Involvement of Calcium-Mediated Reactive Oxygen Species in Inductive GRP78 Expression by Geldanamycin in 9L Rat Brain Tumor Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
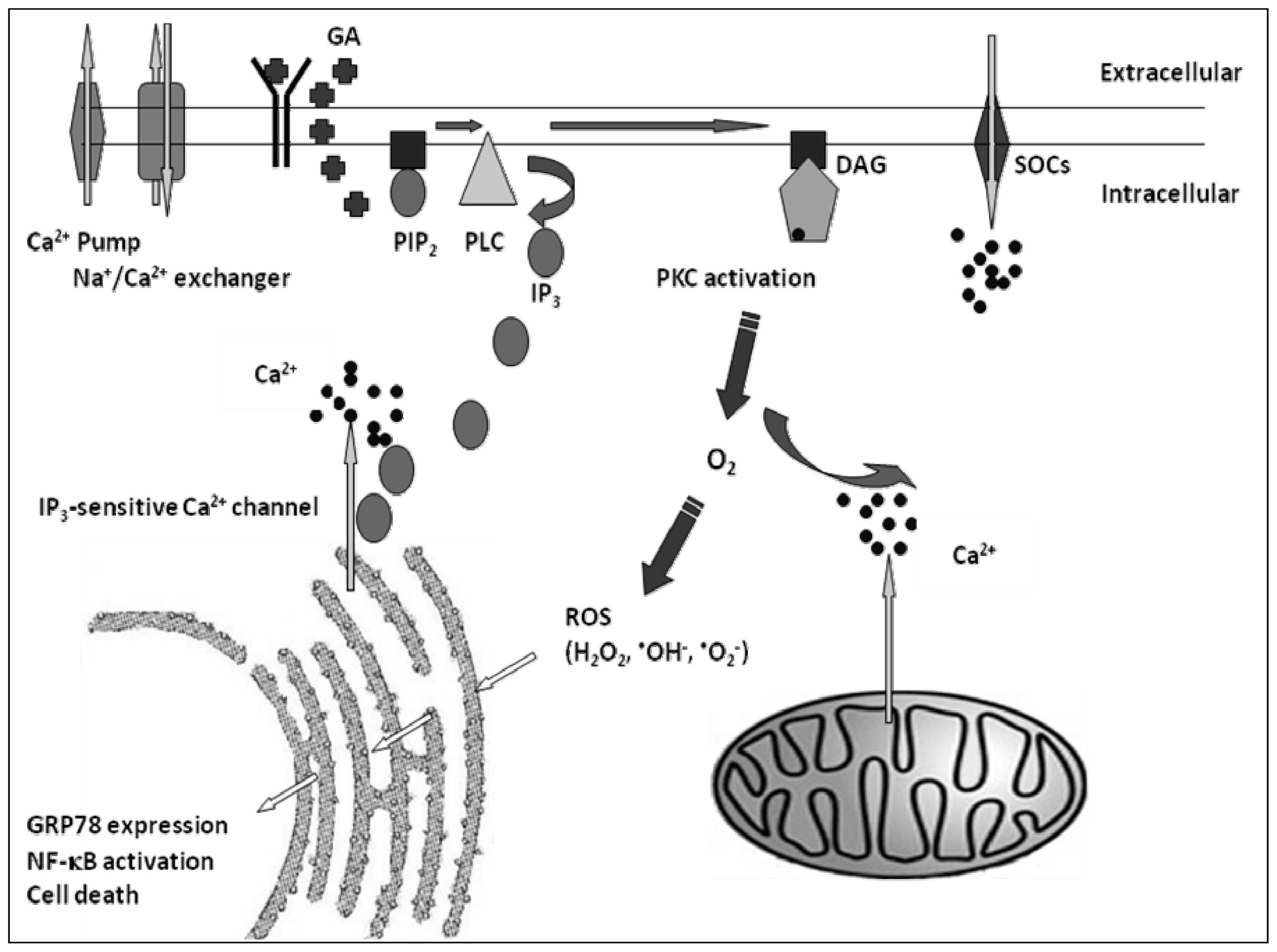
:1. Introduction
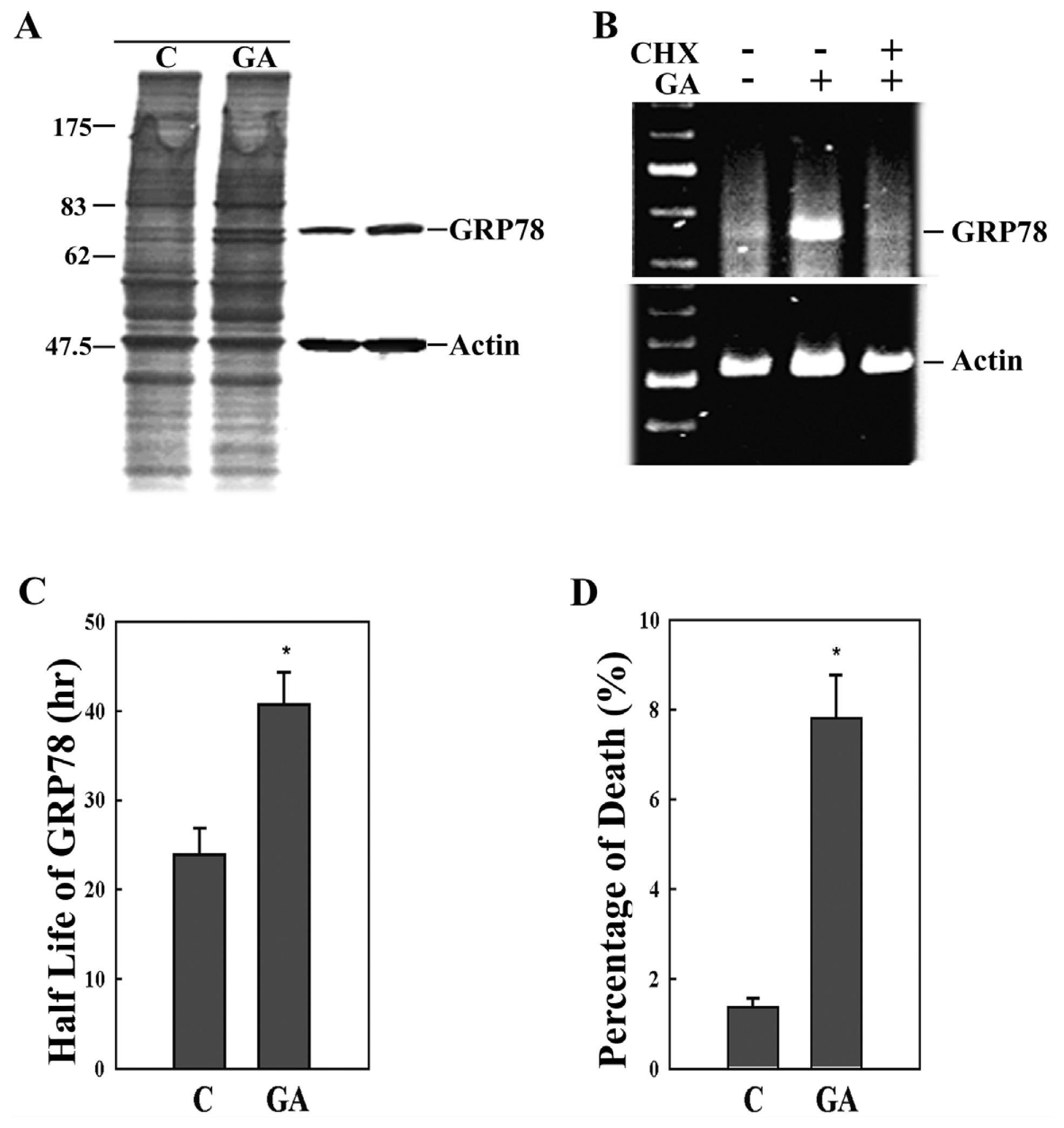
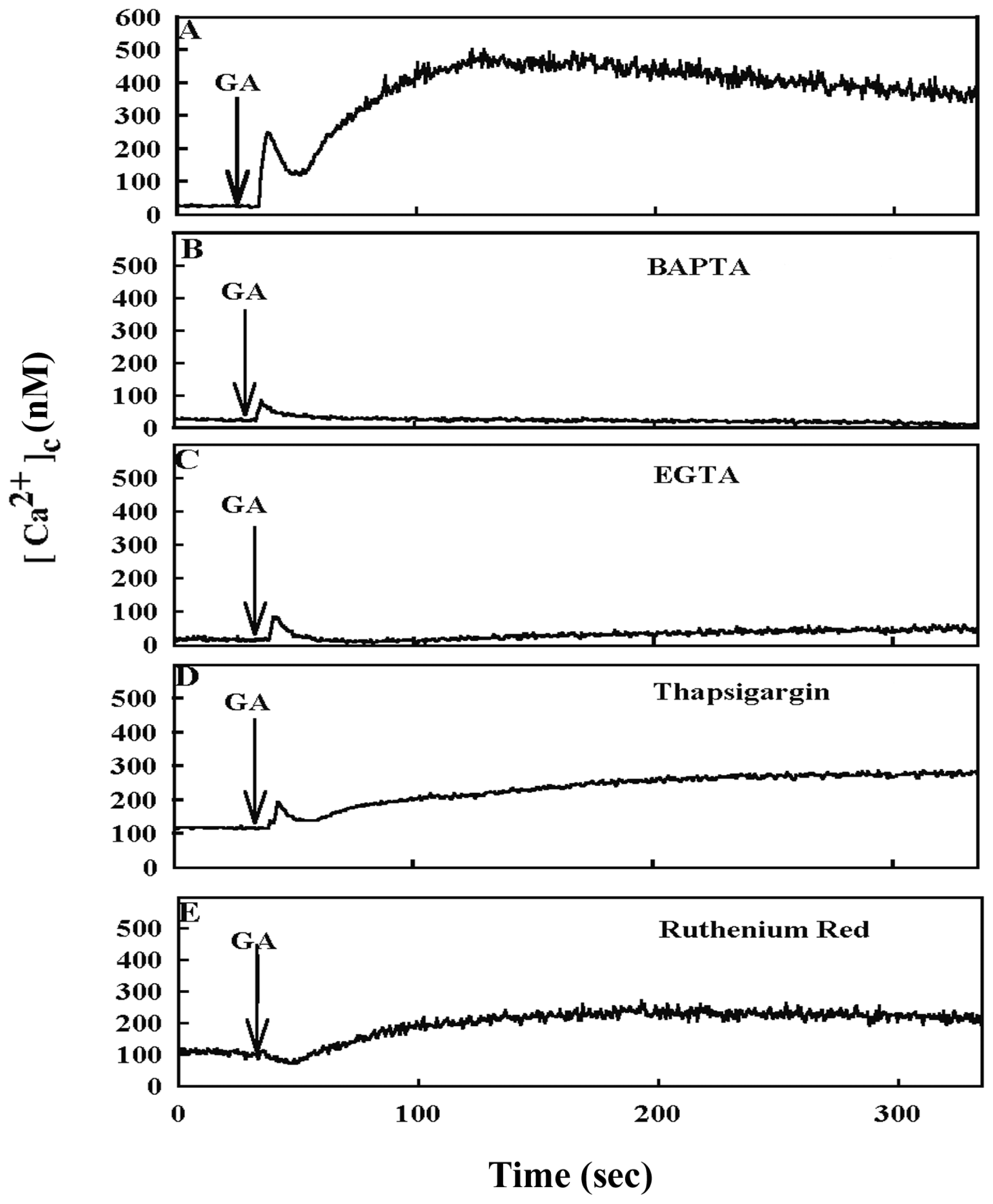
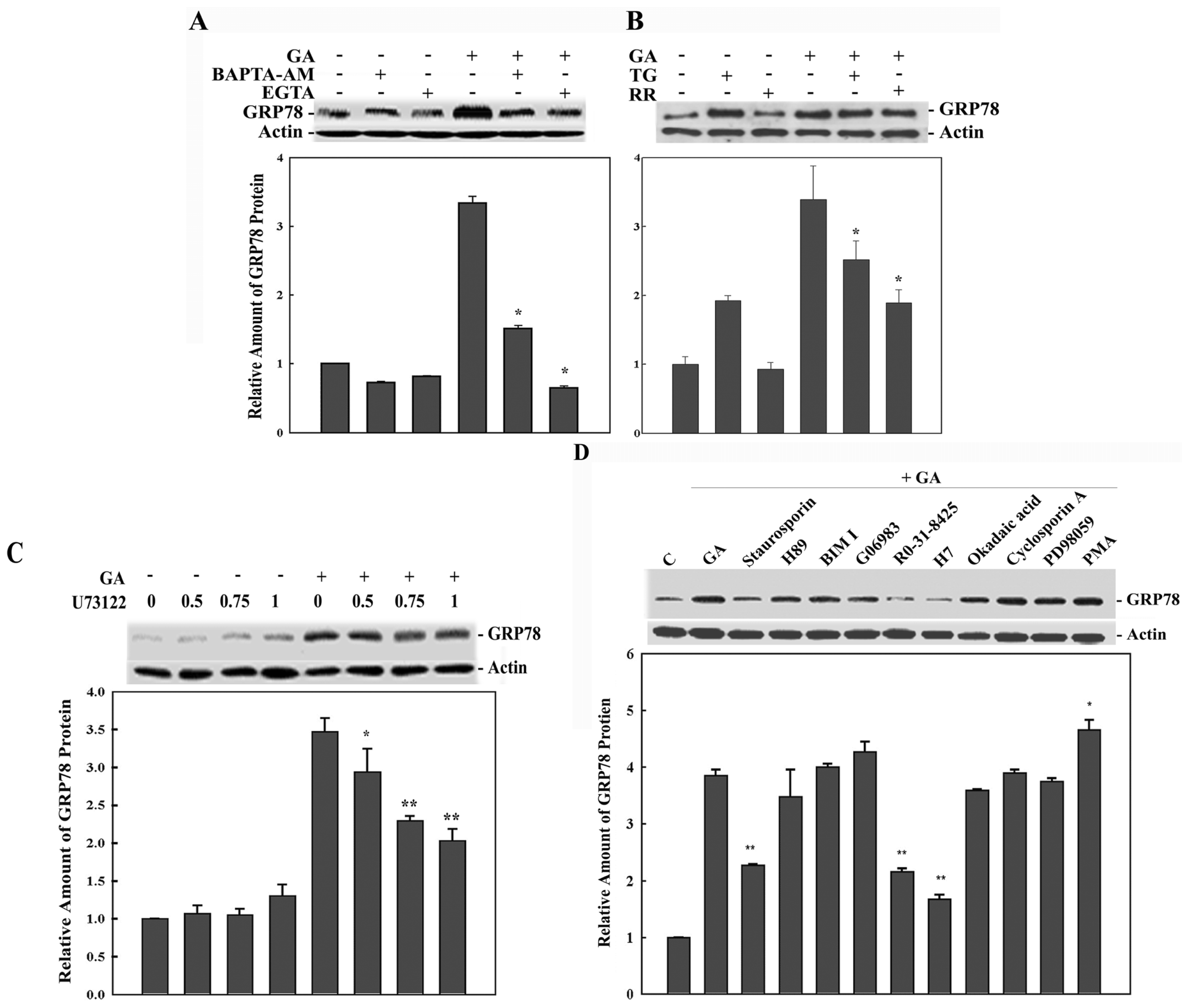
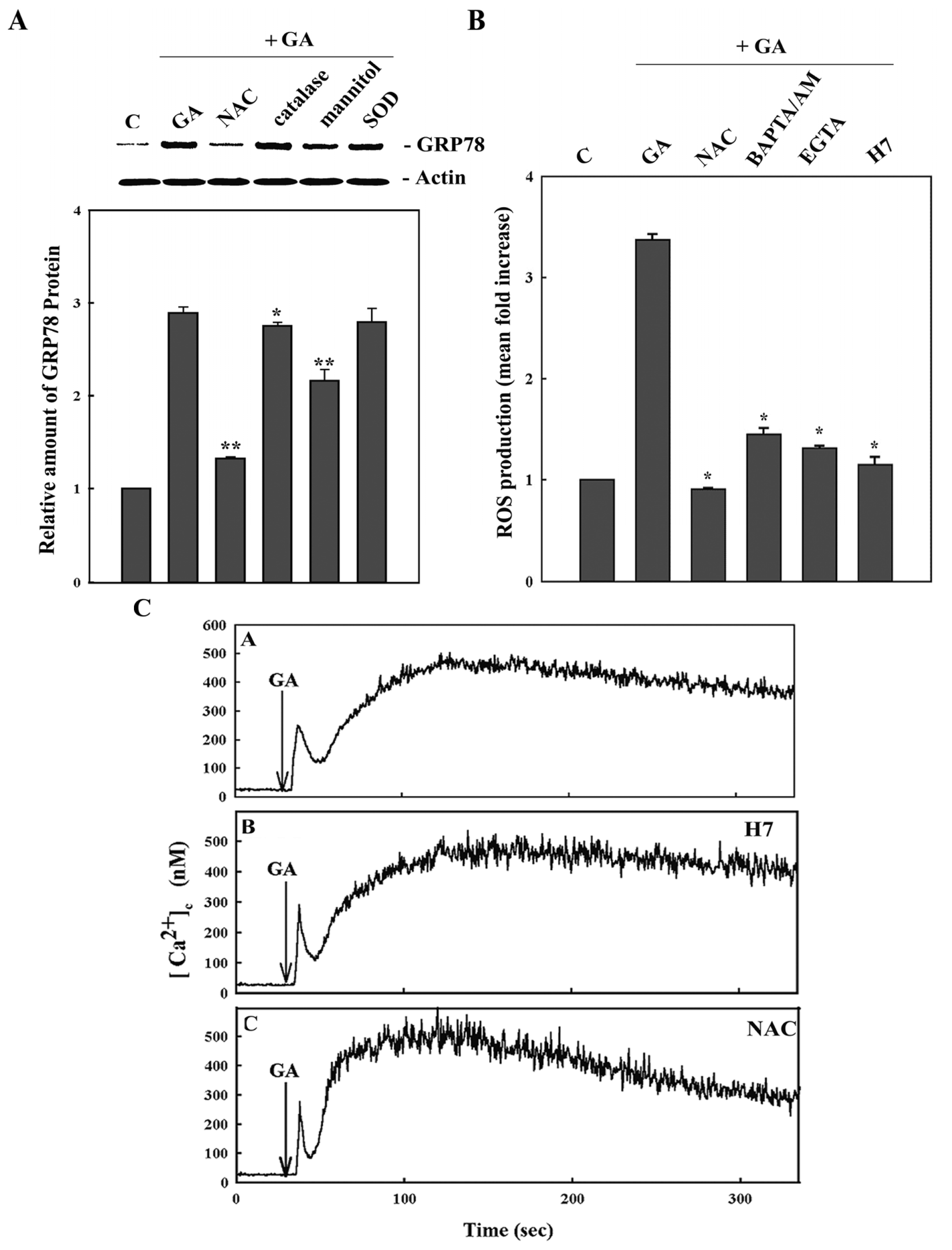
2. Results
3. Discussion
4. Experimental Section
4.1. Cell Cultures and Drug Treatments
4.2. Metabolic Labeling and SDS-PAGE
4.3. Pulse-Chase Analysis of GRP78
4.4. Western Blotting
4.5. Measurement of Intracellular Free Calcium
4.6. Determination of ROS Production
4.7. Statistics Analysis
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Stebbins, C.E.; Russo, A.A.; Schneider, C.; Rosen, N.; Hartl, F.U.; Pavletich, N.P. Crystal structure of an Hsp90-geldanamycin complex: Targeting of a protein chaperone by an antitumor agent. Cell 1997, 89, 239–250. [Google Scholar]
- Schulte, T.W.; Neckers, L.M. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother. Pharmacol 1998, 42, 273–279. [Google Scholar]
- Goetz, M.P.; Toft, D.; Reid, J.; Ames, M.; Stensgard, B.; Safgren, S.; Adjei, A.A.; Sloan, J.; Atherton, P.; Vasile, V.; et al. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J. Clin. Oncol 2005, 23, 1078–1087. [Google Scholar]
- Grem, J.L.; Morrison, G.; Guo, X.D.; Agnew, E.; Takimoto, C.H.; Thomas, R.; Szabo, E.; Grochow, L.; Grollman, F.; Hamilton, J.M.; et al. Phase I and pharmacologic study of 17-(allylamino)-17-demethoxygeldanamycin in adult patients with solid tumors. J. Clin. Oncol 2005, 23, 1885–1893. [Google Scholar]
