First Insights into the Large Genome of Epimedium sagittatum (Sieb. et Zucc) Maxim, a Chinese Traditional Medicinal Plant

Abstract
:
1. Introduction
2. Results
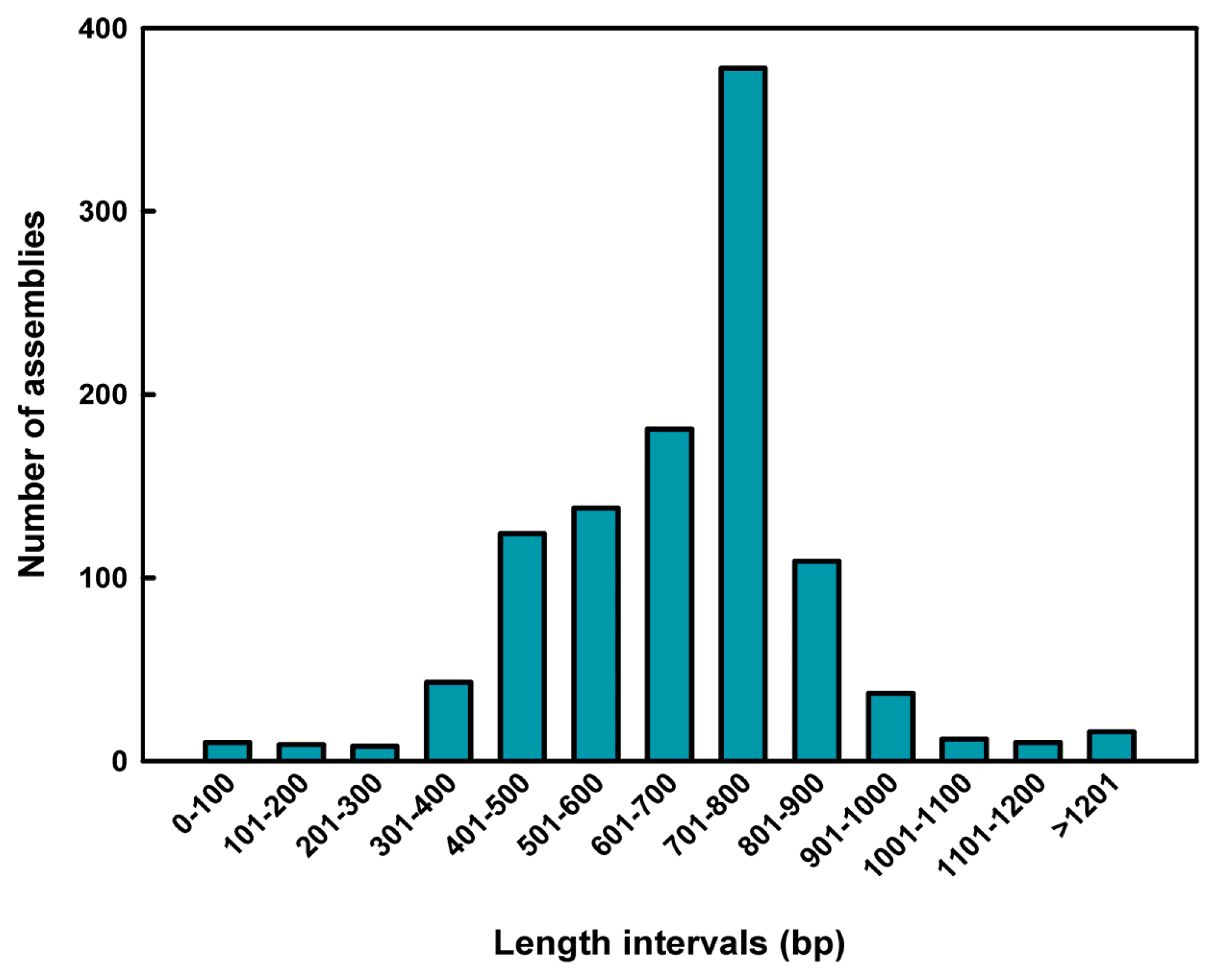
2.1. Sequence Assembly and Composition
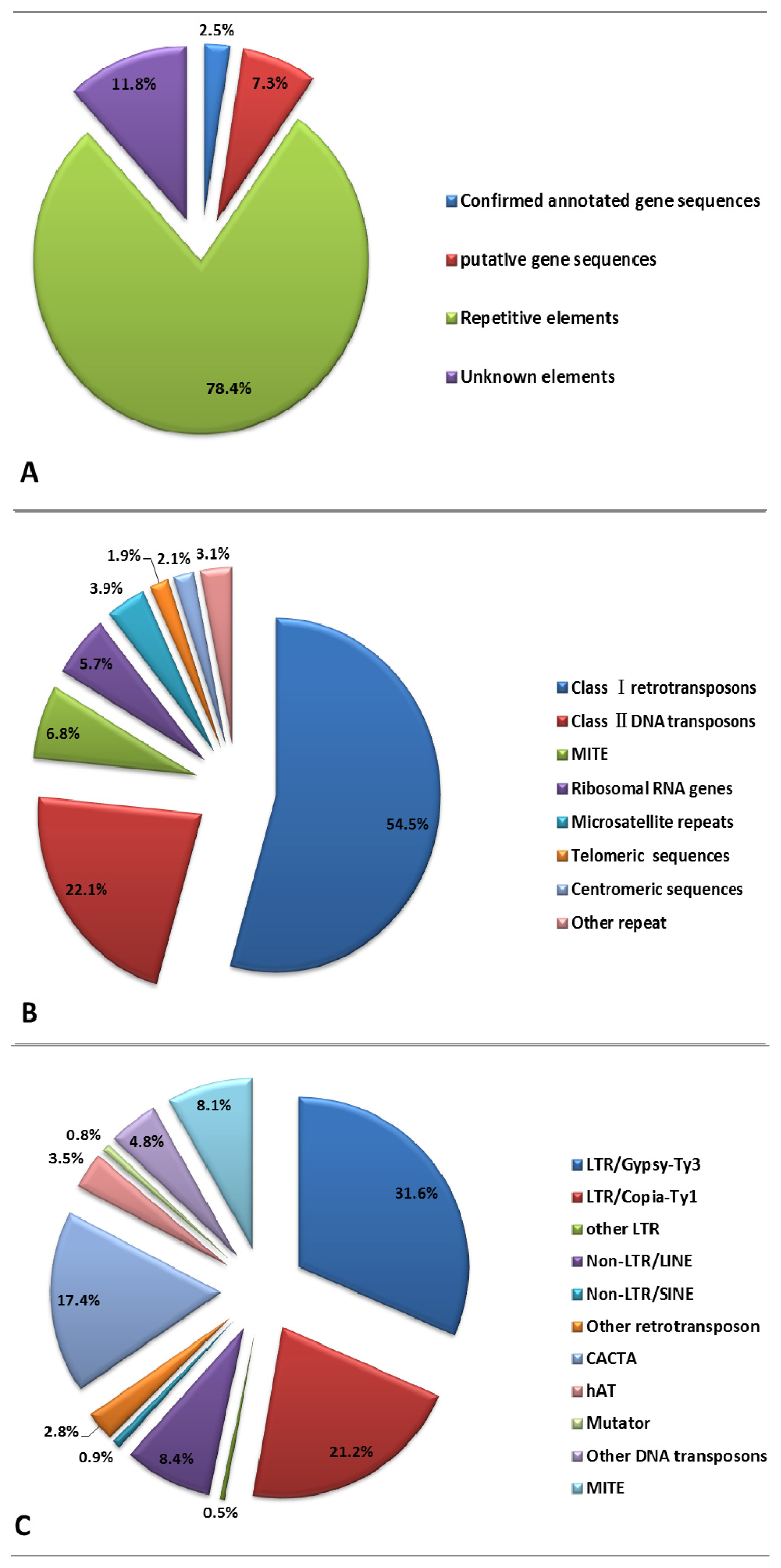
2.2. Repetitive DNA Elements of E. sagittatum Genome
2.3. Gene Content Analysis and Gene Number Estimation
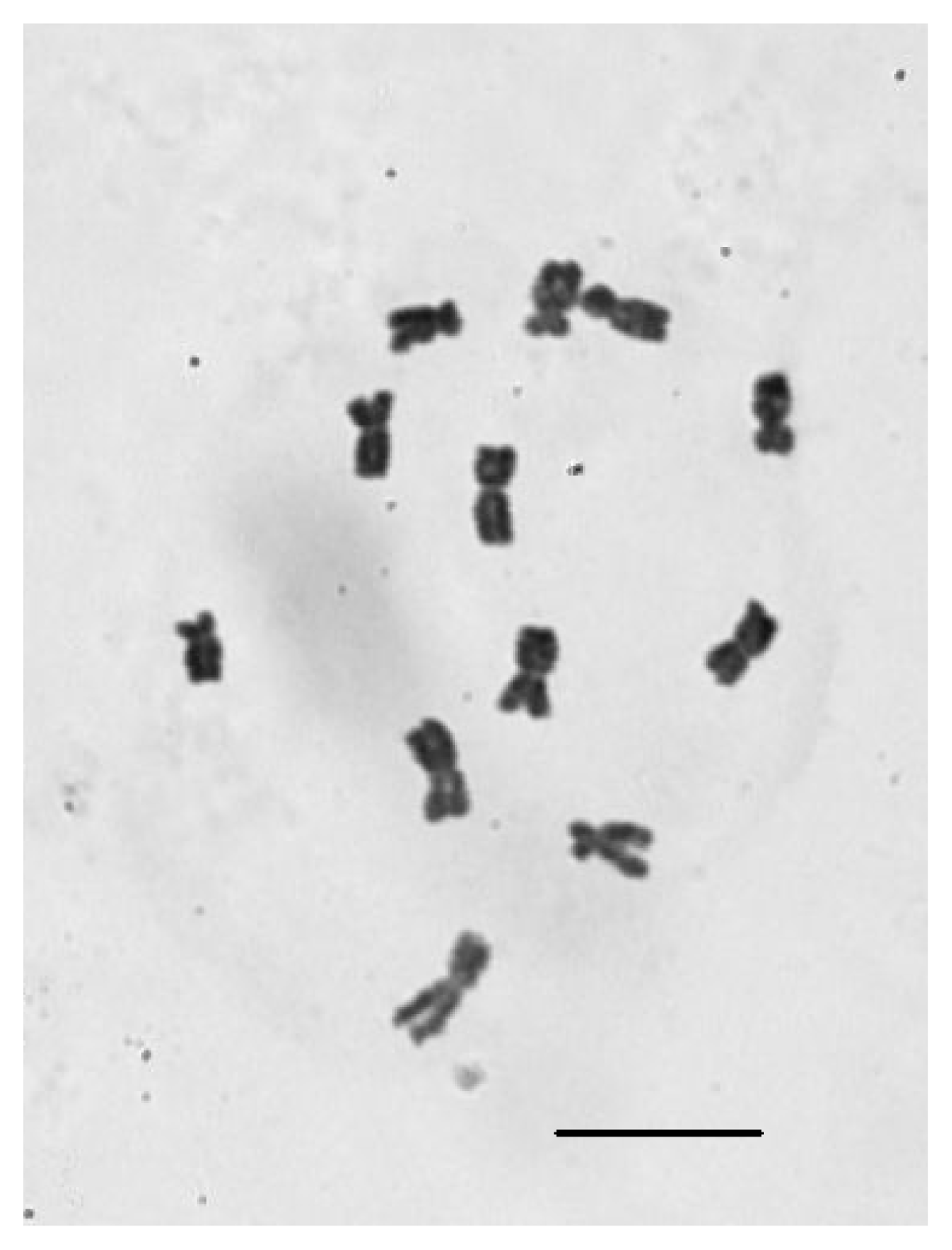
2.4. Chromosomal Number Analysis of E. sagittatum
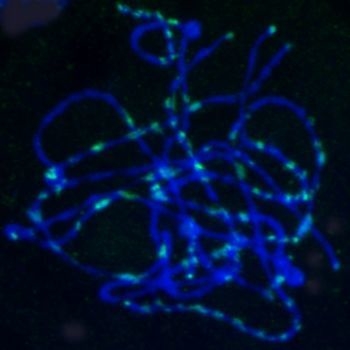
