ACE Inhibition with Captopril Retards the Development of Signs of Neurodegeneration in an Animal Model of Alzheimer’s Disease

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
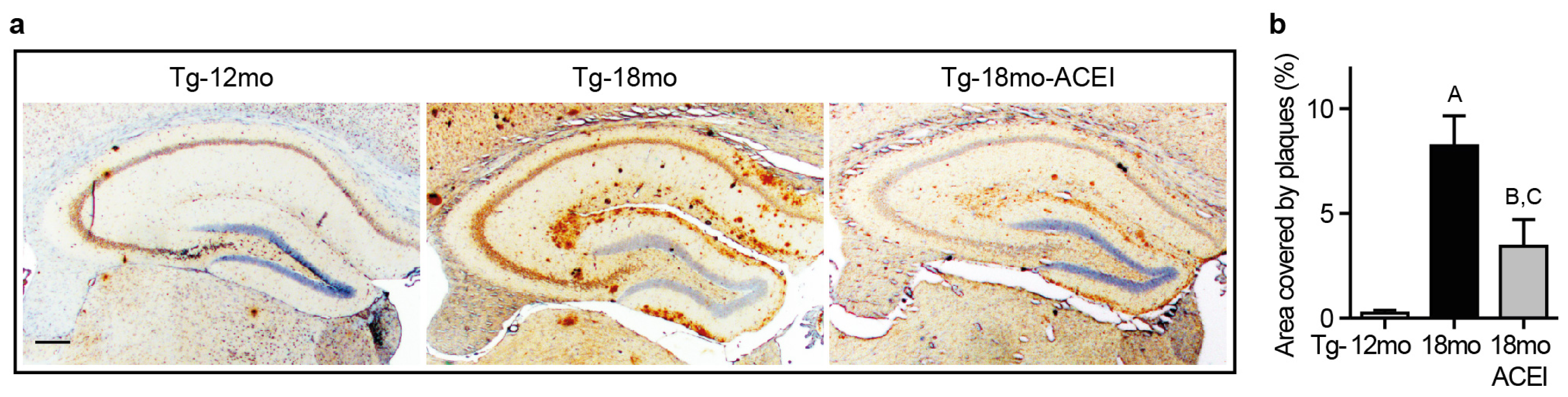
2.1. ACE Inhibition with Captopril Slowed Aβ Plaque Accumulation of AD Mice
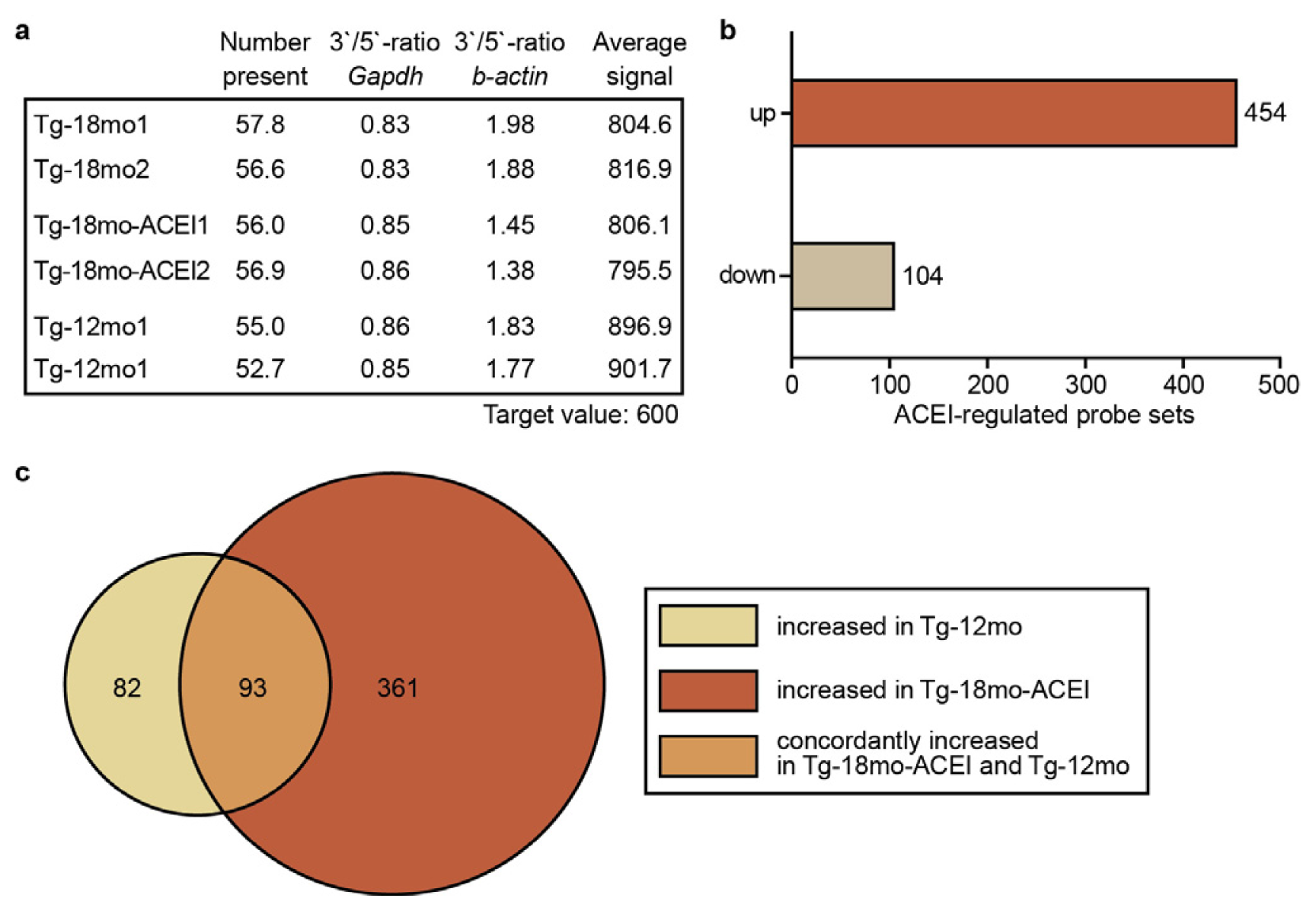
2.2. Whole Genome Microarray Gene Expression Profiling of Hippocampal Genes of AD Mice upon ACE Inhibition with Captopril
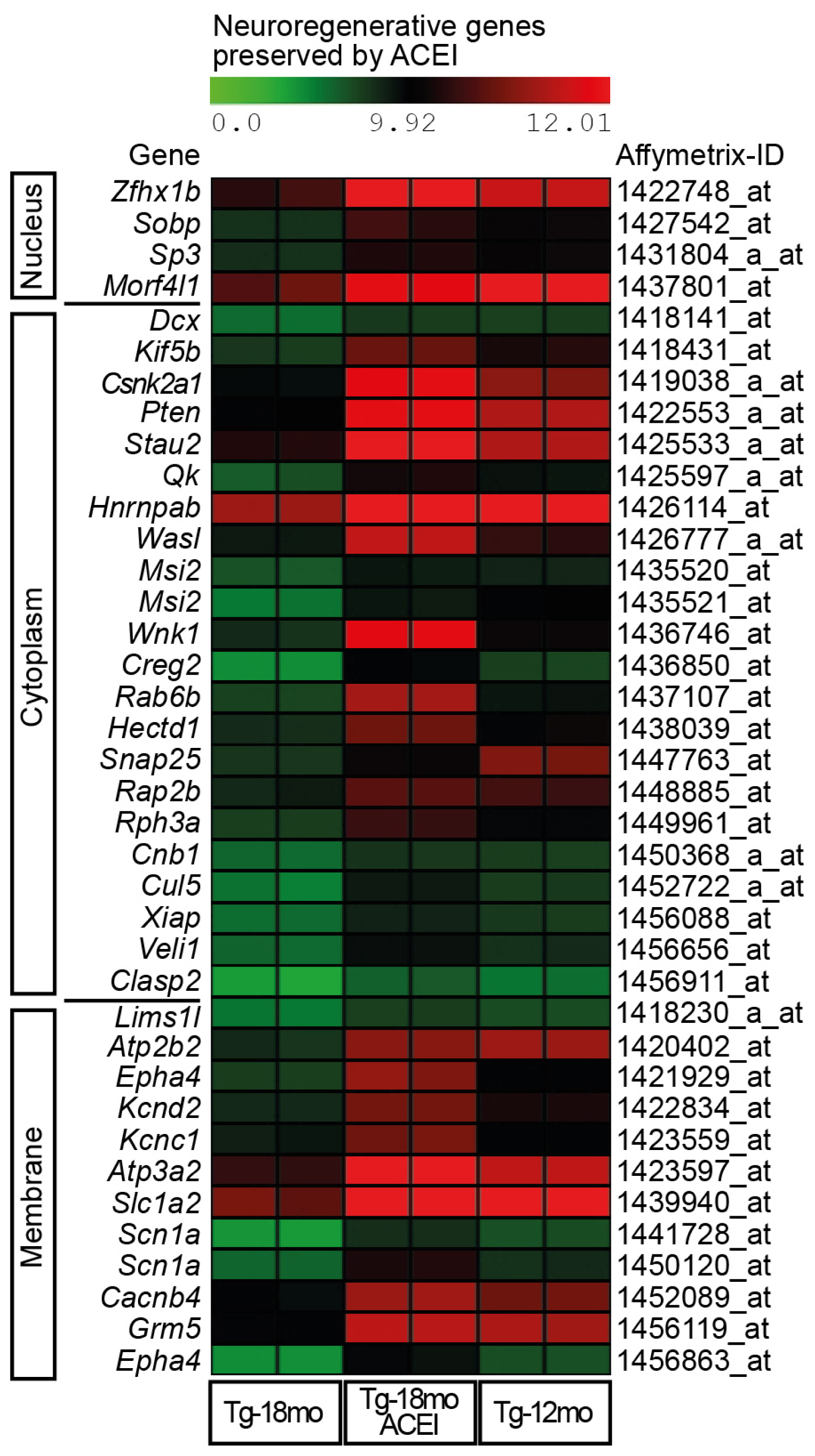
2.3. Captopril Prevented the Down-Regulation of Genes Involved in Neuronal Regeneration and Cognition
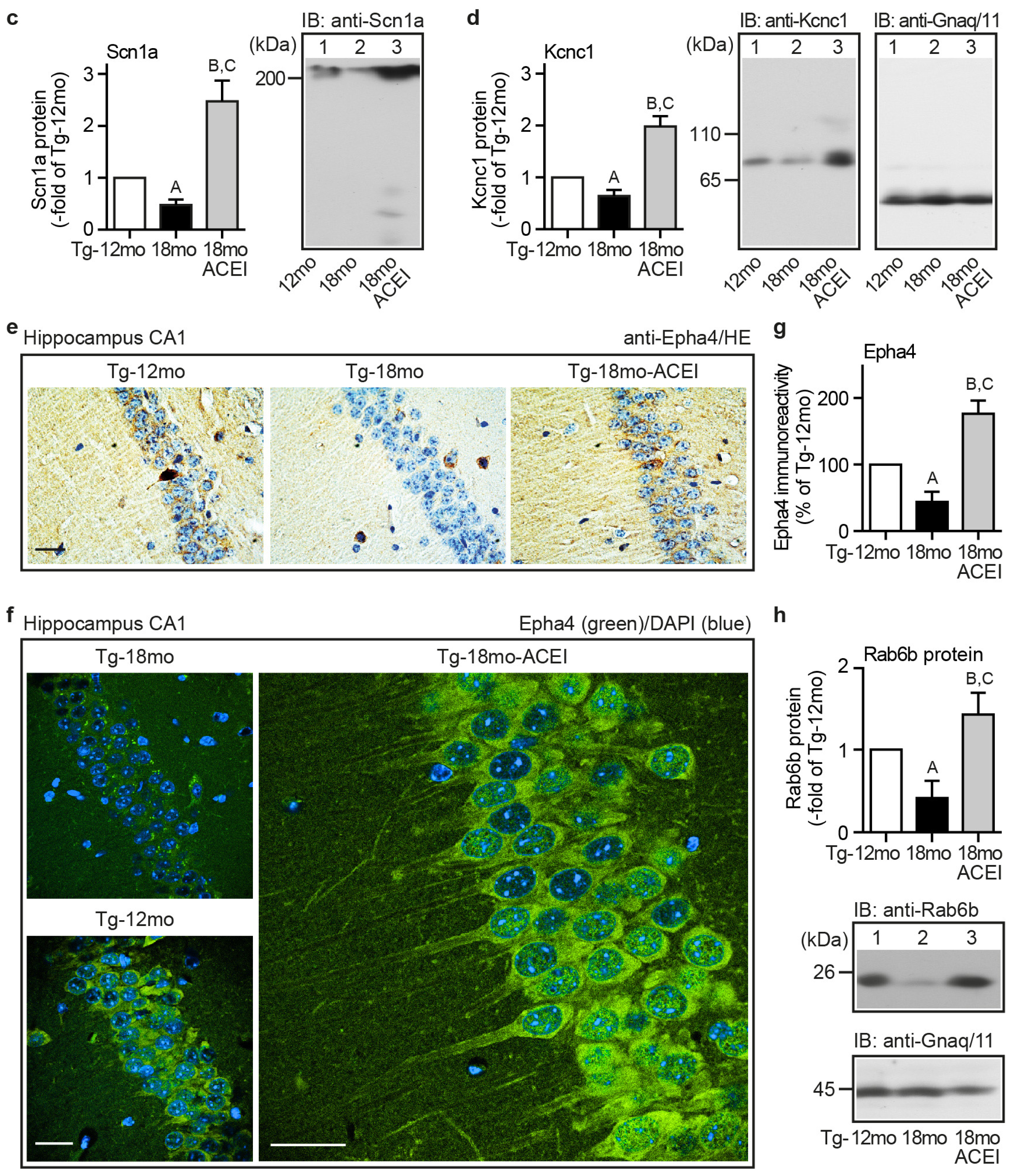
2.4. ACE Inhibition Promoted Signs of Neuronal Regeneration and Up-Regulated the Expression of Genes Involved in the Formation of Neuronal Membrane and Neuronal Processes
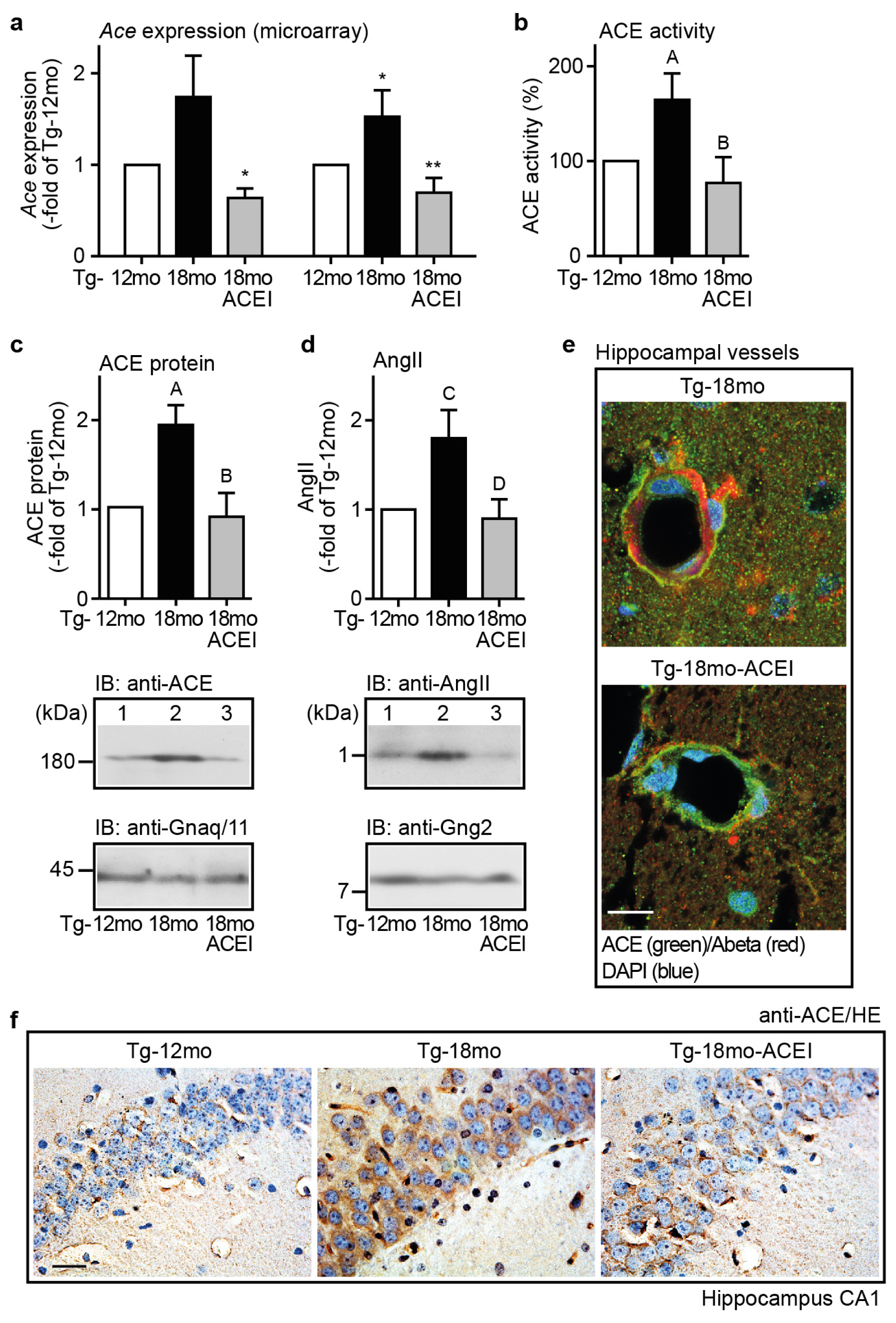
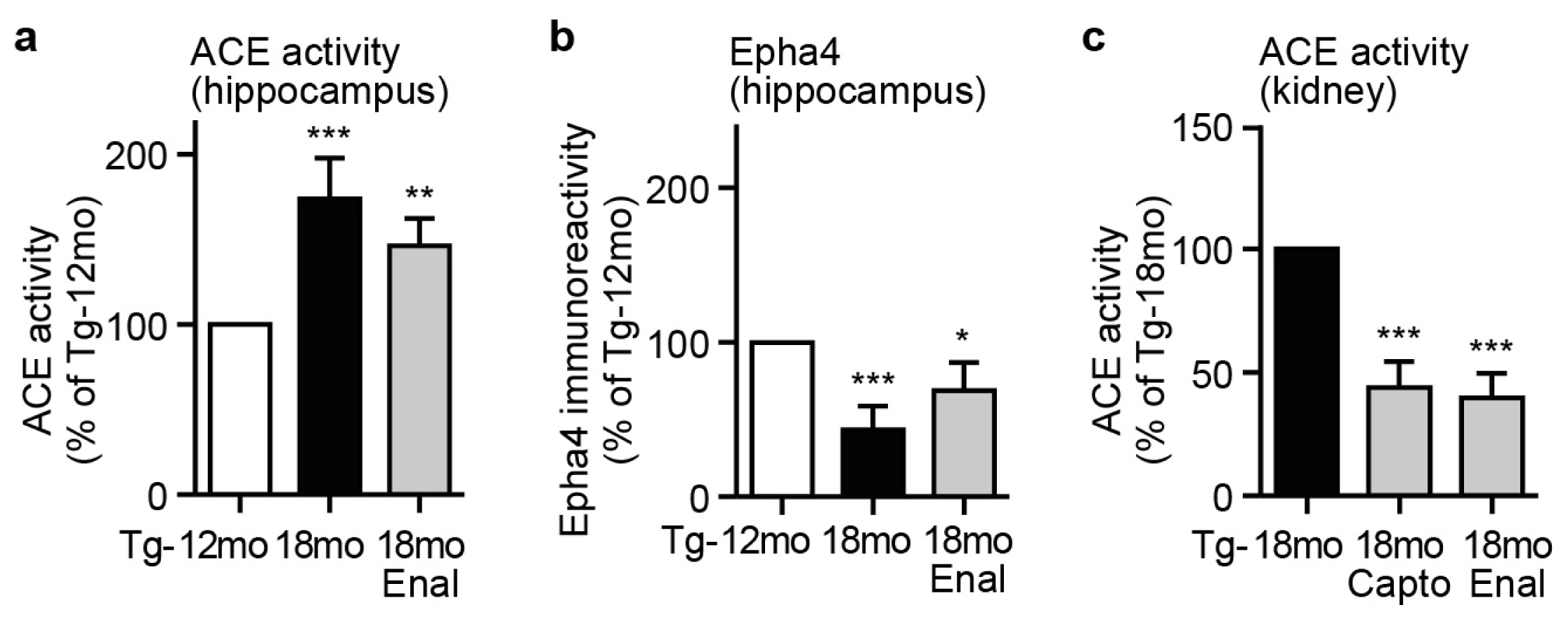
2.5. Captopril Treatment Prevented the AD-Related Up-Regulation of ACE and Reduced the Hippocampal Angiotensin II Level of AD Mice
2.6. ACE Protein in Hippocampal Vessels and Neurons of AD Mice Could Account for a Hippocampal Action of Captopril
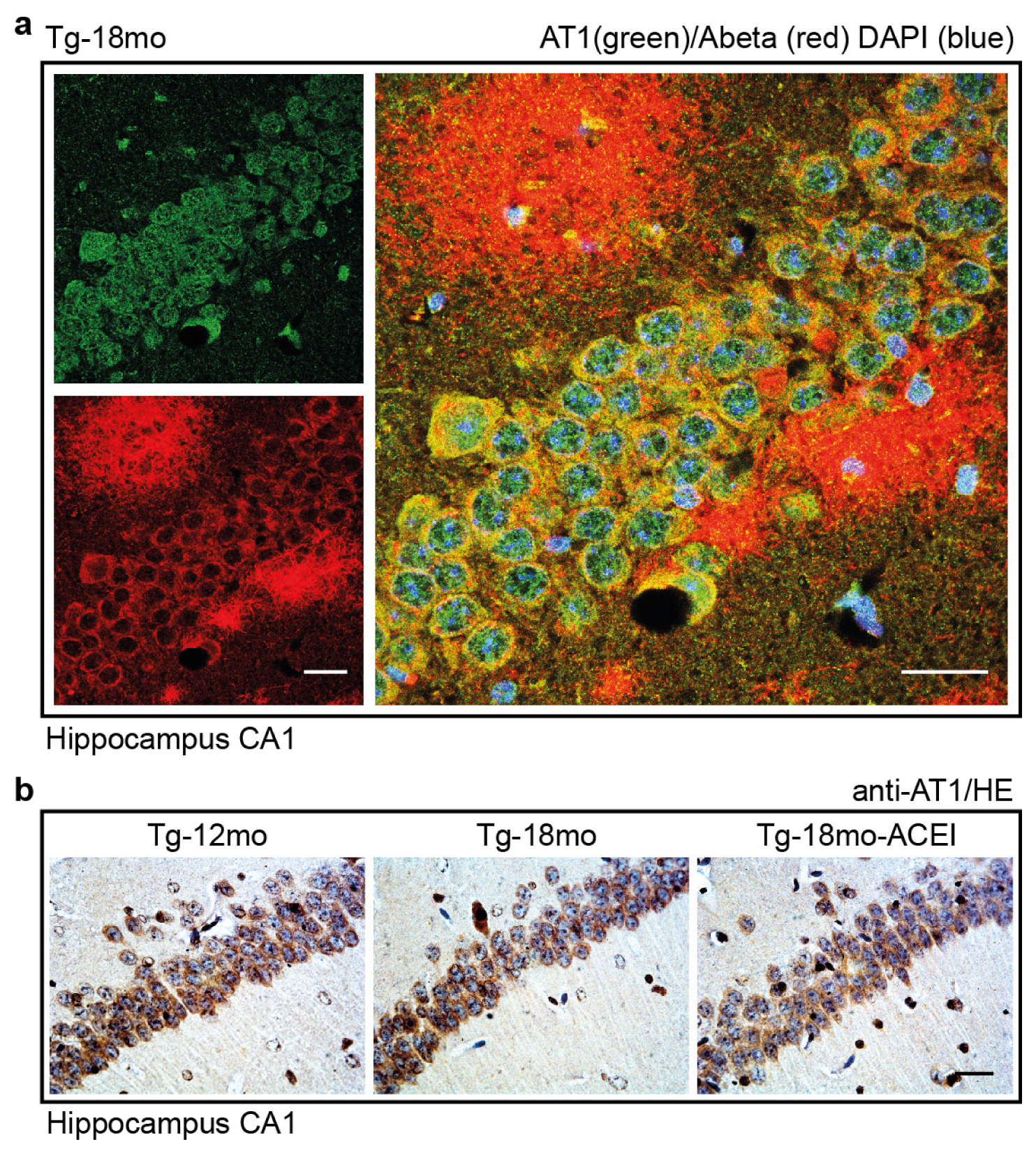
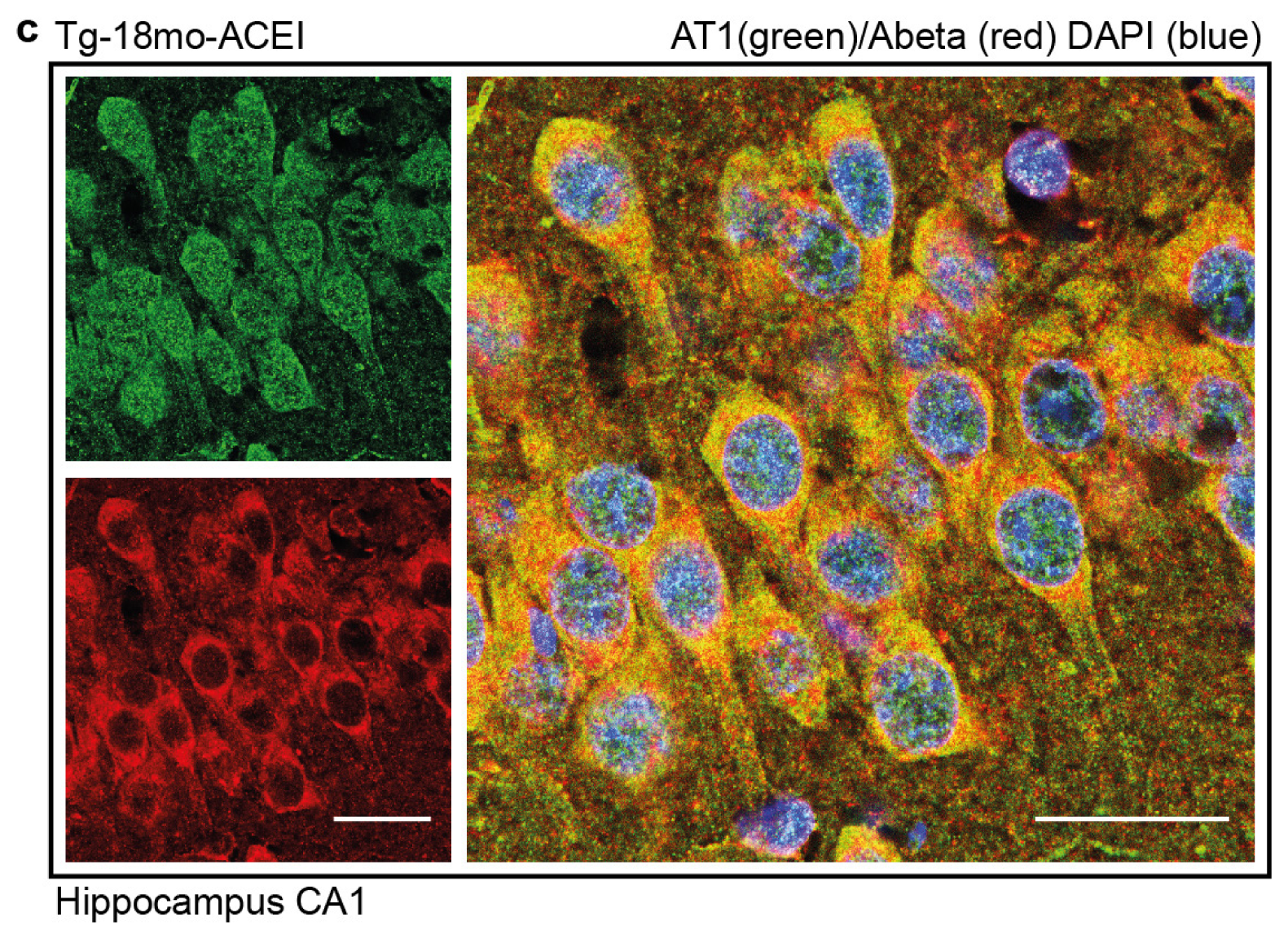
2.7. Co-Localization of the AT1 Receptor with Aβ in Hippocampal Neurons of AD Mice
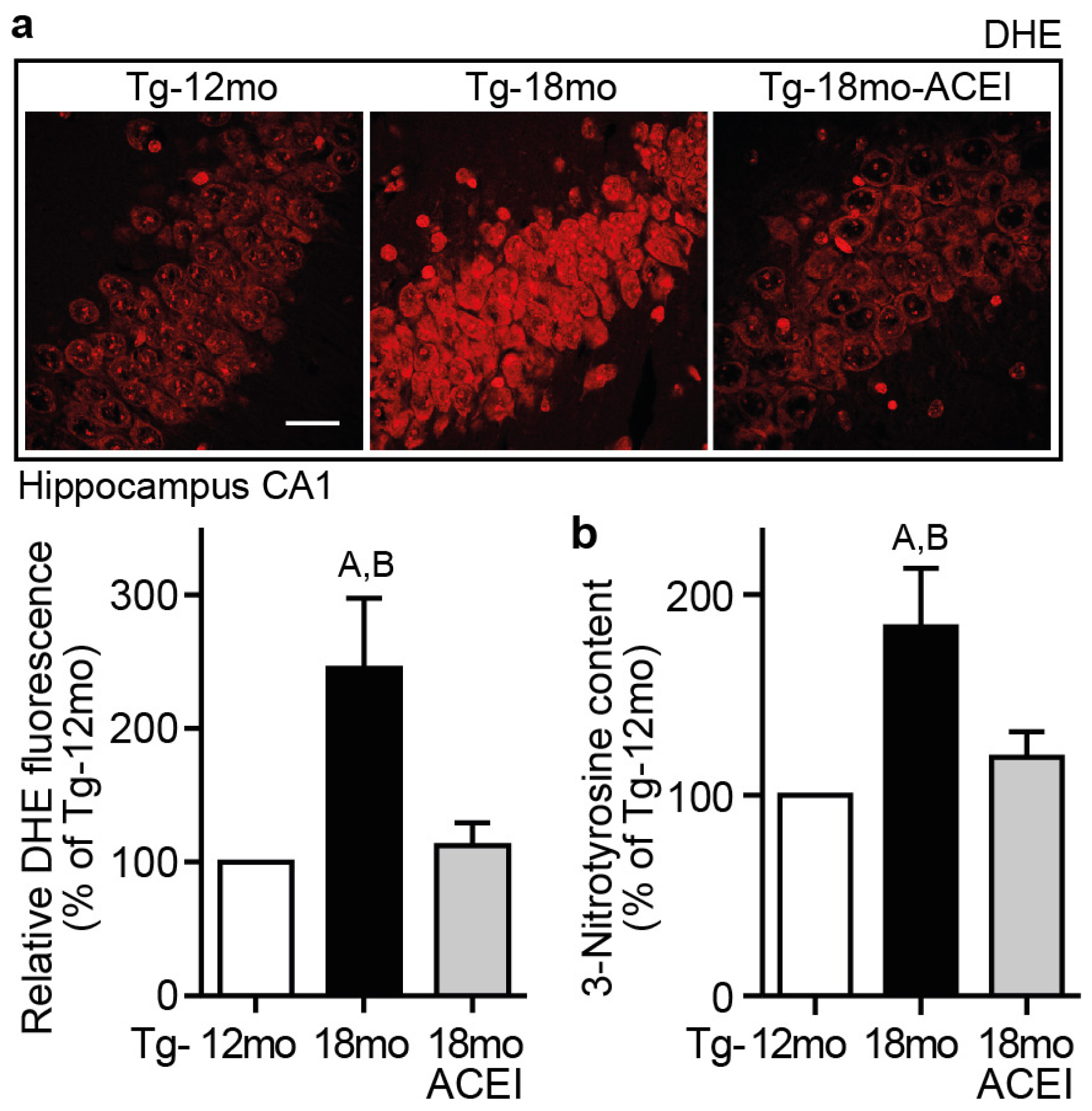
2.8. Captopril Treatment Reduced Hippocampal ROS and Protein Oxidation
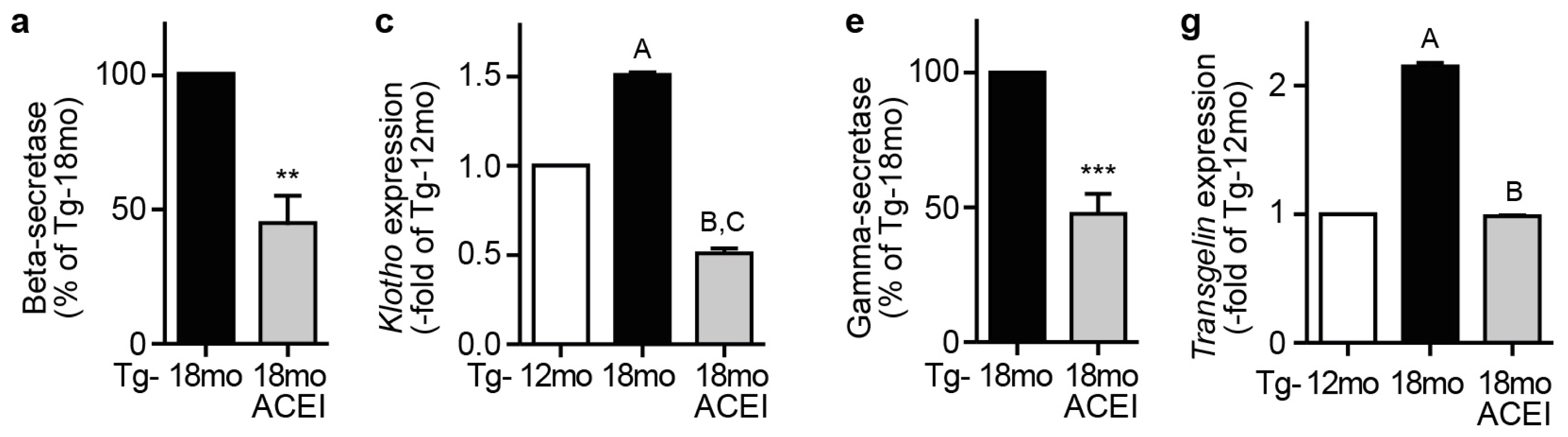
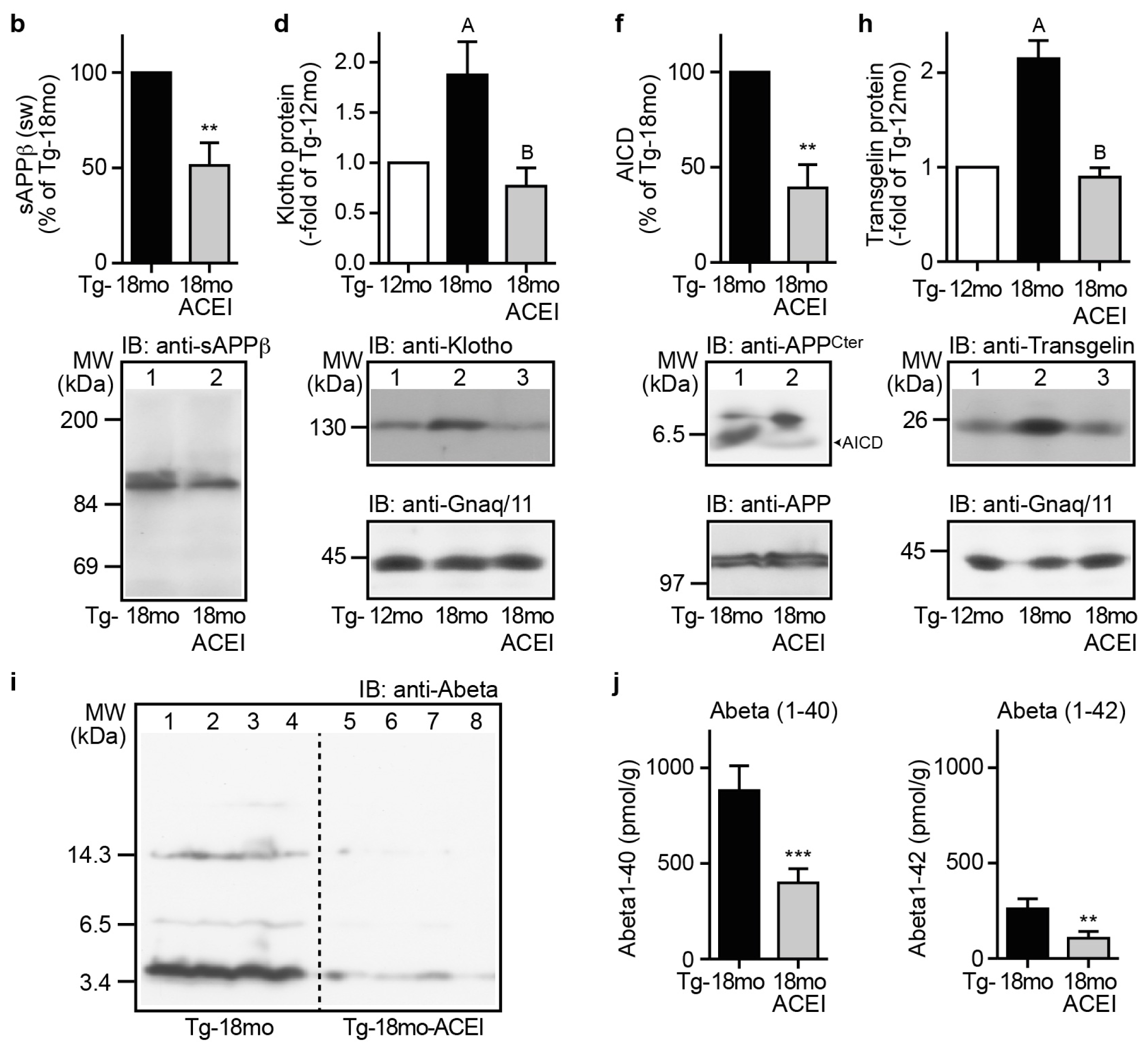
2.9. Captopril Treatment Reduced Markers of Amyloidogenic Processing of APP
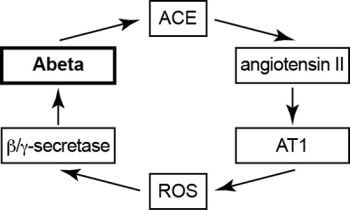
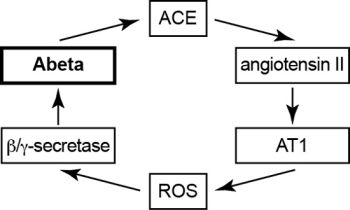
2.10. Model of Vicious Cycle of Angiotensin II-Dependent Aβ Generation
3. Experimental Section
3.1. Animal Model of AD and Captopril Treatment
3.2. Whole Genome Microarray Gene Expression Profiling
3.3. Biochemical Analyses
3.4. Immunohistology, Immunofluorescence and Immunoblotting
3.5. Antibodies
3.6. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Abbott, A. Dementia: A problem of our age. Nature 2011, 475, S2–S4. [Google Scholar]