- Pearl, L.H.; Prodromou, C. Structure and in vivo function of Hsp90. Curr. Opin. Struct. Biol 2000, 10, 46–51. [Google Scholar]
- Scheibel, T.; Buchner, J. The Hsp90 complex—A super-chaperone machine as a novel drug target. Biochem. Pharmacol 1998, 56, 675–682. [Google Scholar]
- Zou, J.; Guo, Y.; Guettouche, T.; Smith, D.F.; Voellmy, R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 1998, 94, 471–480. [Google Scholar]
- Lawson, B.; Brewer, J.W.; Hendershot, L.M. Geldanamycin, an hsp90/GRP94-binding drug, induces increased transcription of endoplasmic reticulum (ER) chaperones via the ER stress pathway. J. Cell. Physiol 1998, 174, 170–178. [Google Scholar]
- Taiyab, A.; Sreedhar, A.S.; Rao Ch, M. Hsp90 inhibitors, GA and 17AAG, lead to ER stress-induced apoptosis in rat histiocytoma. Biochem. Pharmacol 2009, 78, 142–152. [Google Scholar]
- Sreedhar, A.S.; Mihaly, K.; Pato, B.; Schnaider, T.; Stetak, A.; Kis-Petik, K.; Fidy, J.; Simonics, T.; Maraz, A.; Csermely, P. Hsp90 inhibition accelerates cell lysis. Anti-Hsp90 ribozyme reveals a complex mechanism of Hsp90 inhibitors involving both superoxide- and Hsp90-dependent events. J. Biol. Chem 2003, 278, 35231–35240. [Google Scholar]
- Dikalov, S.; Landmesser, U.; Harrison, D.G. Geldanamycin leads to superoxide formation by enzymatic and non-enzymatic redox cycling. Implications for studies of Hsp90 and endothelial cell nitric-oxide synthase. J. Biol. Chem 2002, 277, 25480–25485. [Google Scholar]