2.5. Chromosomal Distribution of Gypsy-Ty3 Retrotransposons in E. sagittatum
3. Discussion
3.1. Characteristics of E. sagittatum Genome
3.2. Genome Size Variation of Epimedium Species
3.3. Repetitive DNA and Retrotransposable Elements
3.4. Gene Content Analysis
4. Experimental Section
4.1. Plant Material
4.2. DNA Preparation and Construction of the Shotgun Insertion Clones
4.3. Sequencing and Sequence Assembly
4.4. Sequence Annotation
4.4.1. Identification of Repetitive DNA Elements
4.4.2. Analysis of Gene Content
4.5. Chromosomal Analysis
4.6. FISH Analysis
5. Conclusions
Acknowledgments
Abbreviations
| ENS | Epimedium sagittatum Nuclear Sequences |
| TE | Transposable Element |
| LTR | Long Terminal Repeat |
| LINE | Long Interspersed Nuclear Element |
| SINE | Short Interspersed Nuclear Element |
| MITE | Miniature Inverted-repeat Transposable Element. |
Conflict of Interest
References
- Ying, T. Petal evolution and distribution patterns of Epimedium L. (Berberidaceae). Acta Phytotaxon. Sin 2002, 40, 481–489. [Google Scholar]
- Pharmacopoeia of the People’s Republic of China; Chinese Pharmacopoeia Commission: Beijing, China, 2005.