- Holtzman, D.M.; Mandelkow, E.; Selkoe, D.J. Alzheimer disease in 2020. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Huang, Y.; Mucke, L. Alzheimer mechanisms and therapeutic strategies. Cell 2012, 148, 1204–1222. [Google Scholar]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun 1984, 120, 885–890. [Google Scholar]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar]
- Butterfield, D.A.; Swomley, A.M.; Sultana, R. Amyloid beta-peptide 1–42-induced oxidative stress in Alzheimer disease: Importance in disease pathogenesis and progression. Antioxid. Redox. Signal. 2013. [Google Scholar] [CrossRef]
- Shen, C.; Chen, Y.; Liu, H.; Zhang, K.; Zhang, T.; Lin, A.; Jing, N. Hydrogen peroxide promotes Abeta production through JNK-dependent activation of gamma-secretase. J. Biol. Chem 2008, 283, 17721–17730. [Google Scholar]
- Murakami, K.; Murata, N.; Noda, Y.; Tahara, S.; Kaneko, T.; Kinoshita, N.; Hatsuta, H.; Murayama, S.; Barnham, K.J.; Irie, K.; et al. SOD1 (copper/zinc superoxide dismutase) deficiency drives amyloid beta protein oligomerization and memory loss in mouse model of Alzheimer disease. J. Biol. Chem 2011, 286, 44557–44568. [Google Scholar]
- Dumont, M.; Wille, E.; Stack, C.; Calingasan, N.Y.; Beal, M.F.; Lin, M.T. Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer’s disease. FASEB J 2009, 23, 2459–2466. [Google Scholar]
- Murakami, K.; Murata, N.; Ozawa, Y.; Kinoshita, N.; Irie, K.; Shirasawa, T.; Shimizu, T. Vitamin C restores behavioral deficits and amyloid-beta oligomerization without affecting plaque formation in a mouse model of Alzheimer’s disease. J. Alzheimers. Dis 2011, 26, 7–18. [Google Scholar]
- Lee, H.P.; Zhu, X.; Casadesus, G.; Castellani, R.J.; Nunomura, A.; Smith, M.A.; Lee, H.G.; Perry, G. Antioxidant approaches for the treatment of Alzheimer’s disease. Expert Rev. Neurother 2010, 10, 1201–1208. [Google Scholar]
- Vina, J.; Lloret, A.; Giraldo, E.; Badia, M.C.; Alonso, M.D. Antioxidant pathways in Alzheimer’s disease: Possibilities of intervention. Curr. Pharm. Des 2011, 17, 3861–3864. [Google Scholar]