- Cysyk, R.L.; Parker, R.J.; Barchi, J.J., Jr; Steeg, P.S.; Hartman, N.R.; Strong, J.M. Reaction of geldanamycin and C17-substituted analogues with glutathione: Product identifications and pharmacological implications. Chem. Res. Toxicol. 2006, 19, 376–381. [Google Scholar]
- Lai, M.T.; Huang, K.L.; Chang, W.M.; Lai, Y.K. Geldanamycin induction of grp78 requires activation of reactive oxygen species via ER stress responsive elements in 9L rat brain tumour cells. Cell. Signal 2003, 15, 585–595. [Google Scholar]
- Shu, C.W.; Cheng, N.L.; Chang, W.M.; Tseng, T.L.; Lai, Y.K. Transactivation of hsp70-1/2 in geldanamycin-treated human non-small cell lung cancer H460 cells: Involvement of intracellular calcium and protein kinase C. J. Cell. Biochem 2005, 94, 1199–1209. [Google Scholar]
- Chang, Y.S.; Lee, L.C.; Sun, F.C.; Chao, C.C.; Fu, H.W.; Lai, Y.K. Involvement of calcium in the differential induction of heat shock protein 70 by heat shock protein 90 inhibitors, geldanamycin and radicicol, in human non-small cell lung cancer H460 cells. J. Cell. Biochem 2006, 97, 156–165. [Google Scholar]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar]
- Steinberg, S.F. Structural basis of protein kinase C isoform function. Physiol. Rev 2008, 88, 1341–1378. [Google Scholar]
- Wilkinson, S.E.; Parker, P.J.; Nixon, J.S. Isoenzyme specificity of bisindolylmaleimides, selective inhibitors of protein kinase C. Biochem. J 1993, 294, 335–337. [Google Scholar]