- Liang, H.; Siren, H.; Jyske, P.; Reikkola, M.; Vuorela, P.; Vuorela, H.; Hiltunen, R. Characterization of flavonoids in extracts from four species of Epimedium by micellar electrokinetic capillary chromatography with diode-array detection. J. Chromatogr. Sci 1997, 35, 117–125. [Google Scholar]
- Hu, Z.W.; Shen, Z.Y.; Huang, J.H. Experimental study on effect of epimedium flavonoids in protecting telomere length of senescence cells HU. Zhongguo Zhong Xi Yi Jie He Za Zhi 2004, 24, 1094–1097. [Google Scholar]
- Huang, J.H.; Shen, Z.Y.; Wu, B. Effect and mechanism of Epimedium flavanoids for aging retardation from viewpoint of transcriptomics and metabonomics. Zhongguo Zhong Xi Yi Jie He Za Zhi 2008, 28, 47–50. [Google Scholar]
- Wu, H.; Lien, E.; Lien, L.; Medicines, C. Chemical and pharmacological investigations of Epimedium species: A survey. Prog. Drug Res 2003, 60, 1–57. [Google Scholar]
- Kim, Y.D.; Kim, S.H.; Landrum, L.R. Taxonomic and phytogeographic implications from ITS phylogeny in Berberis (Berberidaceae). J. Plant Res 2004, 117, 175–182. [Google Scholar]
- Shen, P.; Guo, B.L.; Gong, Y.; Hong, D.Y.; Hong, Y.; Yong, E.L. Taxonomic, genetic, chemical and estrogenic characteristics of Epimedium species. Phytochemistry 2007, 68, 1448–1458. [Google Scholar]
- Sun, Y.; Fung, K.P.; Leung, P.C.; Shaw, P.C. A phylogenetic analysis of Epimedium (Berberidaceae) based on nuclear ribosomal DNA sequences. Mol. Phylogenet. Evol 2005, 35, 287–291. [Google Scholar]
- Zhang, M.L.; Uhink, C.H.; Kadereit, J.W. Phylogeny and biogeography of Epimedium/Vancouveria (Berberidaceae): Western North American-East Asian disjunctions, the origin of European mountain plant taxa, and East Asian species diversity. Syst. Bot 2007, 32, 81–92. [Google Scholar]
- Sun, Y.; Fung, K.; Leung, P.; Shi, D.; Shaw, P. Characterization of medicinal Epimedium species by 5S rRNA gene spacer sequencing. Planta Med 2004, 70, 287–288. [Google Scholar]
- Zeng, S.; Xiao, G.; Guo, J.; Fei, Z.; Xu, Y.; Roe, B.; Wang, Y. Development of a EST dataset and characterization of EST-SSRs in a traditional Chinese medicinal plant, Epimedium sagittatum (Sieb. Et Zucc.) Maxim. BMC Genomics 2010, 11, 94. [Google Scholar]
- Huang, W.; Sun, W.; Wang, Y. Isolation and molecular characterisation of flavonoid 3′-hydroxylase and flavonoid 3′,5′-hydroxylase genes from a traditional Chinese medicinal plant, Epimedium sagittatum. Gene 2012, 497, 125–130. [Google Scholar]
- Huang, W.; Sun, W.; Lv, H.; Xiao, G.; Zeng, S.; Wang, Y. Isolation and molecular characterization of thirteen R2R3-MYB transcription factors from Epimedium sagittatum. Int. J. Mol. Sci 2012, 14, 594–610. [Google Scholar]
- Sun, W.; Huang, W.; Li, Z.; Lv, H.; Huang, H.; Wang, Y. Characterization of a Crabs Claw gene in basal eudicot species Epimedium sagittatum (Berberidaceae). Int. J. Mol. Sci 2013, 14, 1119–1131. [Google Scholar]
- Zhang, Y.; Dang, H.; Meng, A.; Li, J.; Li, X. Karyomorphology of Epimedium (Berberidaceae) and its phylogenetic implications. Caryologia 2008, 61, 283–293. [Google Scholar]
- Chen, J.; Li, L.; Wang, Y. Diversity of genome size and Ty1-copia in Epimedium species used for traditional Chinese medicines. HortScience 2012, 47, 979–984. [Google Scholar]
- Bennett, M.; Leitch, I.; Price, H.; Johnston, J. Comparisons with Caenorhabditis (~100 Mb) and Drosophila (~175 Mb) using flow cytometry show genome size in Arabidopsis to be~157 Mb and thus~25% larger than the Arabidopsis Genome Initiative estimate of ~125 Mb. Ann. Bot 2003, 91, 547–557. [Google Scholar]
