Progressive Changes in Inflammatory and Matrix Adherence of Bronchial Epithelial Cells with Persistent Respiratory Syncytial Virus (RSV) Infection (Progressive Changes in RSV Infection)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
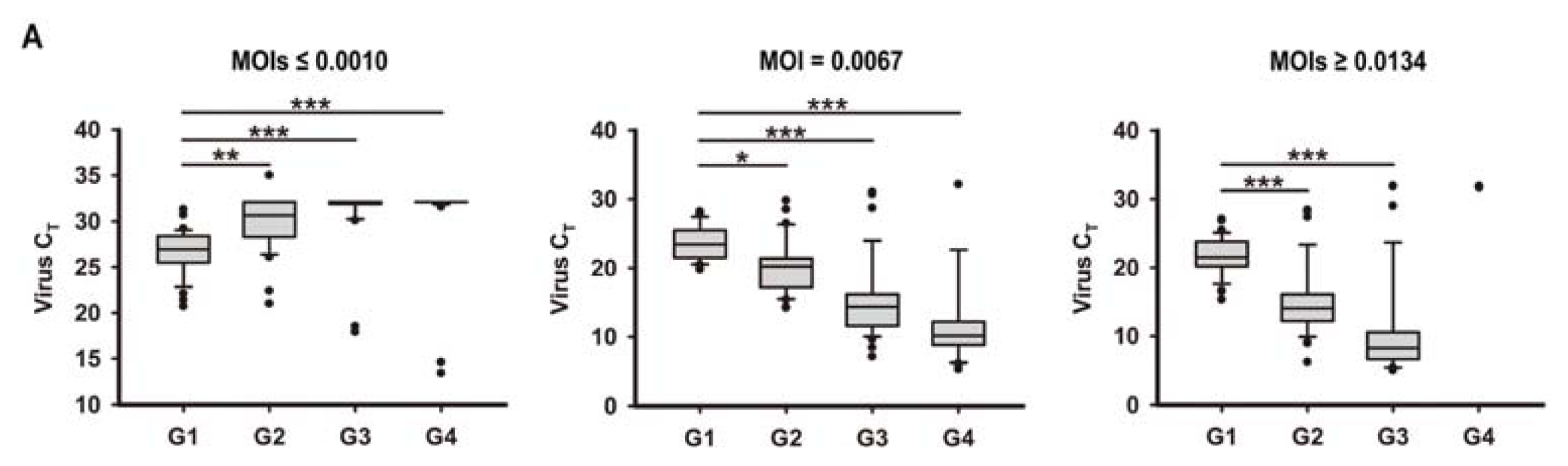
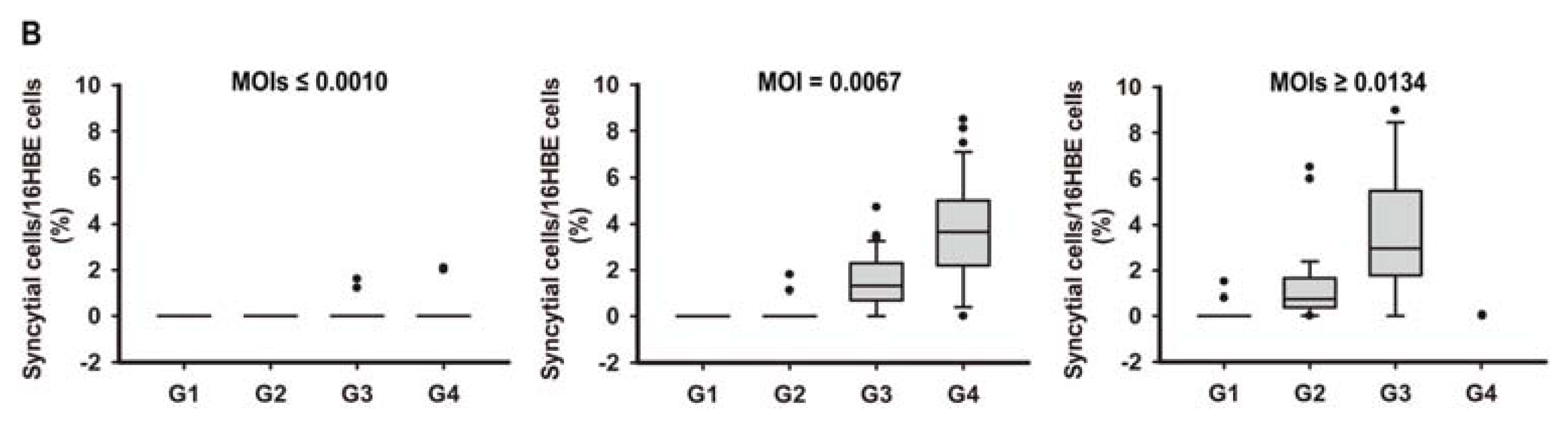
2.1. In Vitro Model of Human Bronchial Epithelial (16HBE) Cells with RSV Infection over Four Generations
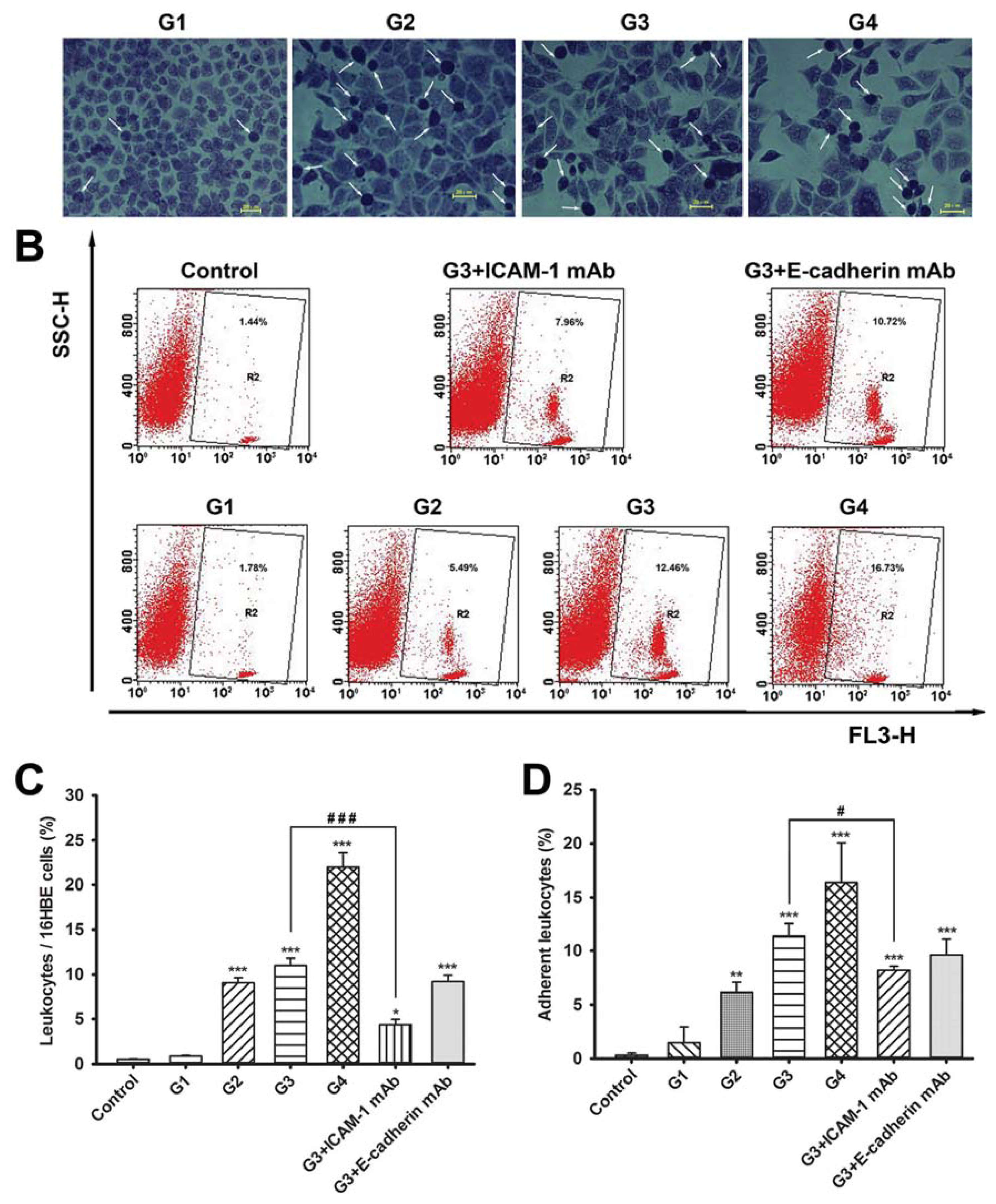
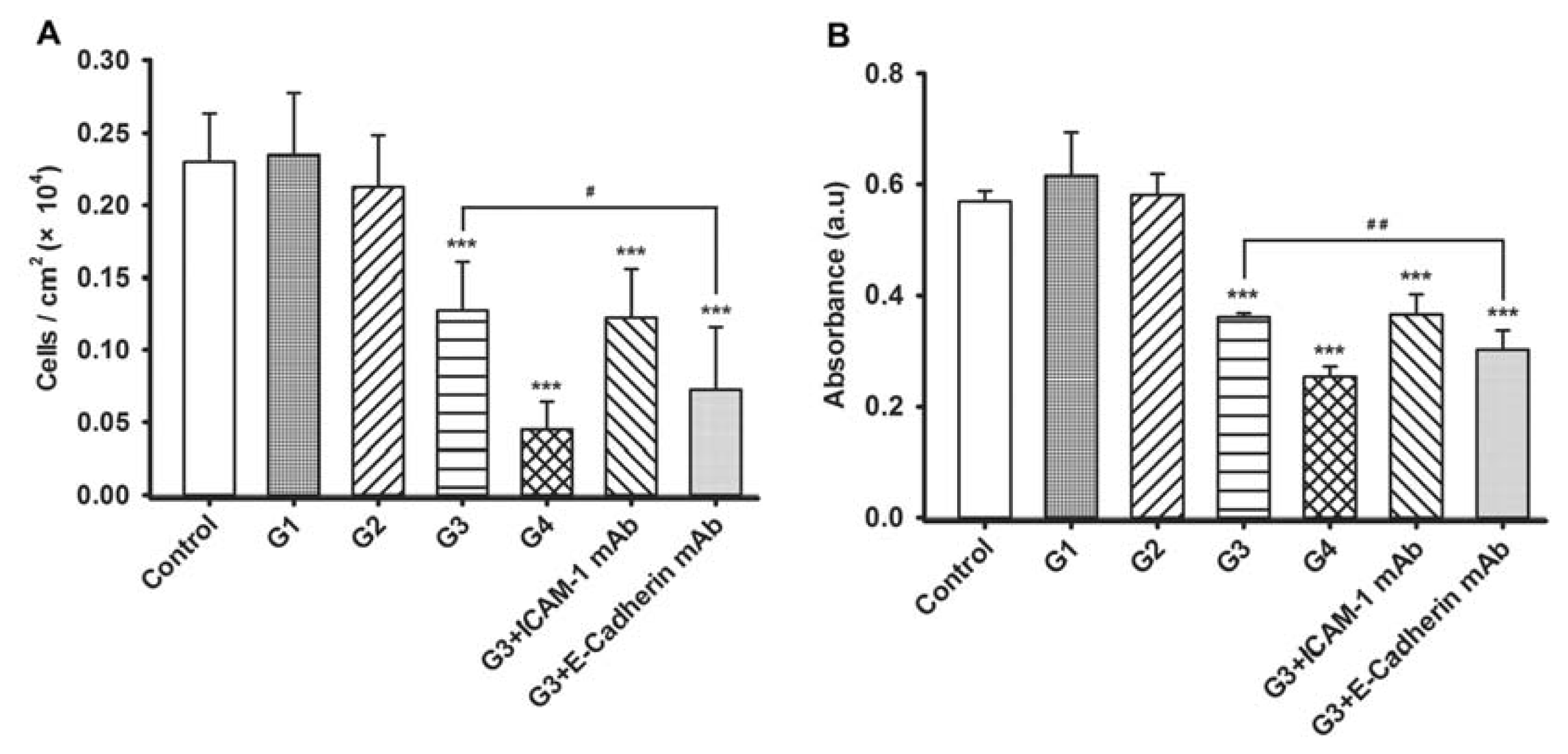
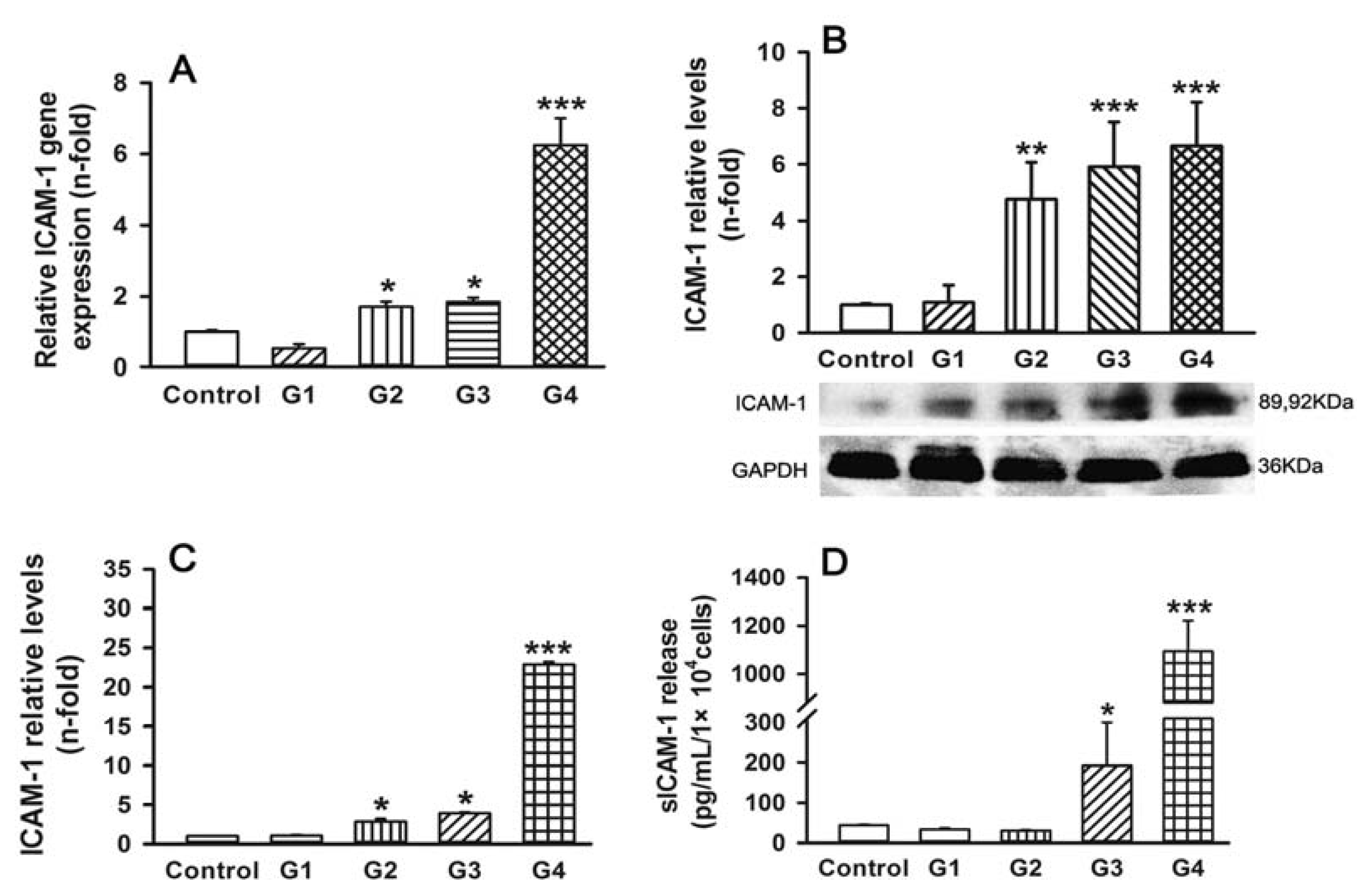
2.2. Dynamic Changes of Leukocyte Adherence to 16HBE Cells with Progressive RSV Infection Involve Intercellular Adhesion Molecule-1 (ICAM-1)
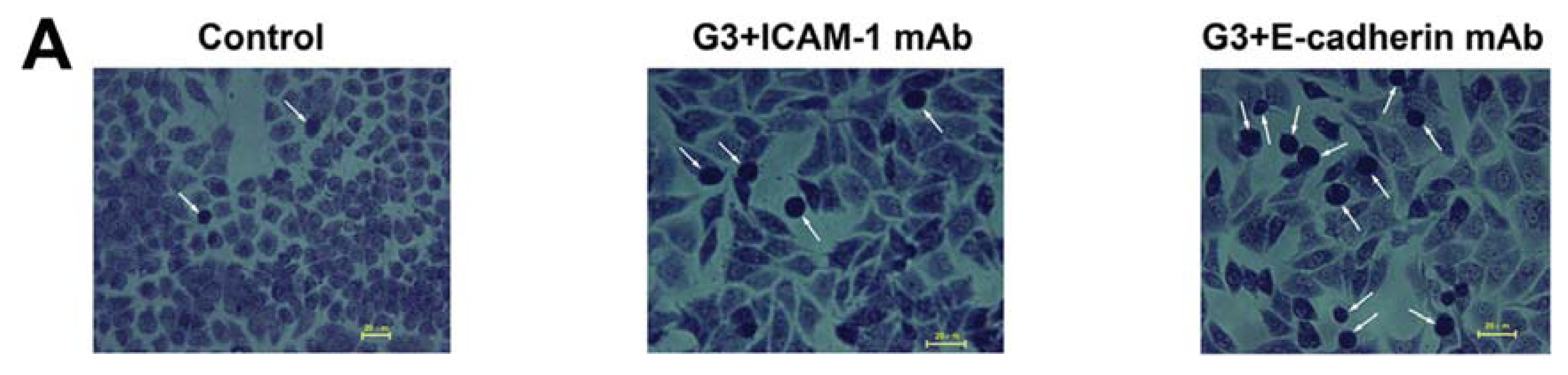
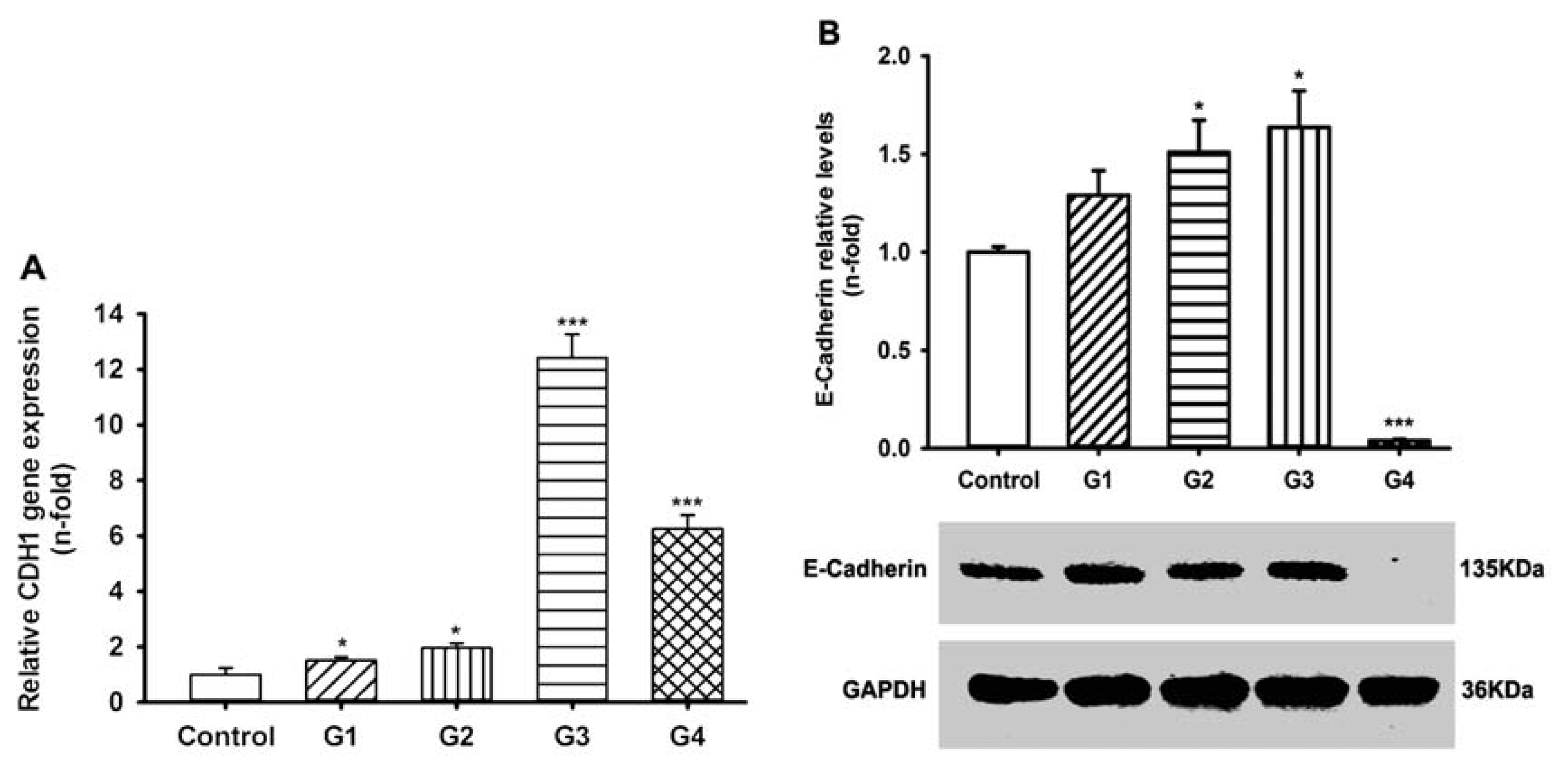
2.3. Dynamic Changes of Extracellular Matrix (ECM) Adherence to 16HBE Cells with Progressive RSV Infection Involve E-cadherin
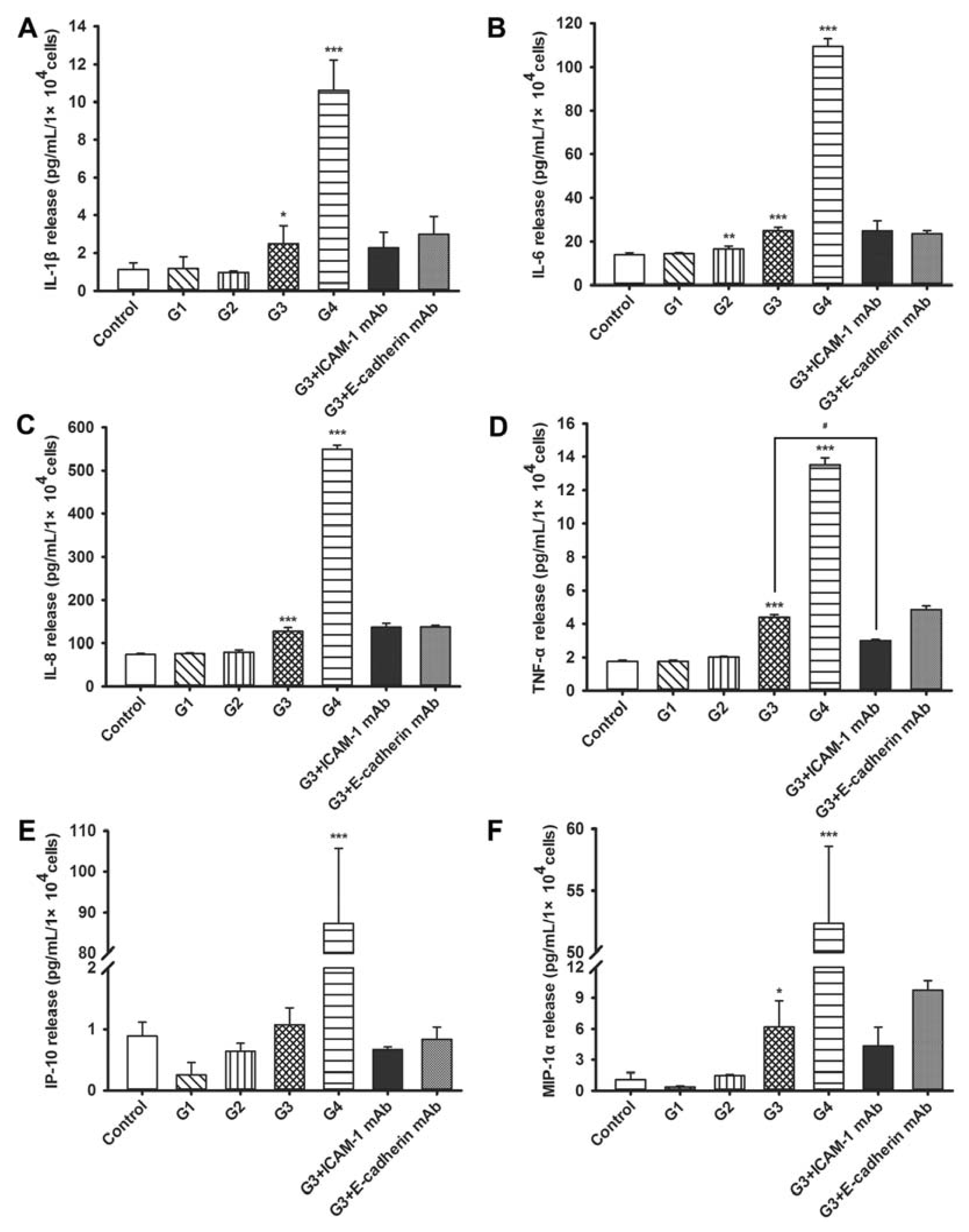
2.4. Dynamic Changes of Cytokine and Chemokine Secretions by 16HBE Cells with Progressive RSV Infection and the Effects of ICAM-1 and E-cadherin Neutralizing mAbs
2.5. Dynamic Changes of Adhesive Molecule Expression or Secretion in 16HBE Cells with Progressive RSV Infection
2.6. Discussion
3. Experimental Section
3.1. RSV Preparation
3.2. 16HBE Cell Culture and RSV Infection
3.3. Assessment of RSV Infection by Real-Time PCR Analysis and Counting the Syncytial Cells
3.4. Isolation of Peripheral Blood Leukocytes
3.5. Leukocyte-16HBE Cell Adherence Assay
3.6. 16HBE Cell-Extracellular Matrix (ECM) Component Adherence Assay
3.7. Assessment of Gene Expression by Real-Time Reverse Transcription (RT)-PCR