- Bergamini, C.M.; Gambetti, S.; Dondi, A.; Cervellati, C. Oxygen, reactive oxygen species and tissue damage. Curr. Pharm. Des 2004, 10, 1611–1626. [Google Scholar]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol 2007, 39, 44–84. [Google Scholar]
- Pinilla, P.J.; Hernandez, A.T.; Camello, M.C.; Pozo, M.J.; Toescu, E.C.; Camello, P.J. Non-stimulated Ca2+ leak pathway in cerebellar granule neurones. Biochem. Pharmacol 2005, 70, 786–793. [Google Scholar]
- Alberdi, E.; Sanchez-Gomez, M.V.; Matute, C. Calcium and glial cell death. Cell Calcium 2005, 38, 417–425. [Google Scholar]
- Knox, C.D.; Belous, A.E.; Pierce, J.M.; Wakata, A.; Nicoud, I.B.; Anderson, C.D.; Pinson, C.W.; Chari, R.S. Novel role of phospholipase C-delta1: Regulation of liver mitochondrial Ca2+ uptake. Am. J. Physiol. Gastrointest. Liver Physiol 2004, 287, G533–G540. [Google Scholar]
- Ermak, G.; Davies, K.J. Calcium and oxidative stress: From cell signaling to cell death. Mol. Immunol 2002, 38, 713–721. [Google Scholar]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6. [Google Scholar] [CrossRef]
- Shiva, S. Mitochondria as metabolizers and targets of nitrite. Nitric Oxide 2010, 22, 64–74. [Google Scholar]
- Peng, T.I.; Jou, M.J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci 2010, 1201, 183–188. [Google Scholar]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res 2008, 102, 488–496. [Google Scholar]
- Ceylan-Isik, A.F.; Guo, K.K.; Carlson, E.C.; Privratsky, J.R.; Liao, S.J.; Cai, L.; Chen, A.F.; Ren, J. Metallothionein abrogates GTP cyclohydrolase I inhibition-induced cardiac contractile and morphological defects: Role of mitochondrial biogenesis. Hypertension 2009, 53, 1023–1031. [Google Scholar]