- Bennett, M.D.; Smith, J.B. Nuclear DNA amounts in Angiosperms. Phil. Trans. Roy. Soc. (Lond. ) B 1991, 334, 309–345. [Google Scholar]
- Arumuganathan, K.; Earle, E.D. Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep 1991, 9, 208–218. [Google Scholar]
- Adam-Blondon, A.; Bernole, A.; Faes, G.; Lamoureux, D.; Pateyron, S.; Grando, M.; Caboche, M.; Velasco, R.; Chalhoub, B. Construction and characterization of BAC libraries from major grapevine cultivars. Theor. Appl. Genet 2005, 110, 1363–1371. [Google Scholar]
- Tuskan, G.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar]
- Hendrix, B.; Stewart, J.M. Estimation of the nuclear DNA content of Gossypium species. Ann. Bot 2005, 95, 789–797. [Google Scholar]
- The Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641.[Green Version]
- Flavell, R.B.; Bennett, M.D.; Smith, J.B.; Smith, D.B. Genome size and the proportion of repeated nucleotide sequence DNA in plants. Biochem. Genet 1974, 12, 257–269. [Google Scholar]
- Brenner, S.; Elgar, G.; Sandford, R.; Macrae, A.; Venkatesh, B.; Aparicio, S. Characterization of the pufferfish (Fugu) genome as a compact model vertebrate genome. Nature 1993, 366, 265–268. [Google Scholar]
- Liu, R.; Vitte, C.; Ma, J.; Mahama, A.A.; Dhliwayo, T.; Lee, M.; Bennetzen, J.L. A GeneTrek analysis of the maize genome. Proc. Natl. Acad. Sci. USA 2007, 104, 11844–11849. [Google Scholar]
- Messing, J.; Bharti, A.K.; Karlowski, W.M.; Gundlach, H.; Kim, H.R.; Yu, Y.; Wei, F.; Fuks, G.; Soderlund, C.A.; Mayer, K.F.; et al. Sequence composition and genome organization of maize. Proc. Natl. Acad. Sci. USA 2004, 101, 14349–14354. [Google Scholar]
- Haberer, G.; Young, S.; Bharti, A.K.; Gundlach, H.; Raymond, C.; Fuks, G.; Butler, E.; Wing, R.A.; Rounsley, S.; Birren, B.; et al. Structure and architecture of the maize genome. Plant Physiol 2005, 139, 1612–1624. [Google Scholar]
- Bartos, J.; Paux, E.; Kofler, R.; Havrankova, M.; Kopecky, D.; Suchankova, P.; Safar, J.; Simkova, H.; Town, C.D.; Lelley, T.; et al. A first survey of the rye (Secale cereale) genome composition through BAC end sequencing of the short arm of chromosome 1R. BMC Plant Biol 2008, 8, 95–106. [Google Scholar]
- Guo, W.; Cai, C.; Wang, C.; Zhao, L.; Wang, L.; Zhang, T. A preliminary analysis of genome structure and composition in Gossypium hirsutum. BMC Genomics 2008, 9, 314–331. [Google Scholar]
- Lai, C.W.; Yu, Q.; Hou, S.; Skelton, R.L.; Jones, M.R.; Lewis, K.L.; Murray, J.; Eustice, M.; Guan, P.; Agbayani, R.; et al. Analysis of papaya BAC end sequences reveals first insights into the organization of a fruit tree genome. Mol. Genet. Genomics 2006, 276, 1–12. [Google Scholar]
- Devos, K.; Ma, J.; Pontaroli, A.; Pratt, L.; Bennetzen, J. Analysis and mapping of randomly chosen bacterial artificial chromosome clones from hexaploid bread wheat. Proc. Natl. Acad. Sci. USA 2005, 102, 19243–19248. [Google Scholar]
- Rajesh, P.N.; O’Bleness, M.; Roe, B.A.; Muehlbauer, F.J. Analysis of genome organization, composition and microsynteny using 500 kb BAC sequences in chickpea. Theor. Appl. Genet 2008, 117, 449–458. [Google Scholar]
- Pontaroli, A.C.; Rogers, R.L.; Zhang, Q.; Shields, M.E.; Davis, T.M.; Folta, K.M.; SanMiguel, P.; Bennetzen, J.L. Gene content and distribution in the nuclear genome of Fragaria vesca. Plant Gen 2009, 2, 93–101. [Google Scholar]
- Cavagnaro, P.; Chung, S.M.; Szklarczyk, M.; Grzebelus, D.; Senalik, D.; Atkins, A.; Simon, P. Characterization of a deep-coverage carrot (Daucus carota L.) BAC library and initial analysis of BAC-end sequences. Mol. Genet. Genomics 2009, 281, 273–288. [Google Scholar]
