Single-Chain Fragment Variable Passive Immunotherapies for Neurodegenerative Diseases

{kind=link}
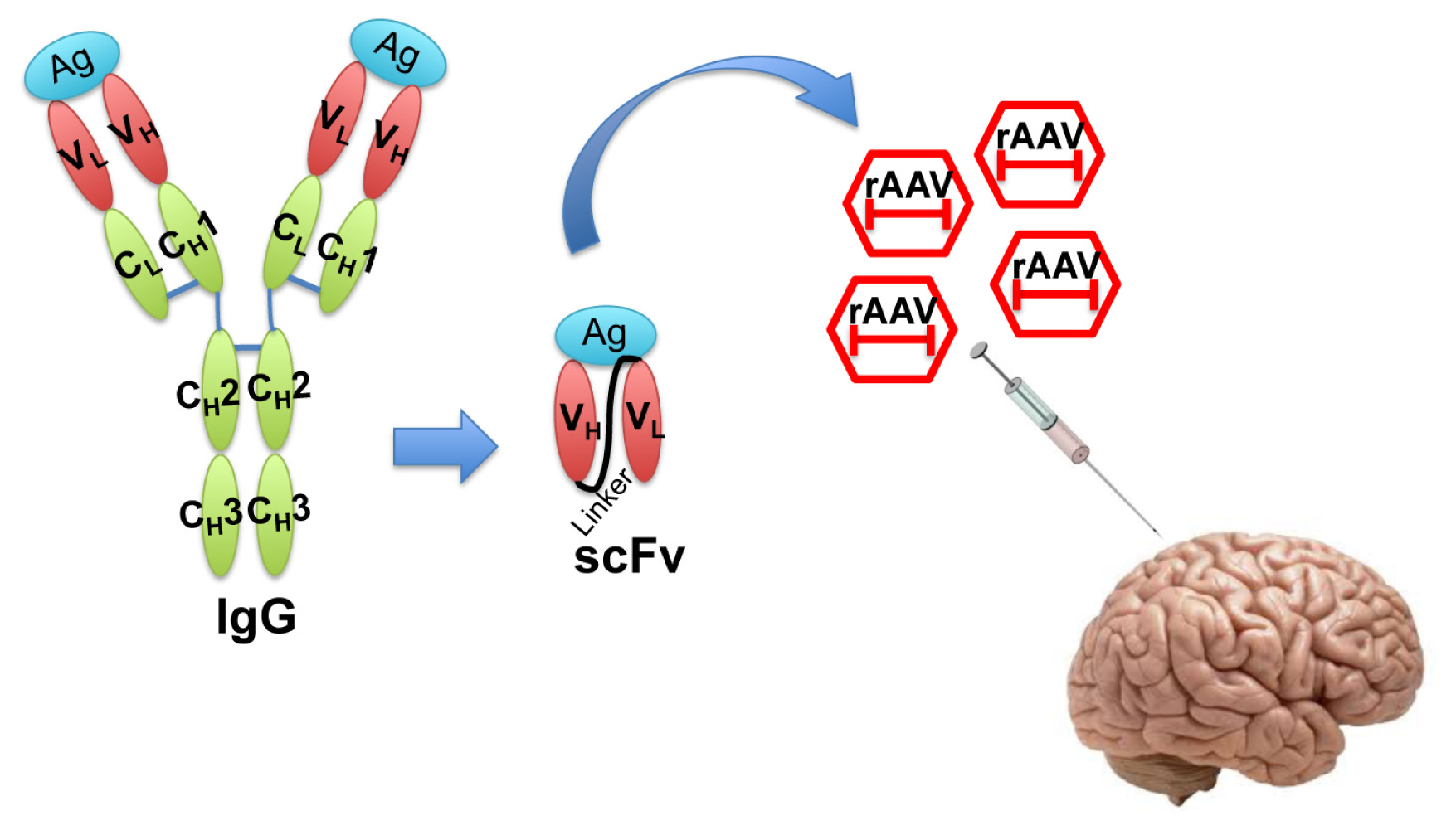{kind=link}
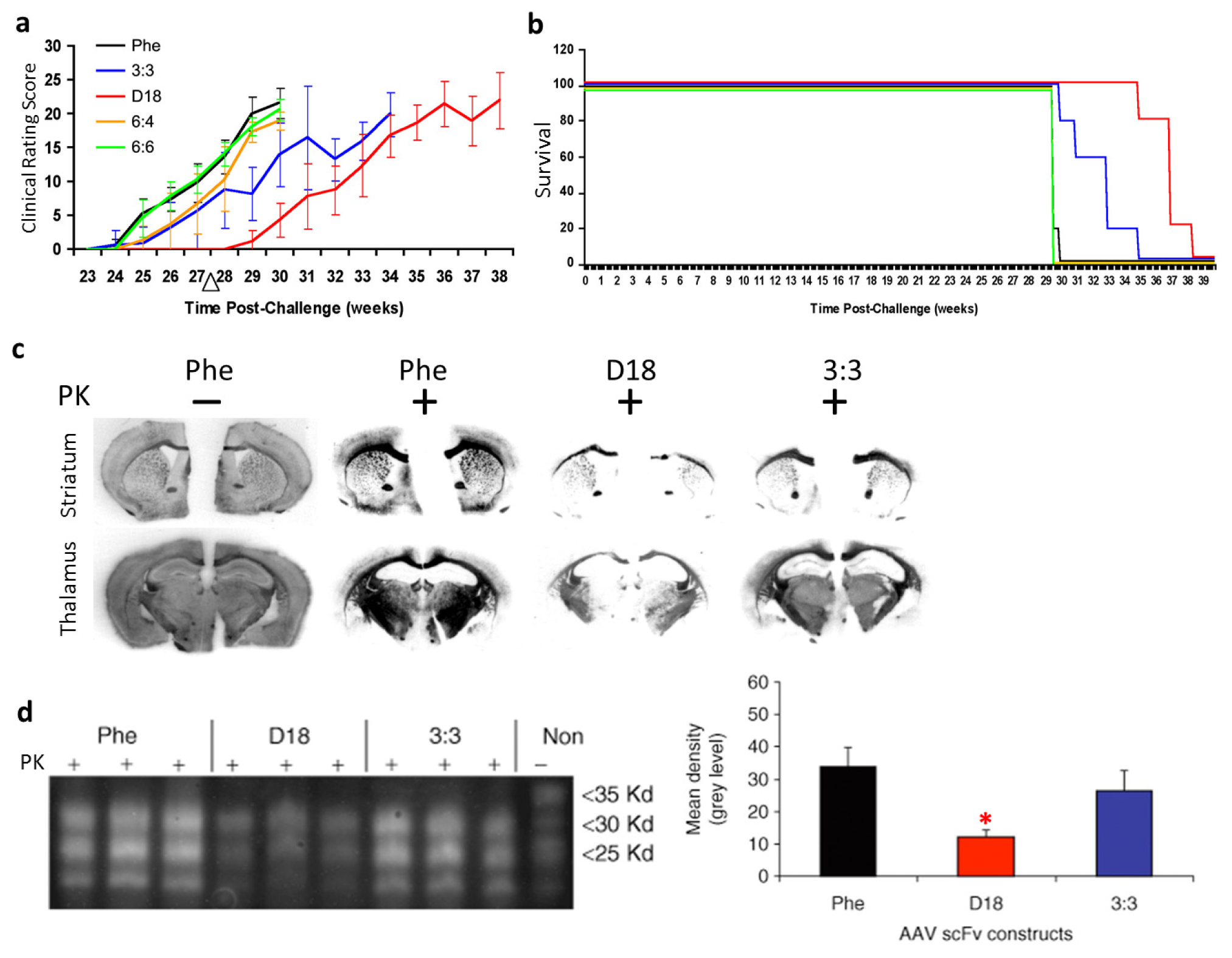{kind=link}
{kind=link}
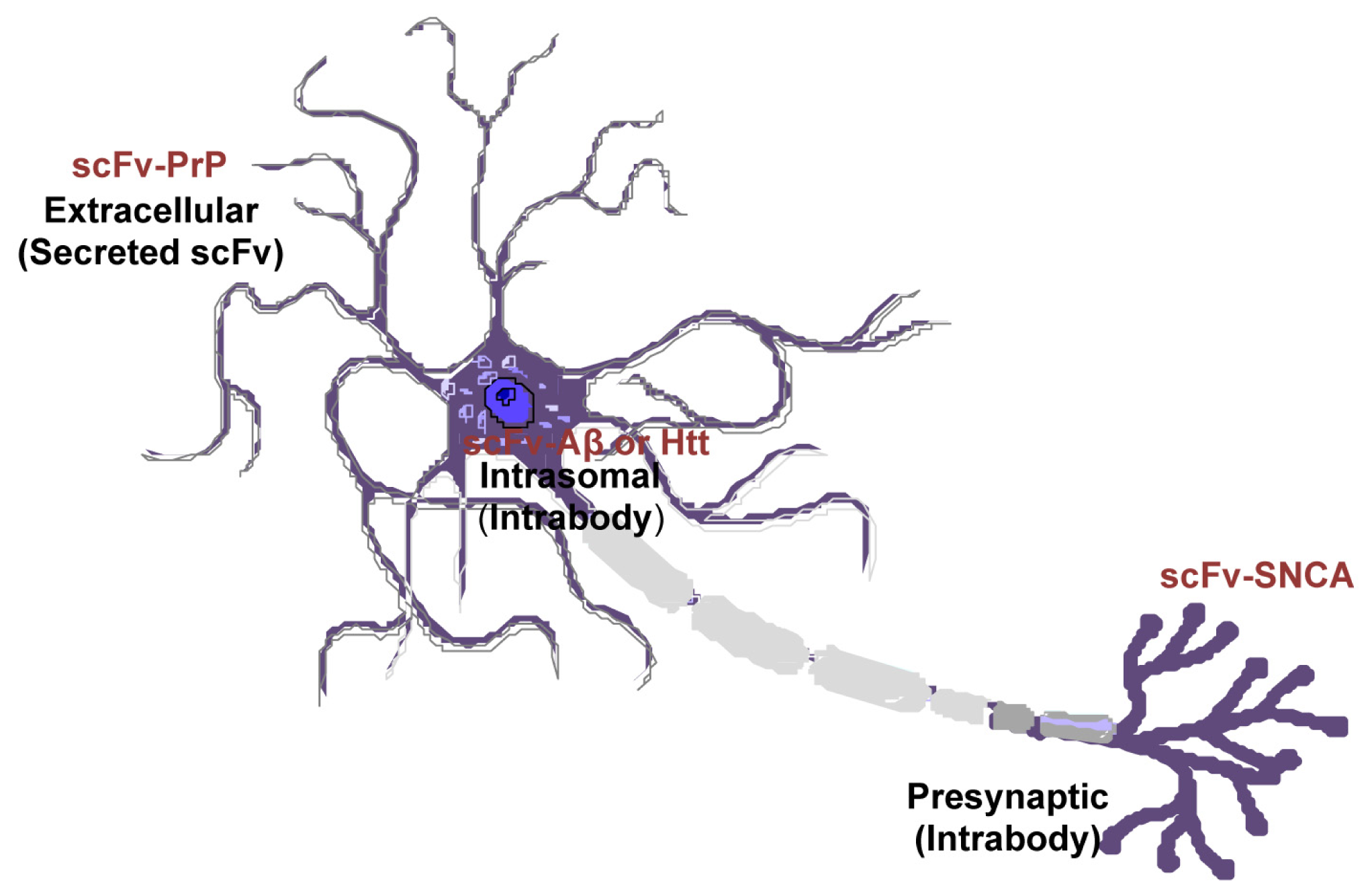{kind=link}
Abstract
:1. Introduction
2. scFv Therapy in Prion Diseases
3. scFv Therapy in Alzheimer’s Diseases
4. scFv Therapy in Huntington’s Diseases
5. scFv Therapy in Parkinson’s Diseases
6. Improvements of scFv for Neuronal Disorder
7. Right Target at the Right Place
8. Conclusions
Conflicts of Interest
References
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar]
- Gilbert, B.J. The role of amyloid beta in the pathogenesis of Alzheimer’s disease. J. Clin. Pathol 2013, 66, 362–366. [Google Scholar]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. (Berl) 2003, 81, 678–699. [Google Scholar]
- Polymenidou, M.; Cleveland, D.W. Prion-like spread of protein aggregates in neurodegeneration. J. Exp. Med 2012, 209, 889–893. [Google Scholar]
- El-Agnaf, O.M.; Irvine, G.B. Review: Formation and properties of amyloid-like fibrils derived from alpha-synuclein and related proteins. J. Struct. Biol 2000, 130, 300–309. [Google Scholar]
- Arrasate, M.; Finkbeiner, S. Protein aggregates in Huntington’s disease. Exp. Neurol 2012, 238, 1–11. [Google Scholar]
- Gadad, B.S.; Britton, G.B.; Rao, K.S. Targeting oligomers in neurodegenerative disorders: Lessons from alpha-synuclein, tau, and amyloid-beta peptide. J. Alzheimers Dis 2011, 24, 223–232. [Google Scholar]
- Ghadge, G.D.; Pavlovic, J.D.; Koduvayur, S.P.; Kay, B.K.; Roos, R.P. Single chain variable fragment antibodies block aggregation and toxicity induced by familial ALS-linked mutant forms of SOD1. Neurobiol. disease 2013, 56, 74–78. [Google Scholar]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar]
- McKinley, M.P.; Bolton, D.C.; Prusiner, S.B. A protease-resistant protein is a structural component of the scrapie prion. Cell 1983, 35, 57–62. [Google Scholar]
- Fernandez-Busquets, X.; de Groot, N.S.; Fernandez, D.; Ventura, S. Recent structural and computational insights into conformational diseases. Curr. Med. Chem 2008, 15, 1336–1349. [Google Scholar]
- Invernizzi, G.; Papaleo, E.; Sabate, R.; Ventura, S. Protein aggregation: Mechanisms and functional consequences. Int. J. Biochem. Cell Biol 2012, 44, 1541–1554. [Google Scholar]
- Peretz, D.; Williamson, R.A.; Kaneko, K.; Vergara, J.; Leclerc, E.; Schmitt-Ulms, G.; Mehlhorn, I.R.; Legname, G.; Wormald, M.R.; Rudd, P.M.; et al. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature 2001, 412, 739–743. [Google Scholar]
- Zhu, M.; Li, J.; Fink, A.L. The association of alpha-synuclein with membranes affects bilayer structure, stability, and fibril formation. J. Biol. Chem 2003, 278, 40186–40197. [Google Scholar]
- Walsh, D.M.; Selkoe, D.J. Oligomers on the brain: The emerging role of soluble protein aggregates in neurodegeneration. Protein Pept. Lett 2004, 11, 213–228. [Google Scholar]
- Miller, T.W.; Messer, A. Intrabody applications in neurological disorders: Progress and future prospects. Mol. Ther 2005, 12, 394–401. [Google Scholar]
- Federoff, H.J. Development of vaccination approaches for the treatment of neurological diseases. J. Comp. Neurol 2009, 515, 4–14. [Google Scholar]
- Wang, Y.J.; Zhou, H.D.; Zhou, X.F. Modified immunotherapies against Alzheimer’s disease: Toward safer and effective amyloid clearance. J. Alzheimers Dis 2010, 21, 1065–1075. [Google Scholar]