- Abramov, A.Y.; Jacobson, J.; Wientjes, F.; Hothersall, J.; Canevari, L.; Duchen, M.R. Expression and modulation of an NADPH oxidase in mammalian astrocytes. J. Neurosci 2005, 25, 9176–9184. [Google Scholar]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev 2007, 87, 245–313. [Google Scholar]
- Billecke, S.S.; Bender, A.T.; Kanelakis, K.C.; Murphy, P.J.; Lowe, E.R.; Kamada, Y.; Pratt, W.B.; Osawa, Y. Hsp90 is required for heme binding and activation of apo-neuronal nitric-oxide synthase: Geldanamycin-mediated oxidant generation is unrelated to any action of hsp90. J. Biol. Chem 2002, 277, 20504–20509. [Google Scholar]
- Pritchard, K.A., Jr; Ackerman, A.W.; Gross, E.R.; Stepp, D.W.; Shi, Y.; Fontana, J.T.; Baker, J.E.; Sessa, W.C. Heat shock protein 90 mediates the balance of nitric oxide and superoxide anion from endothelial nitric-oxide synthase. J. Biol. Chem. 2001, 276, 17621–17624. [Google Scholar]
- Clark, C.B.; Rane, M.J.; El Mehdi, D.; Miller, C.J.; Sachleben, L.R., Jr; Gozal, E. Role of oxidative stress in geldanamycin-induced cytotoxicity and disruption of Hsp90 signaling complex. Free Radic. Biol. Med. 2009, 47, 1440–1449. [Google Scholar]
- Samuni, Y.; Ishii, H.; Hyodo, F.; Samuni, U.; Krishna, M.C.; Goldstein, S.; Mitchell, J.B. Reactive oxygen species mediate hepatotoxicity induced by the Hsp90 inhibitor geldanamycin and its analogs. Free Radic. Biol. Med 2010, 48, 1559–1563. [Google Scholar]