- Hao, D.; Yang, L.; Xiao, P. The first insight into the Taxus genome via fosmid library construction and end sequencing. Mol. Genet. Genomics 2011, 285, 197–205. [Google Scholar]
- Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815.
- SanMiguel, P.; Tikhonov, A.; Jin, Y.K.; Motchoulskaia, N.; Zakharov, D.; Melake-Berhan, A.; Springer, P.S.; Edwards, K.J.; Lee, M.; Avramova, Z.; et al. Nested retrotransposons in the intergenic regions of the maize genome. Science 1996, 274, 765–768. [Google Scholar]
- Piegu, B.; Guyot, R.; Picault, N.; Roulin, A.; Saniyal, A.; Kim, H.; Collura, K.; Brar, D.S.; Jackson, S.; Wing, R.A.; et al. Doubling genome size without polyploidization: Dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res 2006, 16, 1262–1269. [Google Scholar]
- Soltis, D.E.; Soltis, P.S.; Bennett, M.D.; Leitch, I.J. Evolution of genome size in the angiosperms. Am. J. Bot 2003, 90, 1596–1603. [Google Scholar]
- Bennett, M.; Leitch, I. Angiosperm DNA C-Values Database, version 8.0. Available online: http://www.kew.org/cvalues/ (on accessed 7 May 2013).
- Greilhuber, J.; Borsch, T.; Müller, K.; Worberg, A.; Porembski, S.; Barthlott, W. Smallest angiosperm genomes found in Lentibulariaceae, with chromosomes of bacterial size. Plant Biol 2006, 8, 770–777. [Google Scholar]
- Pellicer, J.; Fay, M.F.; Leitch, I.J. The largest eukaryotic genome of them all? Bot. J. Linnean Soc 2010, 164, 10–15. [Google Scholar]
- Veselý, P.; Bureš, P.; Šmarda, P.; Pavlíček, T. Genome size and DNA base composition of geophytes: the mirror of phenology and ecology? Ann. Bot 2012, 109, 65–75. [Google Scholar]
- Olszewska, M.J.; Osiecka, R. The relationship between 2 C DNA content, systematic position, and the level of nuclear DNA endoreplication during differentiation of root parenchyma in some dicotyledonous shrubs and trees. comparison with Herbaceous species. Biochem. Physiol. Pflanzen 1984, 179, 641–657. [Google Scholar]
- Siljak-Yakovlev, S.; Pustahija, F.; Oli, M.E.; Boguni, F.; Muratovi, E.; Ba, N.; Catrice, O.; Brown, S.C. Towards a genome size and chromosome number database of balkan flora: C-values in 343 Taxa with novel values for 242. Adv. Sci. Lett 2010, 3, 190–213. [Google Scholar]
- Ohri, D. Genome size variation and plant systematics. Ann. Bot 1998, 82, 75–83. [Google Scholar]
- Tenaillon, M.I.; Hufford, M.B.; Gaut, B.S.; Ross-Ibarra, J. Genome size and transposable element content as determined by high-throughput sequencing in Maize and Zea luxurians. Genome Biol. Evol 2011, 3, 219–229. [Google Scholar]
- Vitte, C.; Panaud, O. LTR retrotransposons and flowering plant genome size: Emergence of the increase/decrease model. Cytogenet. Genome Res 2005, 110, 91–107. [Google Scholar]
- Vitte, C.; Bennetzen, J. Analysis of retrotransposon structural diversity uncovers properties and propensities in angiosperm genome evolution. Proc. Natl. Acad. Sci. USA 2006, 103, 17638–17643. [Google Scholar]
- Bennetzen, J.; Ma, J.; Devos, K. Mechanisms of recent genome size variation in flowering plants. Ann. Bot 2005, 95, 127–132. [Google Scholar]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar]
- Zuccolo, A.; Sebastian, A.; Talag, J.; Yu, Y.; Kim, H.; Collura, K.; Kudrna, D.; Wing, R. Transposable element distribution, abundance and role in genome size variation in the genus Oryza. BMC Evol. Biol 2007, 7, 152–166. [Google Scholar]
- International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature 2005, 436, 793–800.
