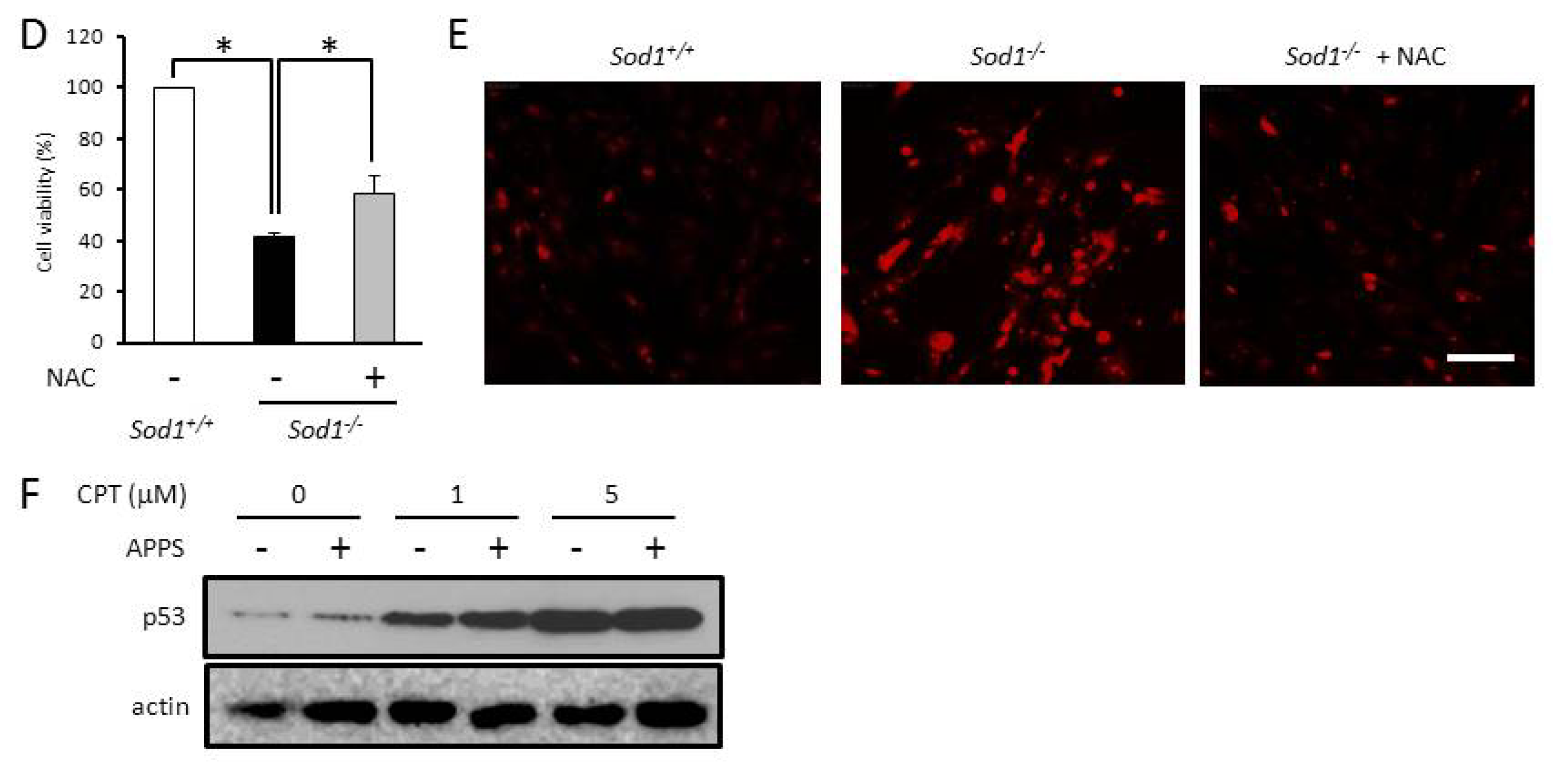
2.3. A Vitamin C Derivative Rescued Viability of Sod1-Deficient Fibroblasts through a Suppression of O2•− Generation and p53 Upregulation
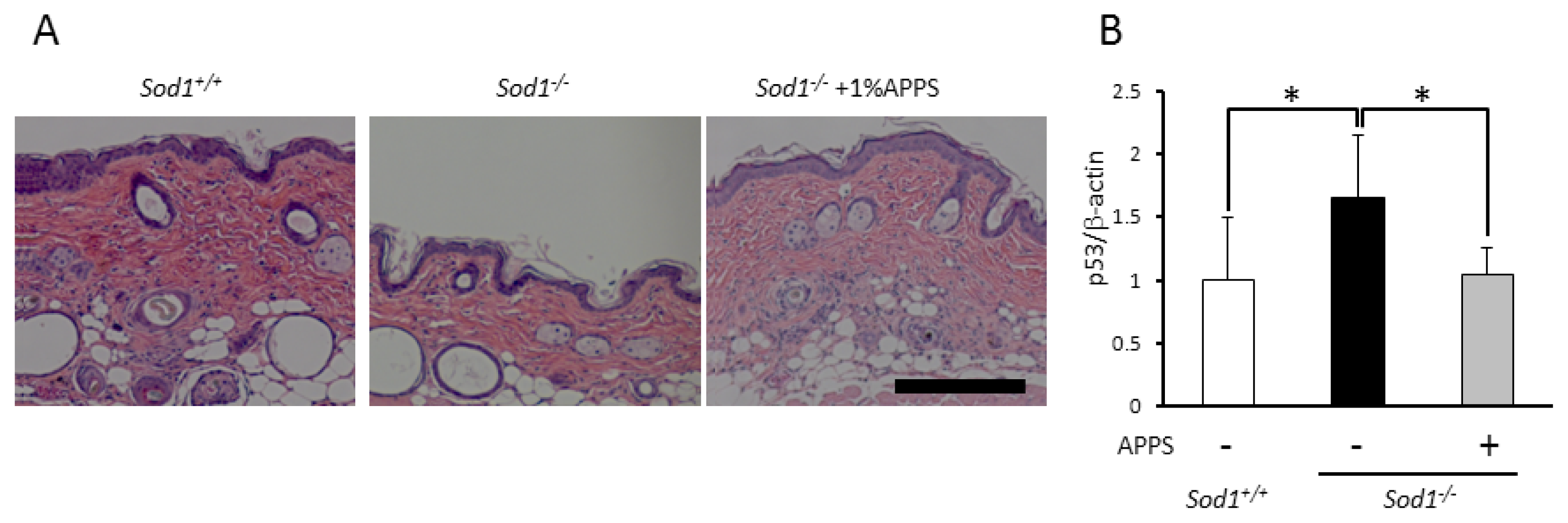
2.4. A Vitamin C Derivative Treatment Reversed Skin Atrophy Induced by Sod1 Loss in Vivo
2.5. Discussion
3. Experimental Section
3.1. Animals
3.2. Cell Culture
3.3. Western Blotting
3.4. Flow Cytometry
3.5. Histology
3.6. Quantitative PCR
3.7. Statics
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar]
- Ditch, S.; Paull, T.T. The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem. Sci 2012, 37, 15–22. [Google Scholar]
- Kumari, U.; Jun, W.Y.; Bay, B.H.; Lyakhovich, A. Evidence of mitochondrial dysfunction and impaired ROS detoxifying machinery in Fanconi Anemia cells. Oncogene 2012. [Google Scholar] [CrossRef]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013. [Google Scholar] [CrossRef]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J. Biol. Chem 2001, 276, 38388–38393. [Google Scholar]
- Imamura, Y.; Noda, S.; Hashizume, K.; Shinoda, K.; Yamaguchi, M.; Uchiyama, S.; Shimizu, T.; Mizushima, Y.; Shirasawa, T.; Tsubota, K. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: A model of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2006, 103, 11282–11287. [Google Scholar]
- Uchiyama, S.; Shimizu, T.; Shirasawa, T. CuZn-SOD deficiency causes ApoB degradation and induces hepatic lipid accumulation by impaired lipoprotein secretion in mice. J. Biol. Chem 2006, 281, 31713–31719. [Google Scholar]
- Murakami, K.; Inagaki, J.; Saito, M.; Ikeda, Y.; Tsuda, C.; Noda, Y.; Kawakami, S.; Shirasawa, T.; Shimizu, T. Skin atrophy in cytoplasmic SOD-deficient mice and its complete recovery using a vitamin C derivative. Biochem. Biophys. Res. Commun 2009, 382, 457–461. [Google Scholar]
- Nojiri, H.; Saita, Y.; Morikawa, D.; Kobayashi, K.; Tsuda, C.; Miyazaki, T.; Saito, M.; Marumo, K.; Yonezawa, I.; Kaneko, K.; et al. Cytoplasmic superoxide causes bone fragility owing to low-turnover osteoporosis and impaired collagen cross-linking. J. Bone Miner. Res 2011, 26, 2682–2694. [Google Scholar]