- Ahmad, Z.A.; Yeap, S.K.; Ali, A.M.; Ho, W.Y.; Alitheen, N.B.; Hamid, M. scFv antibody: Principles and clinical application. Clin. Dev. Immunol 2012, 2012, 980250. [Google Scholar]
- Butler, D.C.; McLear, J.A.; Messer, A. Engineered antibody therapies to counteract mutant huntingtin and related toxic intracellular proteins. Prog. Neurobiol 2012, 97, 190–204. [Google Scholar]
- Robert, R.; Wark, K.L. Engineered antibody approaches for Alzheimer’s disease immunotherapy. Arch. Biochem. Biophys 2012, 526, 132–138. [Google Scholar]
- Roettger, Y.; Du, Y.; Bacher, M.; Zerr, I.; Dodel, R.; Bach, J.P. Immunotherapy in prion disease. Nat. Rev. Neurol 2013, 9, 98–105. [Google Scholar]
- Malone, J.; Sullivan, M.A. Analysis of antibody selection by phage display utilizing anti-phenobarbital antibodies. J. Mol. Recognit 1996, 9, 738–745. [Google Scholar]
- Campana, V.; Zentilin, L.; Mirabile, I.; Kranjc, A.; Casanova, P.; Giacca, M.; Prusiner, S.B.; Legname, G.; Zurzolo, C. Development of antibody fragments for immunotherapy of prion diseases. Biochem. J 2009, 418, 507–515. [Google Scholar]
- Donofrio, G.; Heppner, F.L.; Polymenidou, M.; Musahl, C.; Aguzzi, A. Paracrine inhibition of prion propagation by anti-PrP single-chain Fv miniantibodies. J. Virol 2005, 79, 8330–8338. [Google Scholar]
- Filesi, I.; Cardinale, A.; Mattei, S.; Biocca, S. Selective re-routing of prion protein to proteasomes and alteration of its vesicular secretion prevent PrP(Sc) formation. J. Neurochem 2007, 101, 1516–1526. [Google Scholar]
- Verma, R.; Boleti, E.; George, A.J. Antibody engineering: Comparison of bacterial, yeast, insect and mammalian expression systems. J. Immunol. Methods 1998, 216, 165–181. [Google Scholar]
- Prusiner, S.B. Shattuck lecture-neurodegenerative diseases and prions. N. Engl. J. Med 2001, 344, 1516–1526. [Google Scholar]
- Cardinale, A.; Filesi, I.; Vetrugno, V.; Pocchiari, M.; Sy, M.S.; Biocca, S. Trapping prion protein in the endoplasmic reticulum impairs PrPC maturation and prevents PrPSc accumulation. J. Biol. Chem 2005, 280, 685–694. [Google Scholar]
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E.; et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar]
- Turk, E.; Teplow, D.B.; Hood, L.E.; Prusiner, S.B. Purification and properties of the cellular and scrapie hamster prion proteins. Eur. J. Biochem 1988, 176, 21–30. [Google Scholar]
- Leclerc, E.; Liemann, S.; Wildegger, G.; Vetter, S.W.; Nilsson, F. Selection and characterization of single chain Fv fragments against murine recombinant prion protein from a synthetic human antibody phage display library. Hum. Antibodies 2000, 9, 207–214. [Google Scholar]
- Heppner, F.L.; Musahl, C.; Arrighi, I.; Klein, M.A.; Rulicke, T.; Oesch, B.; Zinkernagel, R.M.; Kalinke, U.; Aguzzi, A. Prevention of scrapie pathogenesis by transgenic expression of anti-prion protein antibodies. Science 2001, 294, 178–182. [Google Scholar]
- Luginbuhl, B.; Kanyo, Z.; Jones, R.M.; Fletterick, R.J.; Prusiner, S.B.; Cohen, F.E.; Williamson, R.A.; Burton, D.R.; Pluckthun, A. Directed evolution of an anti-prion protein scFv fragment to an affinity of 1 pM and its structural interpretation. J. Mol. Biol 2006, 363, 75–97. [Google Scholar]
- Flego, M.; Ascione, A.; Zamboni, S.; Dupuis, M.L.; Imperiale, V.; Cianfriglia, M. Generation of human scFvs antibodies recognizing a prion protein epitope expressed on the surface of human lymphoblastoid cells. BMC Biotechnol 2007, 7, 38. [Google Scholar]