- Liu, H.; Bowes, R.C., 3rd; van de Water, B.; Sillence, C.; Nagelkerke, J.F.; Stevens, J.L. Endoplasmic reticulum chaperones GRP78 and calreticulin prevent oxidative stress, Ca2+ disturbances, and cell death in renal epithelial cells. J. Biol. Chem. 1997, 272, 21751–21759. [Google Scholar]
- Yu, Z.; Luo, H.; Fu, W.; Mattson, M.P. The endoplasmic reticulum stress-responsive protein GRP78 protects neurons against excitotoxicity and apoptosis: Suppression of oxidative stress and stabilization of calcium homeostasis. Exp. Neurol 1999, 155, 302–314. [Google Scholar]
- Shu, C.W.; Sun, F.C.; Cho, J.H.; Lin, C.C.; Liu, P.F.; Chen, P.Y.; Chang, M.D.; Fu, H.W.; Lai, Y.K. GRP78 and Raf-1 cooperatively confer resistance to endoplasmic reticulum stress-induced apoptosis. J. Cell. Physiol 2008, 215, 627–635. [Google Scholar]
- Ouyang, Y.B.; Xu, L.J.; Emery, J.F.; Lee, A.S.; Giffard, R.G. Overexpressing GRP78 influences Ca2+ handling and function of mitochondria in astrocytes after ischemia-like stress. Mitochondrion 2011, 11, 279–286. [Google Scholar]
- Murphy, A.N.; Bredesen, D.E.; Cortopassi, G.; Wang, E.; Fiskum, G. Bcl-2 potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proc. Natl. Acad. Sci. USA 1996, 93, 9893–9898. [Google Scholar]
- Palmer, A.E.; Jin, C.; Reed, J.C.; Tsien, R.Y. Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc. Natl. Acad. Sci. USA 2004, 101, 17404–17409. [Google Scholar]
- Oakes, S.A.; Scorrano, L.; Opferman, J.T.; Bassik, M.C.; Nishino, M.; Pozzan, T.; Korsmeyer, S.J. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2005, 102, 105–110. [Google Scholar]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science 2003, 300, 135–139. [Google Scholar]