3.8. Western Blot Analysis
3.9. Enzyme-Linked Immunosorbent Assay (ELISA)
3.10. Statistical Analysis
3.11. Ethics Statement
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Erickson, J.J.; Gilchuk, P.; Hastings, A.K.; Tollefson, S.J.; Johnson, M.; Downing, M.B.; Boyd, K.L.; Johnson, J.E.; Kim, A.S.; Joyce, S.; et al. Viral acute lower respiratory infections impair CD8+ T cells through PD-1. J. Clin. Invest 2012, 122, 2967–2982. [Google Scholar]
- Shaikh, F.Y.; Cox, R.G.; Lifland, A.W.; Hotard, A.L.; Williams, J.V.; Moore, M.L.; Santangelo, P.J.; Crowe, J.E., Jr. A critical phenylalanine residue in the respiratory syncytial virus fusion protein cytoplasmic tail mediates assembly of internal viral proteins into viral filaments and particles. MBio 2012, 3. [Google Scholar] [CrossRef]
- Caram, L.B.; Chen, J.; Taggart, E.W.; Hillyard, D.R.; She, R.; Polage, C.R.; Twersky, J.; Schmader, K.; Petti, C.A.; Woods, C.W. Respiratory syncytial virus outbreak in a long-term care facility detected using reverse transcriptase polymerase chain reaction: An argument for real-time detection methods. J. Am. Geriatr. Soc 2009, 57, 482–485. [Google Scholar]
- Neu, N.; Plaskett, T.; Hutcheon, G.; Murray, M.; Southwick, K.L.; Saiman, L. Epidemiology of human metapneumovirus in a pediatric long-term care facility. Infect. Control Hosp. Epidemiol 2012, 33, 545–550. [Google Scholar]
- O’Connell, K.; Boo, T.W.; Keady, D.; Niriain, U.; O’Donovan, D.; Commane, M.; Faherty, C.; Cormican, M. Use of palivizumab and infection control measures to control an outbreak of respiratory syncytial virus in a neonatal intensive care unit confirmed by real-time polymerase chain reaction. J. Hosp. Infect 2011, 77, 338–342. [Google Scholar]