- Dikalova, A.; Clempus, R.; Lassegue, B.; Cheng, G.; McCoy, J.; Dikalov, S.; San Martin, A.; Lyle, A.; Weber, D.S.; Weiss, D.; et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 2005, 112, 2668–2676. [Google Scholar]
- Matsuno, K.; Yamada, H.; Iwata, K.; Jin, D.; Katsuyama, M.; Matsuki, M.; Takai, S.; Yamanishi, K.; Miyazaki, M.; Matsubara, H.; et al. Nox1 is involved in angiotensin II-mediated hypertension: A study in Nox1-deficient mice. Circulation 2005, 112, 2677–2685. [Google Scholar]
- Jackman, K.A.; Miller, A.A.; Drummond, G.R.; Sobey, C.G. Importance of NOX1 for angiotensin II-induced cerebrovascular superoxide production and cortical infarct volume following ischemic stroke. Brain Res 2009, 1286, 215–220. [Google Scholar]
- Zimmerman, M.C.; Lazartigues, E.; Lang, J.A.; Sinnayah, P.; Ahmad, I.M.; Spitz, D.R.; Davisson, R.L. Superoxide mediates the actions of angiotensin II in the central nervous system. Circ. Res 2002, 91, 1038–1045. [Google Scholar]
- Campese, V.M.; Shaohua, Y.; Huiquin, Z. Oxidative stress mediates angiotensin II-dependent stimulation of sympathetic nerve activity. Hypertension 2005, 46, 533–539. [Google Scholar]
- Pang, T.; Wang, J.; Benicky, J.; Sanchez-Lemus, E.; Saavedra, J.M. Telmisartan directly ameliorates the neuronal inflammatory response to IL-1β partly through the JNK/c-Jun and NADPH oxidase pathways. J. Neuroinflamm 2012, 9, 102. [Google Scholar]
- Guimond, M.O.; Gallo-Payet, N. The angiotensin type 2 receptor in brain functions: An update. Int. J. Hypertens 2012, 2012, 351758. [Google Scholar]
- AbdAlla, S.; Lother, H.; el Missiry, A.; Langer, A.; Sergeev, P.; el Faramawy, Y.; Quitterer, U. Angiotensin II AT2 receptor oligomers mediate G-protein dysfunction in an animal model of Alzheimer disease. J. Biol. Chem 2009, 284, 6554–6565. [Google Scholar]
- AbdAlla, S.; Lother, H.; el Missiry, A.; Sergeev, P.; Langer, A.; el Faramawy, Y.; Quitterer, U. Dominant-negative AT2 receptor oligomers induce G-protein arrest and symptoms of neurodegeneration. J. Biol. Chem 2009, 284, 6566–6574. [Google Scholar]
- Wright, J.W.; Harding, J.W. The brain RAS and Alzheimer’s disease. Exp. Neurol 2010, 223, 326–333. [Google Scholar]