- Miyamoto, K.; Shimamoto, T.; Aosasa, M.; Kimura, S.; Nakamura, N.; Okubo, Y.; Yokoyama, T.; Horiuchi, H.; Furusawa, S.; Matsuda, H. Development of recombinant chicken IgY from single chain fragment of variable region for diagnosis of BSE. Biologicals 2007, 35, 31–34. [Google Scholar]
- Padiolleau-Lefevre, S.; Alexandrenne, C.; Dkhissi, F.; Clement, G.; Essono, S.; Blache, C.; Couraud, J.Y.; Wijkhuisen, A.; Boquet, D. Expression and detection strategies for an scFv fragment retaining the same high affinity than Fab and whole antibody: Implications for therapeutic use in prion diseases. Mol. Immunol 2007, 44, 1888–1896. [Google Scholar]
- Polymenidou, M.; Moos, R.; Scott, M.; Sigurdson, C.; Shi, Y.Z.; Yajima, B.; Hafner-Bratkovic, I.; Jerala, R.; Hornemann, S.; Wuthrich, K.; et al. The POM monoclonals: A comprehensive set of antibodies to non-overlapping prion protein epitopes. PLoS One 2008, 3, e3872. [Google Scholar]
- Wuertzer, C.A.; Sullivan, M.A.; Qiu, X.; Federoff, H.J. CNS delivery of vectored prion-specific single-chain antibodies delays disease onset. Mol. Ther 2008, 16, 481–486. [Google Scholar]
- Muller-Schiffmann, A.; Petsch, B.; Leliveld, S.R.; Muyrers, J.; Salwierz, A.; Mangels, C.; Schwarzinger, S.; Riesner, D.; Stitz, L.; Korth, C. Complementarity determining regions of an anti-prion protein scFv fragment orchestrate conformation specificity and antiprion activity. Mol. Immunol 2009, 46, 532–540. [Google Scholar]
- Shimizu, Y.; Kaku-Ushiki, Y.; Iwamaru, Y.; Muramoto, T.; Kitamoto, T.; Yokoyama, T.; Mohri, S.; Tagawa, Y. A novel anti-prion protein monoclonal antibody and its single-chain fragment variable derivative with ability to inhibit abnormal prion protein accumulation in cultured cells. Microbiol. Immunol 2010, 54, 112–121. [Google Scholar]
- Skrlj, N.; Serbec, V.C.; Dolinar, M. Single-chain Fv antibody fragments retain binding properties of the monoclonal antibody raised against peptide P1 of the human prion protein. Appl. Biochem. Biotechnol 2010, 160, 1808–1821. [Google Scholar]
- Fujita, K.; Yamaguchi, Y.; Mori, T.; Muramatsu, N.; Miyamoto, T.; Yano, M.; Miyata, H.; Ootsuyama, A.; Sawada, M.; Matsuda, H.; et al. Effects of a brain-engraftable microglial cell line expressing anti-prion scFv antibodies on survival times of mice infected with scrapie prions. Cell Mol. Neurobiol 2011, 31, 999–1008. [Google Scholar]
- Petsch, B.; Muller-Schiffmann, A.; Lehle, A.; Zirdum, E.; Prikulis, I.; Kuhn, F.; Raeber, A.J.; Ironside, J.W.; Korth, C.; Stitz, L. Biological effects and use of PrPSc- and PrP-specific antibodies generated by immunization with purified full-length native mouse prions. J. Virol 2011, 85, 4538–4546. [Google Scholar]
- Skrlj, N.; Vranac, T.; Popovic, M.; Curin Serbec, V.; Dolinar, M. Specific binding of the pathogenic prion isoform: Development and characterization of a humanized single-chain variable antibody fragment. PLoS One 2011, 6, e15783. [Google Scholar]
- Zhang, X.; Sun, X.X.; Xue, D.; Liu, D.G.; Hu, X.Y.; Zhao, M.; Yang, S.G.; Yang, Y.; Xia, Y.J.; Wang, Y.; Liu, R.T. Conformation-dependent scFv antibodies specifically recognize the oligomers assembled from various amyloids and show colocalization of amyloid fibrils with oligomers in patients with amyloidoses. Biochim. Biophys. Acta 2011, 1814, 1703–1712. [Google Scholar]
- Kubota, T.; Hamazoe, Y.; Hashiguchi, S.; Ishibashi, D.; Akasaka, K.; Nishida, N.; Katamine, S.; Sakaguchi, S.; Kuroki, R.; Nakashima, T.; et al. Direct evidence of generation and accumulation of beta-sheet-rich prion protein in scrapie-infected neuroblastoma cells with human IgG1 antibody specific for beta-form prion protein. J. Biol. Chem 2012, 287, 14023–14039. [Google Scholar]