- De, A.; Silva, C.; Dias, L.; Baltieri, S.R.; Rodrigues, T.T.; Takagi, N.B.; Richtmann, R. Respiratory syncytial virus outbreak in neonatal intensive care unit: Impact of infection control measures plus palivizumab use. Antimicrob. Resist. Infect. Control 2012, 1. [Google Scholar] [CrossRef]
- Thorburn, K.; Kerr, S.; Taylor, N.; van Saene, H.K. RSV outbreak in a paediatric intensive care unit. J. Hosp. Infect 2004, 57, 194–201. [Google Scholar]
- Bueno, S.M.; Gonzalez, P.A.; Pacheco, R.; Leiva, E.D.; Cautivo, K.M.; Tobar, H.E.; Mora, J.E.; Prado, C.E.; Zuniga, J.P.; Jimenez, J.; et al. Host immunity during RSV pathogenesis. Int. Immunopharmacol 2008, 8, 1320–1329. [Google Scholar]
- Bian, T.; Gibbs, J.D.; Orvell, C.; Imani, F. Respiratory syncytial virus matrix protein induces lung epithelial cell cycle arrest through a p53 dependent pathway. PLoS One 2012, 7, e38052. [Google Scholar]
- Remot, A.; Roux, X.; Dubuquoy, C.; Fix, J.; Bouet, S.; Moudjou, M.; Eleouet, J.F.; Riffault, S.; Petit-Camurdan, A. Nucleoprotein nanostructures combined with adjuvants adapted to the neonatal immune context: A candidate mucosal RSV vaccine. PLoS One 2012, 7, e37722. [Google Scholar]
- Martinez, I.; Lombardia, L.; Herranz, C.; Garcia-Barreno, B.; Dominguez, O.; Melero, J.A. Cultures of HEp-2 cells persistently infected by human respiratory syncytial virus differ in chemokine expression and resistance to apoptosis as compared to lytic infections of the same cell type. Virology 2009, 388, 31–41. [Google Scholar]