- Kehoe, P.G.; Katzov, H.; Andreasen, N.; Gatz, M.; Wilcock, G.K.; Cairns, N.J.; Palmgren, J.; de Faire, U.; Brookes, A.J.; Pedersen, N.L.; et al. Common variants of ACE contribute to variable age-at-onset of Alzheimer’s disease. Hum. Genet 2004, 114, 478–483. [Google Scholar]
- Nirmal, S.; Tripathi, M.; Shastri, S.S.; Sagar, R.; Vivekanandhan, S. Association of Angiotensin-converting enzyme insertion(I)/deletion (D) genotype in Alzheimer’s disease patients of north Indian population. Int. J. Neurosci 2011, 121, 557–561. [Google Scholar]
- Zhang, Z.; Deng, L.; Yu, H.; Shi, Y.; Bai, F.; Xie, C.; Yuan, Y.; Jia, J.; Zhang, Z. Association of angiotensin-converting enzyme functional gene I/D polymorphism with amnestic mild cognitive impairment. Neurosci. Lett 2012, 514, 131–135. [Google Scholar]
- Arregui, A.; Perry, E.K.; Rossor, M.; Tomlinson, B.E. Angiotensin converting enzyme in Alzheimer’s disease increased activity in caudate nucleus and cortical areas. J. Neurochem 1982, 38, 1490–1492. [Google Scholar]
- Savaskan, E.; Hock, C.; Olivieri, G.; Bruttel, S.; Rosenberg, C.; Hulette, C.; Müller-Spahn, F. Cortical alterations of angiotensin converting enzyme, angiotensin II and AT1 receptor in Alzheimer’s dementia. Neurobiol. Aging 2001, 22, 541–546. [Google Scholar]
- Miners, J.S.; Ashby, E.; Van Helmond, Z.; Chalmers, K.A.; Palmer, L.E.; Love, S.; Kehoe, P.G. Angiotensin-converting enzyme (ACE) levels and activity in Alzheimer’s disease, and relationship of perivascular ACE-1 to cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol 2008, 34, 181–193. [Google Scholar]
- Hemming, M.L.; Selkoe, D.J. Amyloid beta-protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. J. Biol. Chem 2005, 280, 37644–37650. [Google Scholar]
- Eckman, E.A.; Adams, S.K.; Troendle, F.J.; Stodola, B.A.; Kahn, M.A.; Faug, A.H.; Xiao, H.D.; Bernstein, K.E.; Eckman, C.B. Regulation of steady-state beta-amyloid levels in the brain by neprilysin and endothelin-converting enzyme but not angiotensin-converting enzyme. J. Biol. Chem 2006, 281, 30471–30478. [Google Scholar]