- Moda, F.; Vimercati, C.; Campagnani, I.; Ruggerone, M.; Giaccone, G.; Morbin, M.; Zentilin, L.; Giacca, M.; Zucca, I.; Legname, G.; Tagliavini, F. Brain delivery of AAV9 expressing an anti-PrP monovalent antibody delays prion disease in mice. Prion 2012, 6, 383–390. [Google Scholar]
- Kalinke, U.; Krebber, A.; Krebber, C.; Bucher, E.; Pluckthun, A.; Zinkernagel, R.M.; Hengartner, H. Monovalent single-chain Fv fragments and bivalent miniantibodies bound to vesicular stomatitis virus protect against lethal infection. Eur. J. Immunol 1996, 26, 2801–2806. [Google Scholar]
- Krebber, A.; Bornhauser, S.; Burmester, J.; Honegger, A.; Willuda, J.; Bosshard, H.R.; Pluckthun, A. Reliable cloning of functional antibody variable domains from hybridomas and spleen cell repertoires employing a reengineered phage display system. J. Immunol. Methods 1997, 201, 35–55. [Google Scholar]
- White, A.R.; Enever, P.; Tayebi, M.; Mushens, R.; Linehan, J.; Brandner, S.; Anstee, D.; Collinge, J.; Hawke, S. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 2003, 422, 80–83. [Google Scholar]
- Gauczynski, S.; Hundt, C.; Leucht, C.; Weiss, S. Interaction of prion proteins with cell surface receptors, molecular chaperones, and other molecules. Advan. Prot. Chem 2001, 57, 229–272. [Google Scholar]
- Gauczynski, S.; Nikles, D.; El-Gogo, S.; Papy-Garcia, D.; Rey, C.; Alban, S.; Barritault, D.; Lasmezas, C.I.; Weiss, S. The 37-kDa/67-kDa laminin receptor acts as a receptor for infectious prions and is inhibited by polysulfated glycanes. J. Infect. Dis 2006, 194, 702–709. [Google Scholar]
- Ludewigs, H.; Zuber, C.; Vana, K.; Nikles, D.; Zerr, I.; Weiss, S. Therapeutic approaches for prion disorders. Expert Rev. Anti. Infect. Ther 2007, 5, 613–630. [Google Scholar]
- Vana, K.; Weiss, S. A trans-dominant negative 37kDa/67kDa laminin receptor mutant impairs PrP(Sc) propagation in scrapie-infected neuronal cells. J. Mol. Biol. 2006, 358, 57–66. [Google Scholar]
- Morel, E.; Andrieu, T.; Casagrande, F.; Gauczynski, S.; Weiss, S.; Grassi, J.; Rousset, M.; Dormont, D.; Chambaz, J. Bovine prion is endocytosed by human enterocytes via the 37 kDa/67 kDa laminin receptor. Amer. J. Pathol 2005, 167, 1033–1042. [Google Scholar]
- Zuber, C.; Knackmuss, S.; Rey, C.; Reusch, U.; Rottgen, P.; Frohlich, T.; Arnold, G.J.; Pace, C.; Mitteregger, G.; Kretzschmar, H.A.; et al. Single chain Fv antibodies directed against the 37 kDa/67 kDa laminin receptor as therapeutic tools in prion diseases. Mol. Immunol 2008, 45, 144–151. [Google Scholar]
- Zuber, C.; Mitteregger, G.; Schuhmann, N.; Rey, C.; Knackmuss, S.; Rupprecht, W.; Reusch, U.; Pace, C.; Little, M.; Kretzschmar, H.A.; et al. Delivery of single-chain antibodies (scFvs) directed against the 37/67 kDa laminin receptor into mice via recombinant adeno-associated viral vectors for prion disease gene therapy. J. Gen. Virol 2008, 89, 2055–2061. [Google Scholar]
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement 2007, 3, 186–191. [Google Scholar]
- Sosa-Ortiz, A.L.; Acosta-Castillo, I.; Prince, M.J. Epidemiology of dementias and Alzheimer’s disease. Arch. Med. Res 2012, 43, 600–608. [Google Scholar]
- Robert, R.; Dolezal, O.; Waddington, L.; Hattarki, M.K.; Cappai, R.; Masters, C.L.; Hudson, P.J.; Wark, K.L. Engineered antibody intervention strategies for Alzheimer’s disease and related dementias by targeting amyloid and toxic oligomers. Protein Eng. Des. Sel 2009, 22, 199–208. [Google Scholar]