- Othumpangat, S.; Gibson, L.F.; Samsell, L.; Piedimonte, G. NGF is an essential survival factor for bronchial epithelial cells during respiratory syncytial virus infection. PLoS One 2009, 4, e6444. [Google Scholar]
- Hashimoto, S.; Matsumoto, K.; Gon, Y.; Ichiwata, T.; Takahashi, N.; Kobayashi, T. Viral infection in asthma. Allergol. Int 2008, 57, 21–31. [Google Scholar]
- Harcourt, J.L.; Caidi, H.; Anderson, L.J.; Haynes, L.M. Evaluation of the Calu-3 cell line as a model of in vitro respiratory syncytial virus infection. J. Virol. Methods 2011, 174, 144–149. [Google Scholar]
- Fink, K.; Duval, A.; Martel, A.; Soucy-Faulkner, A.; Grandvaux, N. Dual role of NOX2 in respiratory syncytial virus- and sendai virus-induced activation of NF-κB in airway epithelial cells. J. Immunol 2008, 180, 6911–6922. [Google Scholar]
- Mackay, C.R. Chemokines: Immunology’s high impact factors. Nat. Immunol 2001, 2, 95–101. [Google Scholar]
- Miyairi, I.; DeVincenzo, J.P. Human genetic factors and respiratory syncytial virus disease severity. Clin. Microbiol. Rev 2008, 21, 686–703. [Google Scholar]
- Garofalo, R.; Mei, F.; Espejo, R.; Ye, G.; Haeberle, H.; Baron, S.; Ogra, P.L.; Reyes, V.E. Respiratory syncytial virus infection of human respiratory epithelial cells up-regulates class I MHC expression through the induction of IFN-β and IL-1α. J. Immunol 1996, 157, 2506–2513. [Google Scholar]
- Behera, A.K.; Matsuse, H.; Kumar, M.; Kong, X.; Lockey, R.F.; Mohapatra, S.S. Blocking intercellular adhesion molecule-1 on human epithelial cells decreases respiratory syncytial virus infection. Biochem. Biophys. Res. Commun 2001, 280, 188–195. [Google Scholar]