- Hemming, M.L.; Selkoe, D.J.; Farris, W. Effects of prolonged angiotensin-converting enzyme inhibitor treatment on amyloid beta-protein metabolism in mouse models of Alzheimer disease. Neurobiol. Dis 2007, 26, 273–281. [Google Scholar]
- Ferrington, L.; Palmer, L.E.; Love, S.; Horsburgh, K.J.; Kelly, P.A.; Kehoe, P.G. Angiotensin II-inhibition: Effect on Alzheimer’s pathology in the aged triple transgenic mouse. Am. J. Transl. Res 2012, 4, 151–164. [Google Scholar]
- Khachaturian, A.S.; Zandi, P.P.; Lyketsos, C.G.; Hayden, K.M.; Skoog, I.; Norton, M.C.; Tschanz, J.T.; Mayer, L.S.; Welsh-Bohmer, K.A.; Breitner, J.C. Antihypertensive medication use and incident Alzheimer disease: The Cache County Study. Arch. Neurol 2006, 63, 686–692. [Google Scholar]
- Ohrui, T.; Tomita, N.; Sato-Nakagawa, T.; Matsui, T.; Maruyama, M.; Niwa, K.; Arai, H.; Sasaki, H. Effects of brain-penetrating ACE inhibitors on Alzheimer disease progression. Neurology 2004, 63, 1324–1325. [Google Scholar]
- Sink, K.M.; Leng, X.; Williamson, J.; Kritchevsky, S.B.; Yaffe, K.; Kuller, L.; Yasar, S.; Atkinson, H.; Robbins, M.; Psaty, B.; et al. Angiotensin-converting enzyme inhibitors and cognitive decline in older adults with hypertension: Results from the Cardiovascular Health Study. Arch. Intern. Med 2009, 169, 1195–1202. [Google Scholar]
- Dong, Y.F.; Kataoka, K.; Tokutomi, Y.; Nako, H.; Nakamura, T.; Toyama, K.; Sueta, D.; Koibuchi, N.; Yamamoto, E.; Ogawa, H.; Kim-Mitsuyama, S. Perindopril, a centrally active angiotensin-converting enzyme inhibitor, prevents cognitive impairment in mouse models of Alzheimer’s disease. FASEB J 2011, 25, 2911–2920. [Google Scholar]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar]
- Abd Alla, J.; Langer, A.; Elzahwy, S.S.; Arman-Kalcek, G.; Streichert, T.; Quitterer, U. Angiotensin-converting enzyme inhibition down-regulates the pro-atherogenic chemokine receptor 9 (CCR9)-chemokine ligand 25 (CCL25) axis. J. Biol. Chem 2010, 285, 23496–23505. [Google Scholar]