- Guo, F.; Rocha, K.; Bali, P.; Pranpat, M.; Fiskus, W.; Boyapalle, S.; Kumaraswamy, S.; Balasis, M.; Greedy, B.; Armitage, E.S.; et al. Abrogation of heat shock protein 70 induction as a strategy to increase antileukemia activity of heat shock protein 90 inhibitor 17-allylamino-demethoxy geldanamycin. Cancer Res 2005, 65, 10536–10544. [Google Scholar]
- Pyrko, P.; Schonthal, A.H.; Hofman, F.M.; Chen, T.C.; Lee, A.S. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res 2007, 67, 9809–9816. [Google Scholar]
- Schonthal, A.H. Targeting endoplasmic reticulum stress for cancer therapy. Front. Biosci 2012, 4, 412–431. [Google Scholar]
- Schonthal, A.H. Pharmacological targeting of endoplasmic reticulum stress signaling in cancer. Biochem. Pharmacol 2013, 85, 653–666. [Google Scholar]
- Katayama, T.; Imaizumi, K.; Manabe, T.; Hitomi, J.; Kudo, T.; Tohyama, M. Induction of neuronal death by ER stress in Alzheimer’s disease. J. Chem. Neuroanat 2004, 28, 67–78. [Google Scholar]
- Oida, Y.; Izuta, H.; Oyagi, A.; Shimazawa, M.; Kudo, T.; Imaizumi, K.; Hara, H. Induction of BiP, an ER-resident protein, prevents the neuronal death induced by transient forebrain ischemia in gerbil. Brain Res 2008, 1208, 217–224. [Google Scholar]
- Weizsaecker, M.; Deen, D.F.; Rosenblum, M.L.; Hoshino, T.; Gutin, P.H.; Barker, M. The 9L rat brain tumor: Description and application of an animal model. J. Neurol 1981, 224, 183–192. [Google Scholar]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem 1985, 260, 3440–3450. [Google Scholar]