- Bitko, V.; Velazquez, A.; Yang, L.; Yang, Y.C.; Barik, S. Transcriptional induction of multiple cytokines by human respiratory syncytial virus requires activation of NF-κB and is inhibited by sodium salicylate and aspirin. Virology 1997, 232, 369–378. [Google Scholar]
- Elias, J.A.; Zheng, T.; Einarsson, O.; Landry, M.; Trow, T.; Rebert, N.; Panuska, J. Epithelial interleukin-11. Regulation by cytokines, respiratory syncytial virus, and retinoic acid. J. Biol. Chem 1994, 269, 22261–22268. [Google Scholar]
- Stark, J.M.; Godding, V.; Sedgwick, J.B.; Busse, W.W. Respiratory syncytial virus infection enhances neutrophil and eosinophil adhesion to cultured respiratory epithelial cells. Roles of CD18 and intercellular adhesion molecule-1. J. Immunol 1996, 156, 4774–4782. [Google Scholar]
- Chen, Y.T.; Gallup, M.; Nikulina, K.; Lazarev, S.; Zlock, L.; Finkbeiner, W.; McNamara, N. Cigarette smoke induces epidermal growth factor receptor-dependent redistribution of apical MUC1 and junctional β-catenin in polarized human airway epithelial cells. Am. J. Pathol 2010, 177, 1255–1264. [Google Scholar]
- Hulpiau, P.; van Roy, F. Molecular evolution of the cadherin superfamily. Int. J. Biochem. Cell Biol 2009, 41, 349–369. [Google Scholar] [Green Version]
- Kaarteenaho, R.; Lappi-Blanco, E.; Lehtonen, S. Epithelial N-cadherin and nuclear β-catenin are up-regulated during early development of human lung. BMC Dev. Biol. 2010, 10. [Google Scholar] [CrossRef]
- Noah, T.L.; Ivins, S.S.; Murphy, P.; Kazachkova, I.; Moats-Staats, B.; Henderson, F.W. Chemokines and inflammation in the nasal passages of infants with respiratory syncytial virus bronchiolitis. Clin. Immunol 2002, 104, 86–95. [Google Scholar]