- Fu, X.; Koller, S.; Abd Alla, J.; Quitterer, U. Inhibition of G-protein-coupled receptor kinase 2 (GRK2) triggers the growth-promoting mitogen-activated protein kinase (MAPK) pathway. J. Biol. Chem 2013, 288, 7738–7755. [Google Scholar]
- Simon, A.M.; de Maturana, R.L.; Ricobaraza, A.; Escribano, L.; Schiapparelli, L.; Cuadrado-Tejedor, M.; Perez-Mediavilla, A.; Avila, J.; Del Rio, J.; Frechilla, D. Early changes in hippocampal Eph receptors precede the onset of memory decline in mouse models of Alzheimer’s disease. J. Alzheimers. Dis 2009, 17, 773–786. [Google Scholar]
- Verret, L.; Mann, E.O.; Hang, G.B.; Barth, A.M.; Cobos, I.; Ho, K.; Devidze, N.; Masliah, E.; Kreitzer, A.C.; Mody, I.; et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 2012, 149, 708–721. [Google Scholar]
- Boda, E.; Hoxha, E.; Pini, A.; Montarolo, F.; Tempia, F. Brain expression of Kv3 subunits during development, adulthood and aging and in a murine model of Alzheimer’s disease. J. Mol. Neurosci 2012, 46, 606–615. [Google Scholar]
- Miners, S.; Ashby, E.; Baig, S.; Harrison, R.; Tayler, H.; Speedy, E.; Prince, J.A.; Love, S.; Kehoe, P.G. Angiotensin-converting enzyme levels and activity in Alzheimer’s disease: Differences in brain and CSF ACE and association with ACE1 genotypes. Am. J. Transl. Res 2009, 1, 163–177. [Google Scholar]
- Hou, D.R.; Wang, Y.; Zhou, L.; Chen, K.; Tian, Y.; Song, Z.; Bao, J.; Yang, Q.D. Altered angiotensin-converting enzyme and its effects on the brain in a rat model of Alzheimer disease. Chin. Med. J. (Engl. ) 2008, 121, 2320–2323. [Google Scholar]
- Zhu, D.; Shi, J.; Zhang, Y.; Wang, B.; Liu, W.; Chen, Z.; Tong, Q. Central angiotensin II stimulation promotes beta amyloid production in Sprague Dawley rats. PLoS One 2011, 6, e16037. [Google Scholar]
- Castegna, A.; Thongboonkerd, V.; Klein, J.B.; Lynn, B.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of nitrated proteins in Alzheimer’s disease brain. J. Neurochem 2003, 85, 1394–1401. [Google Scholar]
- Tan, J.L.; Li, Q.X.; Ciccotosto, G.D.; Crouch, P.J.; Culvenor, J.G.; White, A.R.; Evin, G. Mild oxidative stress induces redistribution of BACE1 in non-apoptotic conditions and promotes the amyloidogenic processing of Alzheimer’s disease amyloid precursor protein. PLoS One 2013, 8, e61246. [Google Scholar]