- Murakami, K.; Murata, N.; Noda, Y.; Tahara, S.; Kaneko, T.; Kinoshita, N.; Hatsuta, H.; Murayama, S.; Barnham, K.J.; Irie, K.; et al. SOD1 (copper/zinc superoxide dismutase) deficiency drives amyloid β protein oligomerization and memory loss in mouse model of Alzheimer disease. J. Biol. Chem 2011, 286, 44557–44568. [Google Scholar]
- Noda, Y.; Ota, K.; Shirasawa, T.; Shimizu, T. Copper/zinc superoxide dismutase insufficiency impairs progesterone secretion and fertility in female mice. Biol. Reprod 2012, 86, 1–8. [Google Scholar]
- Kojima, T.; Wakamatsu, T.H.; Dogru, M.; Ogawa, Y.; Igarashi, A.; Ibrahim, O.M.; Inaba, T.; Shimizu, T.; Noda, S.; Obata, H.; et al. Age-related dysfunction of the lacrimal gland and oxidative stress: Evidence from the Cu,Zn-superoxide dismutase-1 (Sod1) knockout mice. Am. J. Pathol 2012, 180, 1879–1896. [Google Scholar]
- Elchuri, S.; Oberley, T.D.; Qi, W.; Eisenstein, R.S.; Jackson Roberts, L.; van Remmen, H.; Epstein, C.J.; Huang, T.T. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 2005, 24, 367–380. [Google Scholar]
- Muller, F.L.; Song, W.; Liu, Y.; Chaudhuri, A.; Pieke-Dahl, S.; Strong, R.; Huang, T.T.; Epstein, C.J.; Roberts, L.J.; Csete, M.; et al. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic. Biol. Med 2006, 40, 1993–2004. [Google Scholar]
- Iuchi, Y.; Okada, F.; Onuma, K.; Onoda, T.; Asao, H.; Kobayashi, M.; Fujii, J. Elevated oxidative stress in erythrocytes due to a SOD1 deficiency causes anaemia and triggers autoantibody production. Biochem. J 2007, 402, 219–227. [Google Scholar]
- Huang, T.T.; Yasunami, M.; Carlson, E.J.; Gillespie, A.M.; Reaume, A.G.; Hoffman, E.K.; Chan, P.H.; Scott, R.W.; Epstein, C.J. Superoxide-mediated cytotoxicity in superoxide dismutase-deficient fetal fibroblasts. Arch. Biochem. Biophys 1997, 344, 424–432. [Google Scholar]
- Brenner, C.; Moulin, M. Physiological roles of the permeability transition pore. Circ. Res 2012, 111, 1237–1247. [Google Scholar]