- Bangham, C.R.; McMichael, A.J. Specific human cytotoxic T cells recognize B-cell lines persistently infected with respiratory syncytial virus. Proc. Natl. Acad. Sci. USA 1986, 83, 9183–9187. [Google Scholar]
- Ramaswamy, M.; Shi, L.; Varga, S.M.; Barik, S.; Behlke, M.A.; Look, D.C. Respiratory syncytial virus nonstructural protein 2 specifically inhibits type I interferon signal transduction. Virology 2006, 344, 328–339. [Google Scholar]
- Newcomb, D.C.; Peebles, R.S., Jr. Bugs and asthma: A different disease? Proc. Am. Thorac. Soc. 2009, 6, 266–271. [Google Scholar]
- Campanini, G.; Percivalle, E.; Baldanti, F.; Rovida, F.; Bertaina, A.; Marchi, A.; Stronati, M.; Gerna, G. Human respiratory syncytial virus (hRSV) RNA quantification in nasopharyngeal secretions identifies the hRSV etiologic role in acute respiratory tract infections of hospitalized infants. J. Clin. Virol 2007, 39, 119–124. [Google Scholar]
- Hansdottir, S.; Monick, M.M.; Lovan, N.; Powers, L.; Gerke, A.; Hunninghake, G.W. Vitamin D decreases respiratory syncytial virus induction of NF-κB-linked chemokines and cytokines in airway epithelium while maintaining the antiviral state. J. Immunol 2010, 184, 965–974. [Google Scholar]
- Zerfaoui, M.; Suzuki, Y.; Naura, A.S.; Hans, C.P.; Nichols, C.; Boulares, A.H. Nuclear translocation of p65 NF-κB is sufficient for VCAM-1, but not ICAM-1, expression in TNF-stimulated smooth muscle cells: Differential requirement for PARP-1 expression and interaction. Cell. Signal 2008, 20, 186–194. [Google Scholar]