- Pfeifer, M.; Boncristiano, S.; Bondolfi, L.; Stalder, A.; Deller, T.; Staufenbiel, M.; Mathews, P.M.; Jucker, M. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science 2002, 298, 1379. [Google Scholar]
- Racke, M.M.; Boone, L.I.; Hepburn, D.L.; Parsadainian, M.; Bryan, M.T.; Ness, D.K.; Piroozi, K.S.; Jordan, W.H.; Brown, D.D.; Hoffman, W.P.; et al. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J. Neurosci 2005, 25, 629–636. [Google Scholar]
- Frenkel, D.; Katz, O.; Solomon, B. Immunization against Alzheimer’s beta -amyloid plaques via EFRH phage administration. Proc. Natl. Acad. Sci. USA 2000, 97, 11455–11459. [Google Scholar]
- Liu, R.; Yuan, B.; Emadi, S.; Zameer, A.; Schulz, P.; McAllister, C.; Lyubchenko, Y.; Goud, G.; Sierks, M.R. Single chain variable fragments against beta-amyloid (Abeta) can inhibit Abeta aggregation and prevent abeta-induced neurotoxicity. Biochemistry 2004, 43, 6959–6967. [Google Scholar]
- Fukuchi, K.; Tahara, K.; Kim, H.D.; Maxwell, J.A.; Lewis, T.L.; Accavitti-Loper, M.A.; Kim, H.; Ponnazhagan, S.; Lalonde, R. Anti-Abeta single-chain antibody delivery via adeno-associated virus for treatment of Alzheimer’s disease. Neurobiol. Dis 2006, 23, 502–511. [Google Scholar]
- Levites, Y.; Jansen, K.; Smithson, L.A.; Dakin, R.; Holloway, V.M.; Das, P.; Golde, T.E. Intracranial adeno-associated virus-mediated delivery of anti-pan amyloid beta, amyloid beta40, and amyloid beta42 single-chain variable fragments attenuates plaque pathology in amyloid precursor protein mice. J. Neurosci 2006, 26, 11923–11928. [Google Scholar]
- Wang, Y.J.; Pollard, A.; Zhong, J.H.; Dong, X.Y.; Wu, X.B.; Zhou, H.D.; Zhou, X.F. Intramuscular delivery of a single chain antibody gene reduces brain Abeta burden in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2009, 30, 364–376. [Google Scholar]
- Ryan, D.A.; Mastrangelo, M.A.; Narrow, W.C.; Sullivan, M.A.; Federoff, H.J.; Bowers, W.J. Abeta-directed single-chain antibody delivery via a serotype-1 AAV vector improves learning behavior and pathology in Alzheimer’s disease mice. Mol. Ther 2010, 18, 1471–1481. [Google Scholar]
- Wang, X.P.; Zhang, J.H.; Wang, Y.J.; Feng, Y.; Zhang, X.; Sun, X.X.; Li, J.L.; Du, X.T.; Lambert, M.P.; Yang, S.G.; Zhao, M.; Klein, W.L.; Liu, R.T. Conformation-dependent single-chain variable fragment antibodies specifically recognize beta-amyloid oligomers. FEBS Lett 2009, 583, 579–584. [Google Scholar]
- Finkbeiner, S. Huntington’s Disease. Cold Spring Harb. Perspect. Biol 2011, 3, a007476. [Google Scholar]
- La Spada, A.R.; Taylor, J.P. Repeat expansion disease: Progress and puzzles in disease pathogenesis. Nat. Rev. Genet 2010, 11, 247–258. [Google Scholar]
- Wexler, N.S.; Lorimer, J.; Porter, J.; Gomez, F.; Moskowitz, C.; Shackell, E.; Marder, K.; Penchaszadeh, G.; Roberts, S.A.; Gayan, J.; et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc. Natl. Acad. Sci. USA 2004, 101, 3498–3503. [Google Scholar]
- Lecerf, J.M.; Shirley, T.L.; Zhu, Q.; Kazantsev, A.; Amersdorfer, P.; Housman, D.E.; Messer, A.; Huston, J.S. Human single-chain Fv intrabodies counteract in situ huntingtin aggregation in cellular models of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 4764–4769. [Google Scholar]
- Murphy, R.C.; Messer, A. A single-chain Fv intrabody provides functional protection against the effects of mutant protein in an organotypic slice culture model of Huntington’s disease. Mol. Brain Res 2004, 121, 141–145. [Google Scholar]