- Li, H.; Wang, B.; Wang, Z.; Guo, Q.; Tabuchi, K.; Hammer, R.E.; Südhof, T.C.; Zheng, H. Soluble amyloid precursor protein (APP) regulates transthyretin and Klotho gene expression without rescuing the essential function of APP. Proc. Natl. Acad. Sci. USA 2010, 107, 17362–17367. [Google Scholar]
- Ghosal, K.; Vogt, D.L.; Liang, M.; Shen, Y.; Lamb, B.T.; Pimplikar, S.W. Alzheimer’s disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc. Natl. Acad. Sci. USA 2009, 106, 18367–18372. [Google Scholar]
- Müller, T.; Concannon, C.G.; Ward, M.W.; Walsh, C.M.; Tirniceriu, A.L.; Tribl, F.; Kögel, D.; Prehn, J.H.; Egensperger, R. Modulation of gene expression and cytoskeletal dynamics by the amyloid precursor protein intracellular domain (AICD). Mol. Biol. Cell 2007, 18, 201–210. [Google Scholar]
- Da Cunha, V.; Tham, D.M.; Martin-McNulty, B.; Deng, G.; Ho, J.J.; Wilson, D.W.; Rutledge, J.C.; Vergona, R.; Sullivan, M.E.; Wang, Y.X. Enalapril attenuates angiotensin II-induced atherosclerosis and vascular inflammation. Atherosclerosis 2005, 178, 9–17. [Google Scholar]
- Reddy, P.H.; McWeeney, S.; Park, B.S.; Manczak, M.; Gutala, R.V.; Partovi, D.; Jung, Y.; Yau, V.; Searles, R.; Mori, M.; et al. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: Up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum. Mol. Genet 2004, 13, 1225–1240. [Google Scholar]
- Stein, T.D.; Anders, N.J.; DeCarli, C.; Chan, S.L.; Mattson, M.P.; Johnson, J.A. Neutralization of transthyretin reverses the neuroprotective effects of secreted amyloid precursor protein (APP) in APPSW mice resulting in tau phosphorylation and loss of hippocampal neurons: Support for the amyloid hypothesis. J. Neurosci 2004, 24, 7707–7717. [Google Scholar]
- Tan, L.; Wang, X.; Ni, Z.F.; Zhu, X.; Wu, W.; Zhu, L.Q.; Liu, D. A systematic analysis of genomic changes in Tg2576 mice. Mol. Neurobiol 2013, 47, 883–891. [Google Scholar]