- Reynaud, S.; Duchiron, C.; Deschaux, P. 3-Methylcholanthrene increases phorbol 12-myristate 13-acetate-induced respiratory burst activity and intracellular calcium levels in common carp (Cyprinus carpio L) macrophages. Toxicol. Appl. Pharmacol 2001, 175, 1–9. [Google Scholar]





© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sun, F.-C.; Shyu, H.-Y.; Lee, M.-S.; Lee, M.-S.; Lai, Y.-K. Involvement of Calcium-Mediated Reactive Oxygen Species in Inductive GRP78 Expression by Geldanamycin in 9L Rat Brain Tumor Cells. Int. J. Mol. Sci. 2013, 14, 19169-19185. https://doi.org/10.3390/ijms140919169
Sun F-C, Shyu H-Y, Lee M-S, Lee M-S, Lai Y-K. Involvement of Calcium-Mediated Reactive Oxygen Species in Inductive GRP78 Expression by Geldanamycin in 9L Rat Brain Tumor Cells. International Journal of Molecular Sciences. 2013; 14(9):19169-19185. https://doi.org/10.3390/ijms140919169
Chicago/Turabian StyleSun, Fang-Chun, Hsin-Yi Shyu, Meng-Shiou Lee, Meng-Shiunn Lee, and Yiu-Kay Lai. 2013. "Involvement of Calcium-Mediated Reactive Oxygen Species in Inductive GRP78 Expression by Geldanamycin in 9L Rat Brain Tumor Cells" International Journal of Molecular Sciences 14, no. 9: 19169-19185. https://doi.org/10.3390/ijms140919169
APA StyleSun, F. -C., Shyu, H. -Y., Lee, M. -S., Lee, M. -S., & Lai, Y. -K. (2013). Involvement of Calcium-Mediated Reactive Oxygen Species in Inductive GRP78 Expression by Geldanamycin in 9L Rat Brain Tumor Cells. International Journal of Molecular Sciences, 14(9), 19169-19185. https://doi.org/10.3390/ijms140919169