- Wang, Y.; Tang, X.; Cheng, Z.; Mueller, L.; Giovannoni, J.; Tanksley, S. Euchromatin and pericentromeric heterochromatin: comparative composition in the tomato genome. Genetics 2006, 172, 2529–2540. [Google Scholar]
- Natali, L.; Santini, S.; Giordani, T.; Minelli, S.; Maestrini, P.; Cionini, P.; Cavallini, A. Distribution of Ty3-gypsy-and Ty1-copia-like DNA sequences in the genus Helianthus and other Asteraceae. Genome 2006, 49, 64–72. [Google Scholar]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.; Kanamori, H.; McCombie, W.; Ouyang, S.; Schwartz, D.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 1–10. [Google Scholar]
- Ming, R.; Hou, S.; Feng, Y.; Yu, Q.; Dionne-Laporte, A.; Albert, H.; Suzuki, J.; Tripathi, S.; Moore, P.; Gonsalves, D. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 2008, 452, 991–996. [Google Scholar]
- Jaillon, O.; Aury, J.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar]
- Shulaev, V.; Sargent, D.J.; Crowhurst, R.N.; Mockler, T.C.; Folkerts, O.; Delcher, A.L.; Jaiswal, P.; Mockaitis, K.; Liston, A.; Mane, S.P.; et al. The genome of woodland strawberry (Fragaria vesca). Nat. Genet 2011, 43, 109–116. [Google Scholar]
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S.; et al. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet 2012, 44, 1098–1103. [Google Scholar]
- Bennetzen, J.L.; Coleman, C.; Liu, R.; Ma, J.; Ramakrishna, W. Consistent over-estimation of gene number in complex plant genomes. Curr. Opin. Plant Biol 2004, 7, 732–736. [Google Scholar]
- Rabinowicz, P. Constructing gene-enriched plant genomic libraries using methylation filtration technology. In Methods in Molecular Biology: Plant Functional Genomics; Grotewold, E., Ed.; Humana Press: Totowa, NJ, USA, 2003; Volume 236, pp. 21–36. [Google Scholar]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory press: New York, NY, USA, 2001. [Google Scholar]
- Ewing, B.; Hillier, L.; Wendl, M.; Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 1998, 8, 175–185. [Google Scholar]
- Chou, H.H.; Holmes, M.H. DNA sequence quality trimming and vector removal. Bioinformatics 2001, 17, 1093. [Google Scholar]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res 1999, 9, 868–877. [Google Scholar]
- RepeatMasker, Version 4.0.2. Available online: http://www.repeatmasker.org/cgi-bin/WEBRepeatMasker (on accessed 8 May 2013).
- Kohany, O.; Gentles, A.; Hankus, L.; Jurka, J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinforma 2006, 7, 474–480. [Google Scholar]
- Ouyang, S.; Buell, C.R. The TIGR Plant Repeat Databases: A collective resource for the identification of repetitive sequences in plants. Nucleic Acids Res 2004, 32, D360–D363. [Google Scholar]
- Altschul, S.; Madden, T.; Schaffer, A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 1997, 25, 3389–3402. [Google Scholar]
- MolQuest, Version 2.1.1. Available online: http://www.molquest.com/molquest.phtml (on accessed 25 March 2010).