- Vurusaner, B.; Poli, G.; Basaga, H. Tumor suppressor genes and ROS: Complex networks of interactions. Free Radic. Biol. Med 2012, 52, 7–18. [Google Scholar]
- Shibuya, S.; Kinoshita, K.; Shimizu, T. Handbook of Diet, Nutrition and the Skin, 2nd ed.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2012; pp. 351–366. [Google Scholar]
- Muller, F.L.; Liu, Y.; van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem 2004, 279, 49064–49073. [Google Scholar]
- Jang, Y.C.; Liu, Y.; Hayworth, C.R.; Bhattacharya, A.; Lustgarten, M.S.; Muller, F.L.; Chaudhuri, A.; Qi, W.; Li, Y.; Huang, J.Y.; et al. Dietary restriction attenuates age-associated muscle atrophy by lowering oxidative stress in mice even in complete absence of CuZnSOD. Aging Cell 2012, 11, 770–782. [Google Scholar]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar]
- Gajjar, M.; Candeias, M.M.; Malbert-Colas, L.; Mazars, A.; Fujita, J.; Olivares-Illana, V.; Fåhraeus, R. The p53 mRNA-Mdm2 interaction controls Mdm2 nuclear trafficking and is required for p53 activation following DNA damage. Cancer Cell 2012, 21, 25–35. [Google Scholar]
- Hamard, P.J.; Manfredi, J.J. Mdm2’s dilemma: To degrade or to translate p53? Cancer Cell 2012, 21, 3–5. [Google Scholar]
- Mbaya, E.; Oulès, B.; Caspersen, C.; Tacine, R.; Massinet, H.; Pennuto, M.; Chrétien, D.; Munnich, A.; Rötig, A.; Rizzuto, R.; et al. Calcium signalling-dependent mitochondrial dysfunction and bioenergetics regulation in respiratory chain Complex II deficiency. Cell Death Differ 2010, 17, 1855–1866. [Google Scholar]
- Jiang, P.; Du, W.; Mancuso, A.; Wellen, K.E.; Yang, X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 2013, 493, 689–693. [Google Scholar]
- Tyner, S.D.; Venkatachalam, S.; Choi, J.; Jones, S.; Ghebranious, N.; Igelmann, H.; Lu, X.; Soron, G.; Cooper, B.; Brayton, C.; et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 2002, 415, 45–53. [Google Scholar]
- Gannon, H.S.; Donehower, L.A.; Lyle, S.; Jones, S.N. Mdm2-p53 signaling regulates epidermal stem cell senescence and premature aging phenotypes in mouse skin. Dev. Biol 2011, 353, 1–9. [Google Scholar]