- Whiteman, S.C.; Bianco, A.; Knight, R.A.; Spiteri, M.A. Human rhinovirus selectively modulates membranous and soluble forms of its intercellular adhesion molecule-1 (ICAM-1) receptor to promote epithelial cell infectivity. J. Biol. Chem 2003, 278, 11954–11961. [Google Scholar]
- Lidington, E.A.; Moyes, D.L.; McCormack, A.M.; Rose, M.L. A comparison of primary endothelial cells and endothelial cell lines for studies of immune interactions. Transpl. Immunol 1999, 7, 239–246. [Google Scholar]
- Liu, P.; Jamaluddin, M.; Li, K.; Garofalo, R.P.; Casola, A.; Brasier, A.R. Retinoic acid-inducible gene I mediates early antiviral response and Toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J. Virol 2007, 81, 1401–1411. [Google Scholar]
- Dey, N.; Liu, T.; Garofalo, R.P.; Casola, A. TAK1 regulates NF-κB and AP-1 activation in airway epithelial cells following RSV infection. Virology 2011, 418, 93–101. [Google Scholar]
- Saeij, J.P.; Verburg-van Kemenade, L.B.; van Muiswinkel, W.B.; Wiegertjes, G.F. Daily handling stress reduces resistance of carp to Trypanoplasma borreli: In vitro modulatory effects of cortisol on leukocyte function and apoptosis. Dev. Comp. Immunol 2003, 27, 233–245. [Google Scholar]
- Floreani, A.A.; Wyatt, T.A.; Stoner, J.; Sanderson, S.D.; Thompson, E.G.; Allen-Gipson, D.; Heires, A.J. Smoke and C5a induce airway epithelial intercellular adhesion molecule-1 and cell adhesion. Am. J. Respir. Cell Mol. Biol 2003, 29, 472–482. [Google Scholar]
- Schroth, M.K.; Grimm, E.; Frindt, P.; Galagan, D.M.; Konno, S.I.; Love, R.; Gern, J.E. Rhinovirus replication causes RANTES production in primary bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol 1999, 20, 1220–1228. [Google Scholar]








© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, X.; Qin, X.; Xiang, Y.; Liu, H.; Gao, G.; Qin, L.; Liu, C.; Qu, X. Progressive Changes in Inflammatory and Matrix Adherence of Bronchial Epithelial Cells with Persistent Respiratory Syncytial Virus (RSV) Infection (Progressive Changes in RSV Infection). Int. J. Mol. Sci. 2013, 14, 18024-18040. https://doi.org/10.3390/ijms140918024
Liu X, Qin X, Xiang Y, Liu H, Gao G, Qin L, Liu C, Qu X. Progressive Changes in Inflammatory and Matrix Adherence of Bronchial Epithelial Cells with Persistent Respiratory Syncytial Virus (RSV) Infection (Progressive Changes in RSV Infection). International Journal of Molecular Sciences. 2013; 14(9):18024-18040. https://doi.org/10.3390/ijms140918024
Chicago/Turabian StyleLiu, Xiaoai, Xiaoqun Qin, Yang Xiang, Huijun Liu, Ge Gao, Ling Qin, Chi Liu, and Xiangping Qu. 2013. "Progressive Changes in Inflammatory and Matrix Adherence of Bronchial Epithelial Cells with Persistent Respiratory Syncytial Virus (RSV) Infection (Progressive Changes in RSV Infection)" International Journal of Molecular Sciences 14, no. 9: 18024-18040. https://doi.org/10.3390/ijms140918024
APA StyleLiu, X., Qin, X., Xiang, Y., Liu, H., Gao, G., Qin, L., Liu, C., & Qu, X. (2013). Progressive Changes in Inflammatory and Matrix Adherence of Bronchial Epithelial Cells with Persistent Respiratory Syncytial Virus (RSV) Infection (Progressive Changes in RSV Infection). International Journal of Molecular Sciences, 14(9), 18024-18040. https://doi.org/10.3390/ijms140918024