- Miller, T.W.; Zhou, C.; Gines, S.; MacDonald, M.E.; Mazarakis, N.D.; Bates, G.P.; Huston, J.S.; Messer, A. A human single-chain Fv intrabody preferentially targets amino-terminal Huntingtin’s fragments in striatal models of Huntington’s disease. Neurobiol. Dis 2005, 19, 47–56. [Google Scholar]
- Bortvedt, S.F.; McLear, J.A.; Messer, A.; Ahern-Rindell, A.J.; Wolfgang, W.J. Cystamine and intrabody co-treatment confers additional benefits in a fly model of Huntington’s disease. Neurobiol. Dis 2010, 40, 130–134. [Google Scholar]
- Wolfgang, W.J.; Miller, T.W.; Webster, J.M.; Huston, J.S.; Thompson, L.M.; Marsh, J.L.; Messer, A. Suppression of Huntington’s disease pathology in Drosophila by human single-chain Fv antibodies. Proc. Natl. Acad. Sci. USA 2005, 102, 11563–11568. [Google Scholar]
- Snyder-Keller, A.; McLear, J.A.; Hathorn, T.; Messer, A. Early or late-stage anti-N-terminal Huntingtin intrabody gene therapy reduces pathological features in B6.HDR6/1 mice. J. Neuropathol. Exp. Neurol 2010, 69, 1078–1085. [Google Scholar]
- Butler, D.C.; Messer, A. Bifunctional anti-huntingtin proteasome-directed intrabodies mediate efficient degradation of mutant huntingtin exon 1 protein fragments. PLoS One 2011, 6, e29199. [Google Scholar]
- Khoshnan, A.; Ko, J.; Patterson, P.H. Effects of intracellular expression of anti-huntingtin antibodies of various specificities on mutant huntingtin aggregation and toxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 1002–1007. [Google Scholar]
- Hornykiewicz, O. Biochemical aspects of Parkinson’s disease. Neurology 1998, 51, S2–S9. [Google Scholar]
- Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol 1996, 55, 259–272. [Google Scholar]
- Vance, J.M.; Ali, S.; Bradley, W.G.; Singer, C.; Di Monte, D.A. Gene-environment interactions in Parkinson’s disease and other forms of parkinsonism. Neurotoxicology 2010, 31, 598–602. [Google Scholar]
- Gao, H.M.; Hong, J.S. Gene-environment interactions: Key to unraveling the mystery of Parkinson’s disease. Prog. Neurobiol 2011, 94, 1–19. [Google Scholar]
- Dickson, D.W. Parkinson’s disease and parkinsonism: Neuropathology. Cold Spring Harb. Perspect. Med 2012, 2, a009258. [Google Scholar]
- Kalia, L.V.; Kalia, S.K.; McLean, P.J.; Lozano, A.M.; Lang, A.E. alpha-Synuclein oligomers and clinical implications for Parkinson disease. Ann. Neurol 2013, 73, 155–169. [Google Scholar]
- Mezey, E.; Dehejia, A.M.; Harta, G.; Tresser, N.; Suchy, S.F.; Nussbaum, R.L.; Brownstein, M.J.; Polymeropoulos, M.H. Alpha synuclein is present in Lewy bodies in sporadic Parkinson’s disease. Mol. Psychiatry 1998, 3, 493–499. [Google Scholar]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar]
- Maguire-Zeiss, K.A.; Yehling, E.; Giuliano, R.; Sullivan, M.; J.F.H. HSV Amplicon Expression of Single Chain Antibodies Directed Against α-Synuclein Conformers. Mol. Ther 2004, 9, S86. [Google Scholar]
- Emadi, S.; Liu, R.; Yuan, B.; Schulz, P.; McAllister, C.; Lyubchenko, Y.; Messer, A.; Sierks, M.R. Inhibiting aggregation of alpha-synuclein with human single chain antibody fragments. Biochemistry 2004, 43, 2871–2878. [Google Scholar]
- Lynch, S.M.; Zhou, C.; Messer, A. An scFv intrabody against the nonamyloid component of alpha-synuclein reduces intracellular aggregation and toxicity. J. Mol. Biol 2008, 377, 136–147. [Google Scholar]
- Zhou, C.; Emadi, S.; Sierks, M.R.; Messer, A. A human single-chain Fv intrabody blocks aberrant cellular effects of overexpressed alpha-synuclein. Mol. Ther 2004, 10, 1023–1031. [Google Scholar]