- Mei, N.; Fuscoe, J.C.; Lobenhofer, E.K.; Guo, L. Application of microarray-based analysis of gene expression in the field of toxicogenomics. Methods Mol. Biol 2010, 597, 227–241. [Google Scholar]
- Guo, L.; Lobenhofer, E.K.; Wang, C.; Shippy, R.; Harris, S.C.; Zhang, L.; Mei, N.; Chen, T.; Herman, D.; Goodsaid, F.M.; et al. Rat toxicogenomic study reveals analytical consistency across microarray plaforms. Nat. Biotechnol 2006, 24, 1162–1169. [Google Scholar]
- AbdAlla, S.; Lother, H.; Langer, A.; el Faramawy, Y.; Quitterer, U. Factor XIIIA transglutaminase crosslinks AT1 receptor dimers of monocytes at the onset of atherosclerosis. Cell 2004, 119, 343–354. [Google Scholar]
- Butterfield, D.A.; Reed, T.T.; Perluigi, M.; de Marco, C.; Coccia, R.; Keller, J.N.; Markesbery, W.R.; Sultana, R. Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: Implications for the role of nitration in the progression of Alzheimer‘s disease. Brain Res 2007, 1148, 243–248. [Google Scholar]













Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
AbdAlla, S.; Langer, A.; Fu, X.; Quitterer, U. ACE Inhibition with Captopril Retards the Development of Signs of Neurodegeneration in an Animal Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2013, 14, 16917-16942. https://doi.org/10.3390/ijms140816917
AbdAlla S, Langer A, Fu X, Quitterer U. ACE Inhibition with Captopril Retards the Development of Signs of Neurodegeneration in an Animal Model of Alzheimer’s Disease. International Journal of Molecular Sciences. 2013; 14(8):16917-16942. https://doi.org/10.3390/ijms140816917
Chicago/Turabian StyleAbdAlla, Said, Andreas Langer, Xuebin Fu, and Ursula Quitterer. 2013. "ACE Inhibition with Captopril Retards the Development of Signs of Neurodegeneration in an Animal Model of Alzheimer’s Disease" International Journal of Molecular Sciences 14, no. 8: 16917-16942. https://doi.org/10.3390/ijms140816917
APA StyleAbdAlla, S., Langer, A., Fu, X., & Quitterer, U. (2013). ACE Inhibition with Captopril Retards the Development of Signs of Neurodegeneration in an Animal Model of Alzheimer’s Disease. International Journal of Molecular Sciences, 14(8), 16917-16942. https://doi.org/10.3390/ijms140816917