- Jiang, J.; Gill, B.; Wang, G.; Ronald, P.; Ward, D. Metaphase and interphase fluorescence in situ hybridization mapping of the rice genome with bacterial artificial chromosomes. Proc. Natl. Acad. Sci. USA 1995, 92, 4487–4491. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total number of assemblies | 1075 |
|---|---|
| Contigs | 126 |
| Singlets | 949 |
| Total sequence length | 725,008 bp |
| Average insert length | 674.43 bp |
| GC content | 39.00% |
| Nuclear DNA insert | 1023 |
| Organellic DNA insert (cp, mt) a | 50 |
| Bacterial DNA insert | 2 |
| Nuclear DNA insert | Number of assemblies | Calculated sequence length (bp) |
|---|---|---|
| Confirmed annotated gene sequences | 23 | 17,360 |
| Putative gene sequences | 75 | 50,466 |
| Repetitive DNA elements | 795 | 541,625 |
| Unknown elements | 130 | 81,353 |
| Total ENS dataset | 1023 | 690,804 |
| Number of assemblies | Calculated length of repetitive DNA (bp) | Percentage of total repetitive DNA (%) | |
|---|---|---|---|
| Total TE a repeats | 656 | 451,355 | 83.33 |
| Classi retrotransposons | 431 | 295,036 | 54.47 |
| Class ii DNA transposons | 171 | 119,536 | 22.07 |
| MITE b | 54 | 36,783 | 6.79 |
| Ribosomal RNA genes | 47 | 31,002 | 5.72 |
| Microsatellite repeats | 34 | 21,065 | 3.89 |
| Telomeric sequences | 16 | 10,249 | 1.89 |
| Centromeric sequences | 17 | 11,105 | 2.05 |
| Other repeat | 25 | 16,849 | 3.11 |
| Total repetitive DNA elements | 795 | 541,625 | 100 |
| Number of assemblies | Calculated length of TE repeats (bp) | Percentage of TE repeats (%) | |
|---|---|---|---|
| Classiretrotransposons | 431 | 295,036 | 65.37 |
| LTR a/Gypsy-Ty3 | 211 | 142,593 | 31.59 |
| Copia-Ty1 | 136 | 95,698 | 21.20 |
| Other LTR | 3 | 2,176 | 0.48 |
| Non-LTR LINE b/RTE | 26 | 17,700 | 3.92 |
| LINE/L1 | 16 | 11,985 | 2.66 |
| Other LINE | 13 | 8261 | 1.83 |
| SINE c | 6 | 3840 | 0.85 |
| Other retrotransposon | 20 | 12,783 | 2.83 |
| Classii DNA transposons | 171 | 119,536 | 26.48 |
| CACTA | 110 | 78,494 | 17.39 |
| hAT | 21 | 15,805 | 3.50 |
| Mutator | 6 | 3760 | 0.83 |
| Other DNA transposons | 34 | 21,477 | 4.76 |
| MITE | 54 | 36,783 | 8.15 |
| Total TE repeats | 656 | 451,355 | 100 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, D.; Zeng, S.-H.; Chen, J.-J.; Zhang, Y.-J.; Xiao, G.; Zhu, L.-Y.; Wang, Y. First Insights into the Large Genome of Epimedium sagittatum (Sieb. et Zucc) Maxim, a Chinese Traditional Medicinal Plant. Int. J. Mol. Sci. 2013, 14, 13559-13576. https://doi.org/10.3390/ijms140713559
Liu D, Zeng S-H, Chen J-J, Zhang Y-J, Xiao G, Zhu L-Y, Wang Y. First Insights into the Large Genome of Epimedium sagittatum (Sieb. et Zucc) Maxim, a Chinese Traditional Medicinal Plant. International Journal of Molecular Sciences. 2013; 14(7):13559-13576. https://doi.org/10.3390/ijms140713559
Chicago/Turabian StyleLiu, Di, Shao-Hua Zeng, Jian-Jun Chen, Yan-Jun Zhang, Gong Xiao, Lin-Yao Zhu, and Ying Wang. 2013. "First Insights into the Large Genome of Epimedium sagittatum (Sieb. et Zucc) Maxim, a Chinese Traditional Medicinal Plant" International Journal of Molecular Sciences 14, no. 7: 13559-13576. https://doi.org/10.3390/ijms140713559
APA StyleLiu, D., Zeng, S. -H., Chen, J. -J., Zhang, Y. -J., Xiao, G., Zhu, L. -Y., & Wang, Y. (2013). First Insights into the Large Genome of Epimedium sagittatum (Sieb. et Zucc) Maxim, a Chinese Traditional Medicinal Plant. International Journal of Molecular Sciences, 14(7), 13559-13576. https://doi.org/10.3390/ijms140713559