- Emadi, S.; Barkhordarian, H.; Wang, M.S.; Schulz, P.; Sierks, M.R. Isolation of a human single chain antibody fragment against oligomeric alpha-synuclein that inhibits aggregation and prevents alpha-synuclein-induced toxicity. J. Mol. Biol 2007, 368, 1132–1144. [Google Scholar]
- Hudson, P.J.; Kortt, A.A. High avidity scFv multimers; diabodies and triabodies. J. Immunol. Methods 1999, 231, 177–189. [Google Scholar]
- Joshi, S.N.; Butler, D.C.; Messer, A. Fusion to a highly charged proteasomal retargeting sequence increases soluble cytoplasmic expression and efficacy of diverse anti-synuclein intrabodies. mAbs 2012, 4, 686–693. [Google Scholar]
- Whitlow, M.; Bell, B.A.; Feng, S.L.; Filpula, D.; Hardman, K.D.; Hubert, S.L.; Rollence, M.L.; Wood, J.F.; Schott, M.E.; Milenic, D.E.; et al. An improved linker for single-chain Fv with reduced aggregation and enhanced proteolytic stability. Protein Eng 1993, 6, 989–995. [Google Scholar]
- Fisher, A.C.; DeLisa, M.P. Efficient isolation of soluble intracellular single-chain antibodies using the twin-arginine translocation machinery. J. Mol. Biol 2009, 385, 299–311. [Google Scholar]
- Chames, P.; Fieschi, J.; Baty, D. Production of a soluble and active MBP-scFv fusion: Favorable effect of the leaky tolR strain. FEBS Lett 1997, 405, 224–228. [Google Scholar]
- Sun, W.; Xie, J.; Lin, H.; Mi, S.; Li, Z.; Hua, F.; Hu, Z. A combined strategy improves the solubility of aggregation-prone single-chain variable fragment antibodies. Protein Expr. Purif 2012, 83, 21–29. [Google Scholar]
- Boado, R.J.; Lu, J.Z.; Hui, E.K.; Pardridge, W.M. IgG-single chain Fv fusion protein therapeutic for Alzheimer’s disease: Expression in CHO cells and pharmacokinetics and brain delivery in the rhesus monkey. Biotechnol. Bioeng 2010, 105, 627–635. [Google Scholar]
- Skrlj, N.; Drevensek, G.; Hudoklin, S.; Romih, R.; Curin Serbec, V.; Dolinar, M. Recombinant single-chain antibody with the Trojan peptide penetratin positioned in the linker region enables cargo transfer across the blood-brain barrier. Appl. Biochem. Biotechnol 2013, 169, 159–169. [Google Scholar]
- Wei, X.; Roettger, Y.; Tan, B.; He, Y.; Dodel, R.; Hampel, H.; Wei, G.; Haney, J.; Gu, H.; Johnstone, B.H.; et al. Human anti-prion antibodies block prion peptide fibril formation and neurotoxicity. J. Biol. Chem 2012, 287, 12858–12866. [Google Scholar]
- Hoffner, G.; Soues, S.; Djian, P. Aggregation of expanded huntingtin in the brains of patients with Huntington disease. Prion 2007, 1, 26–31. [Google Scholar]
- Huang, P.; Lian, F.; Wen, Y.; Guo, C.; Lin, D. Prion protein oligomer and its neurotoxicity. Acta Biochim. Biophys. Sin. (Shanghai) 2013, 45, 442–451. [Google Scholar]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of alpha-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci 2013, 14, 38–48. [Google Scholar]
- Danzer, K.M.; Haasen, D.; Karow, A.R.; Moussaud, S.; Habeck, M.; Giese, A.; Kretzschmar, H.; Hengerer, B.; Kostka, M. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J. Neurosci 2007, 27, 9220–9232. [Google Scholar]
- Volles, M.J.; Lee, S.J.; Rochet, J.C.; Shtilerman, M.D.; Ding, T.T.; Kessler, J.C.; Lansbury, P.T., Jr. Vesicle permeabilization by protofibrillar alpha-synuclein: Implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry 2001, 40, 7812–7819. [Google Scholar]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Amer. J. Pathol 2000, 157, 401–410. [Google Scholar]