- Peterkofsky, B. Ascorbate requirement for hydroxylation and secretion of procollagen: Relationship to inhibition of collagen synthesis in scurvy. Am. J. Clin. Nutr 1991, 54, S1135–S1140. [Google Scholar]
- Murakami, K.; Murata, N; Ozawa, Y.; Kinoshita, N.; Irie, K.; Shirasawa, T.; Shimizu, T. Vitamin C restores behavioral deficits and amyloid-β oligomerization without affecting plaque formation in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2011, 26, 7–18. [Google Scholar]
- Kalyanaraman, B.; Darley-Usmar, V.; Davies, K.J.; Dennery, P.A.; Forman, H.J.; Grisham, M.B.; Mann, G.E.; Moore, K.; Roberts, L.J.; Ischiropoulos, H. Measuring reactive oxygen and nitrogen species with fluorescent probes: Challenges and limitations. Free Radic. Biol. Med 2012, 52, 1–6. [Google Scholar]






© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Watanabe, K.; Shibuya, S.; Koyama, H.; Ozawa, Y.; Toda, T.; Yokote, K.; Shimizu, T. Sod1 Loss Induces Intrinsic Superoxide Accumulation Leading to p53-Mediated Growth Arrest and Apoptosis. Int. J. Mol. Sci. 2013, 14, 10998-11010. https://doi.org/10.3390/ijms140610998
Watanabe K, Shibuya S, Koyama H, Ozawa Y, Toda T, Yokote K, Shimizu T. Sod1 Loss Induces Intrinsic Superoxide Accumulation Leading to p53-Mediated Growth Arrest and Apoptosis. International Journal of Molecular Sciences. 2013; 14(6):10998-11010. https://doi.org/10.3390/ijms140610998
Chicago/Turabian StyleWatanabe, Kenji, Shuichi Shibuya, Hirofumi Koyama, Yusuke Ozawa, Toshihiko Toda, Koutaro Yokote, and Takahiko Shimizu. 2013. "Sod1 Loss Induces Intrinsic Superoxide Accumulation Leading to p53-Mediated Growth Arrest and Apoptosis" International Journal of Molecular Sciences 14, no. 6: 10998-11010. https://doi.org/10.3390/ijms140610998
APA StyleWatanabe, K., Shibuya, S., Koyama, H., Ozawa, Y., Toda, T., Yokote, K., & Shimizu, T. (2013). Sod1 Loss Induces Intrinsic Superoxide Accumulation Leading to p53-Mediated Growth Arrest and Apoptosis. International Journal of Molecular Sciences, 14(6), 10998-11010. https://doi.org/10.3390/ijms140610998