- Alim, M.A.; Ma, Q.L.; Takeda, K.; Aizawa, T.; Matsubara, M.; Nakamura, M.; Asada, A.; Saito, T.; Kaji, H.; Yoshii, M.; et al. Demonstration of a role for alpha-synuclein as a functional microtubule-associated protein. J. Alzheimer’s Dis 2004, 6, 435–442. [Google Scholar]
- Brown, D.R. Oligomeric alpha-synuclein and its role in neuronal death. IUBMB Life 2010, 62, 334–339. [Google Scholar]
- Kudo, W.; Lee, H.P.; Zou, W.Q.; Wang, X.; Perry, G.; Zhu, X.; Smith, M.A.; Petersen, R.B.; Lee, H.G. Cellular prion protein is essential for oligomeric amyloid-beta-induced neuronal cell death. Hum. Mol. Genet 2012, 21, 1138–1144. [Google Scholar]
- Kudo, W.; Petersen, R.B.; Lee, H.G. Cellular prion protein and Alzheimer disease: Link to oligomeric amyloid-beta and neuronal cell death. Prion 2013, 7, 114–116. [Google Scholar]
- Silveira, J.R.; Raymond, G.J.; Hughson, A.G.; Race, R.E.; Sim, V.L.; Hayes, S.F.; Caughey, B. The most infectious prion protein particles. Nature 2005, 437, 257–261. [Google Scholar]
- Simoneau, S.; Rezaei, H.; Sales, N.; Kaiser-Schulz, G.; Lefebvre-Roque, M.; Vidal, C.; Fournier, J.G.; Comte, J.; Wopfner, F.; Grosclaude, J.; et al. In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog 2007, 3, e125. [Google Scholar]
- Bueler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar]
- Nannenga, B.L.; Zameer, A.; Sierks, M.R. Anti-oligomeric single chain variable domain antibody differentially affects huntingtin and alpha-synuclein aggregates. FEBS Lett 2008, 582, 517–522. [Google Scholar]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 2010, 65, 66–79. [Google Scholar]
- Lundblad, M.; Decressac, M.; Mattsson, B.; Bjorklund, A. Impaired neurotransmission caused by overexpression of alpha-synuclein in nigral dopamine neurons. Proc. Nat. Acad. Sci. USA 2012, 109, 3213–3219. [Google Scholar]
- Bellucci, A.; Zaltieri, M.; Navarria, L.; Grigoletto, J.; Missale, C.; Spano, P. From alpha-synuclein to synaptic dysfunctions: New insights into the pathophysiology of Parkinson’s disease. Brain Res 2012, 1476, 183–202. [Google Scholar]
- Bellucci, A.; Navarria, L.; Zaltieri, M.; Missale, C.; Spano, P. alpha-Synuclein synaptic pathology and its implications in the development of novel therapeutic approaches to cure Parkinson’s disease. Brain Res 2012, 1432, 95–113. [Google Scholar]
- Atwal, R.S.; Desmond, C.R.; Caron, N.; Maiuri, T.; Xia, J.; Sipione, S.; Truant, R. Kinase inhibitors modulate huntingtin cell localization and toxicity. Nat. Chem. Biol 2011, 7, 453–460. [Google Scholar]





© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Huang, L.; Su, X.; Federoff, H.J. Single-Chain Fragment Variable Passive Immunotherapies for Neurodegenerative Diseases. Int. J. Mol. Sci. 2013, 14, 19109-19127. https://doi.org/10.3390/ijms140919109
Huang L, Su X, Federoff HJ. Single-Chain Fragment Variable Passive Immunotherapies for Neurodegenerative Diseases. International Journal of Molecular Sciences. 2013; 14(9):19109-19127. https://doi.org/10.3390/ijms140919109
Chicago/Turabian StyleHuang, Liang, Xiaomin Su, and Howard J. Federoff. 2013. "Single-Chain Fragment Variable Passive Immunotherapies for Neurodegenerative Diseases" International Journal of Molecular Sciences 14, no. 9: 19109-19127. https://doi.org/10.3390/ijms140919109
APA StyleHuang, L., Su, X., & Federoff, H. J. (2013). Single-Chain Fragment Variable Passive Immunotherapies for Neurodegenerative Diseases. International Journal of Molecular Sciences, 14(9), 19109-19127. https://doi.org/10.3390/ijms140919109