Study on the Characteristics of Gas Molecular Mean Free Pathin Nanopores by Molecular Dynamics Simulations

Abstract
:1. Introduction
2. Results and Discussion
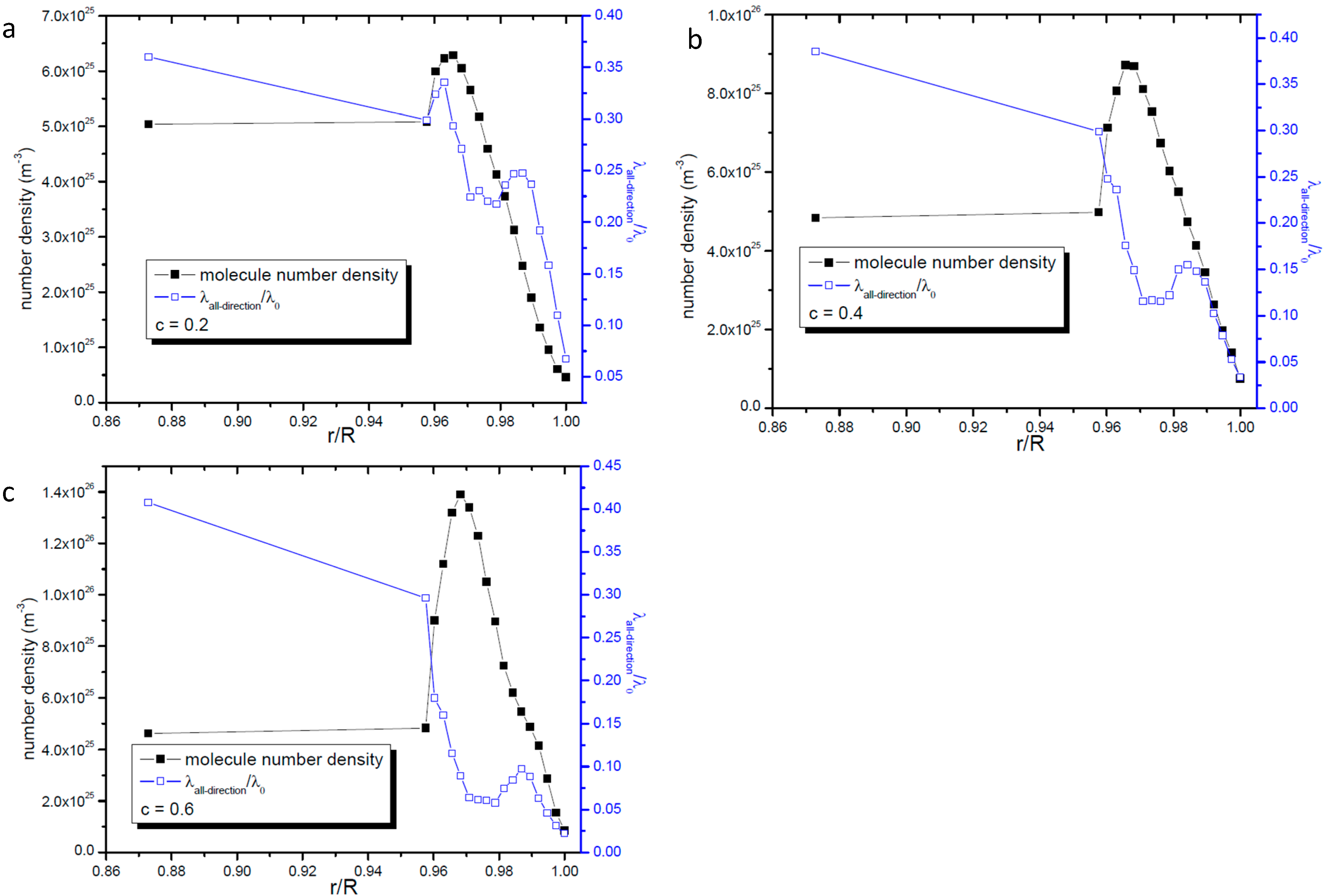
2.1. Mean Free Path of All Molecules in Nanopore



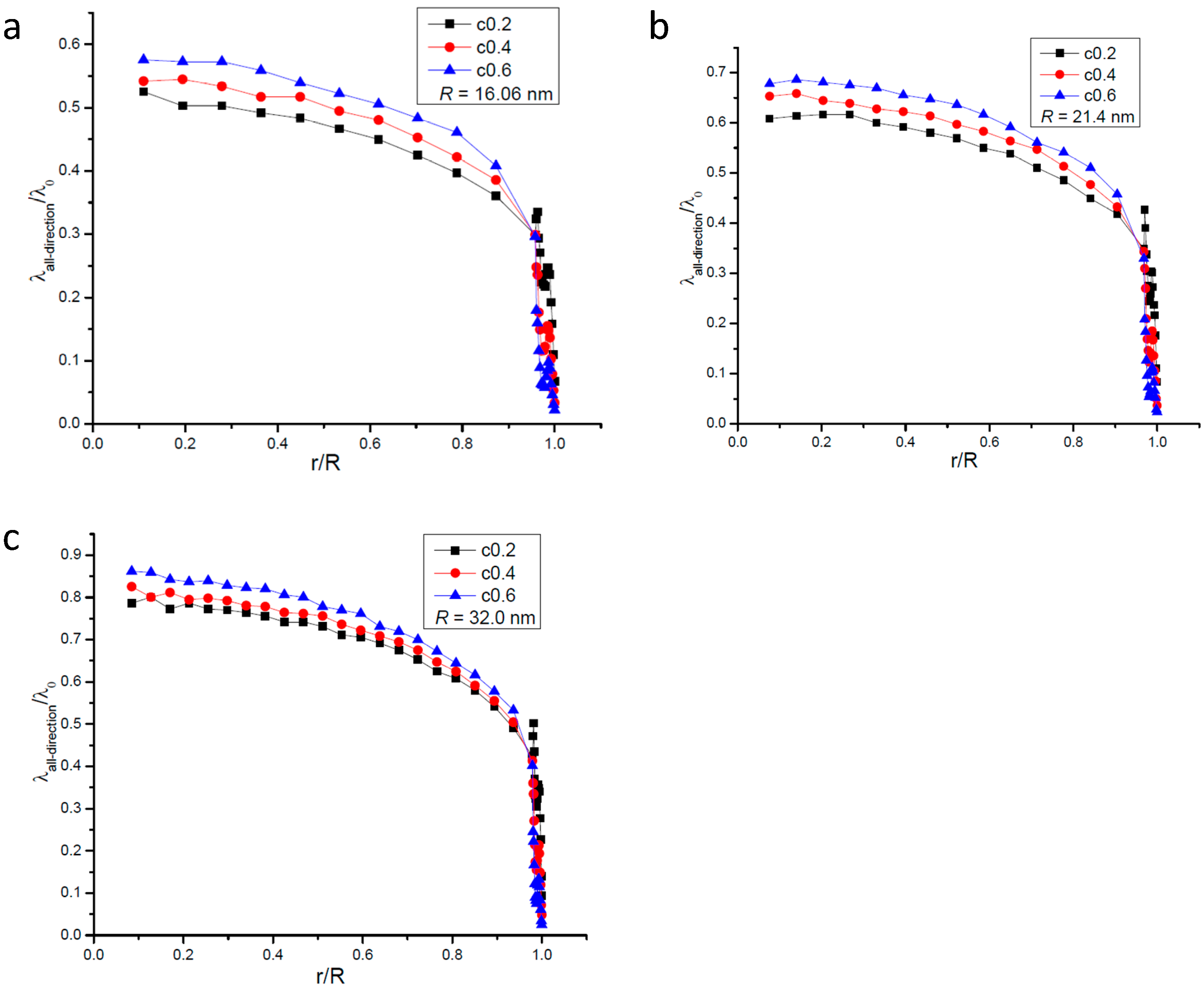{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| λall/λ0 | R = 16.06 nm | R = 21.4 nm | R = 32.0 nm |
|---|---|---|---|
| c = 0.2 | 0.321 | 0.388 | 0.519 |
| c = 0.4 | 0.209 | 0.248 | 0.371 |
| c = 0.6 | 0.128 | 0.179 | 0.237 |
| Actual Kn | R = 16.06 nm | R = 21.4 nm | R = 32.0 nm |
|---|---|---|---|
| Kn0 = 1.116 | Kn0 = 0.837 | Kn0 = 0.560 | |
| c = 0.2 | 0.358 | 0.325 | 0.291 |
| c =0.4 | 0.233 | 0.208 | 0.208 |
| c = 0.6 | 0.143 | 0.150 | 0.133 |
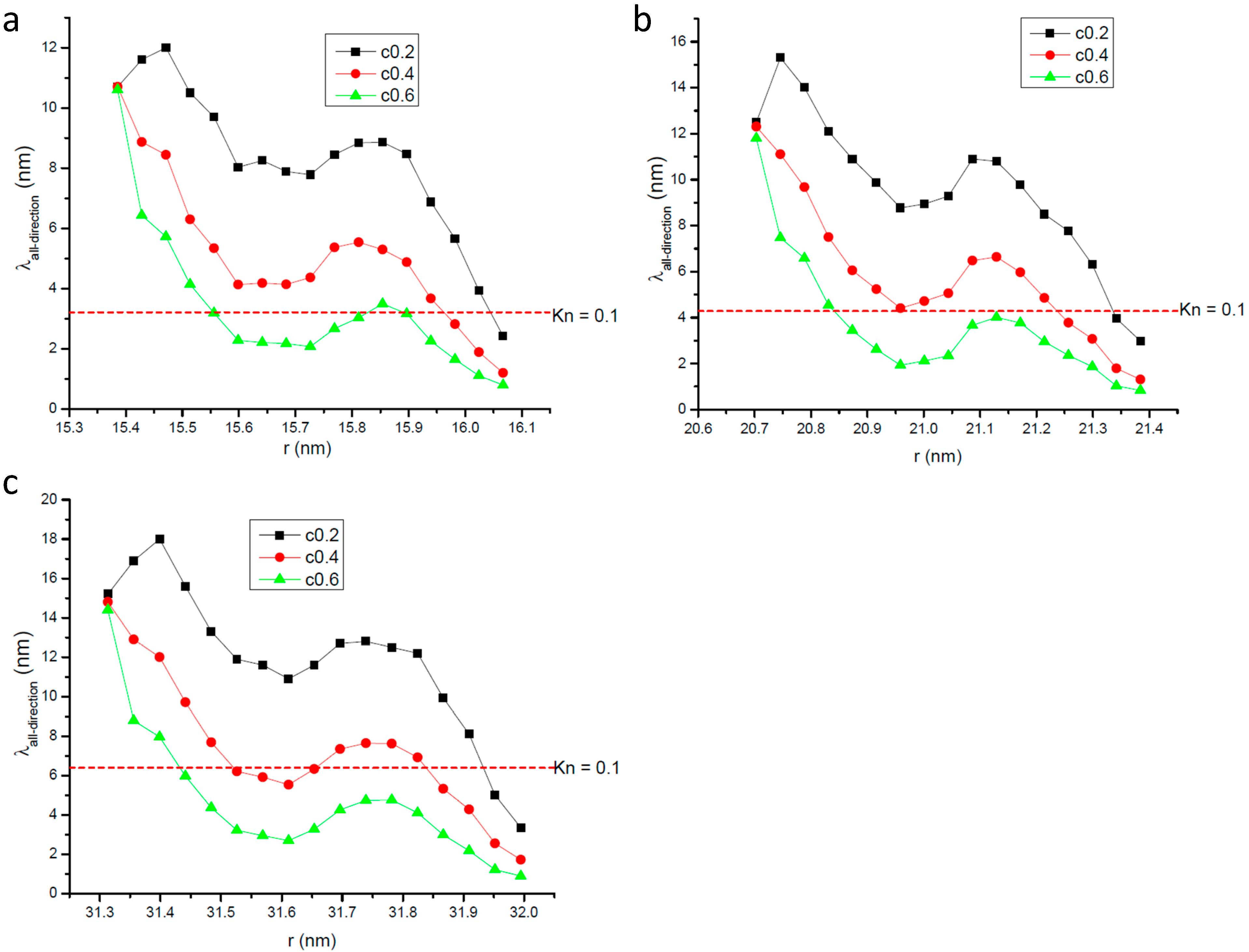
2.2. Profile of Gas Molecular Mean Free Path

| Comparison of Molecule Collision Times | R = 16.06 nm | R = 21.4 nm | R = 32.0 nm |
|---|---|---|---|
| c = 0.2 | 1 | 1 | 1 |
| c = 0.4 | 0.937 | 0.968 | 0.981 |
| c = 0.6 | 0.893 | 0.911 | 0.947 |

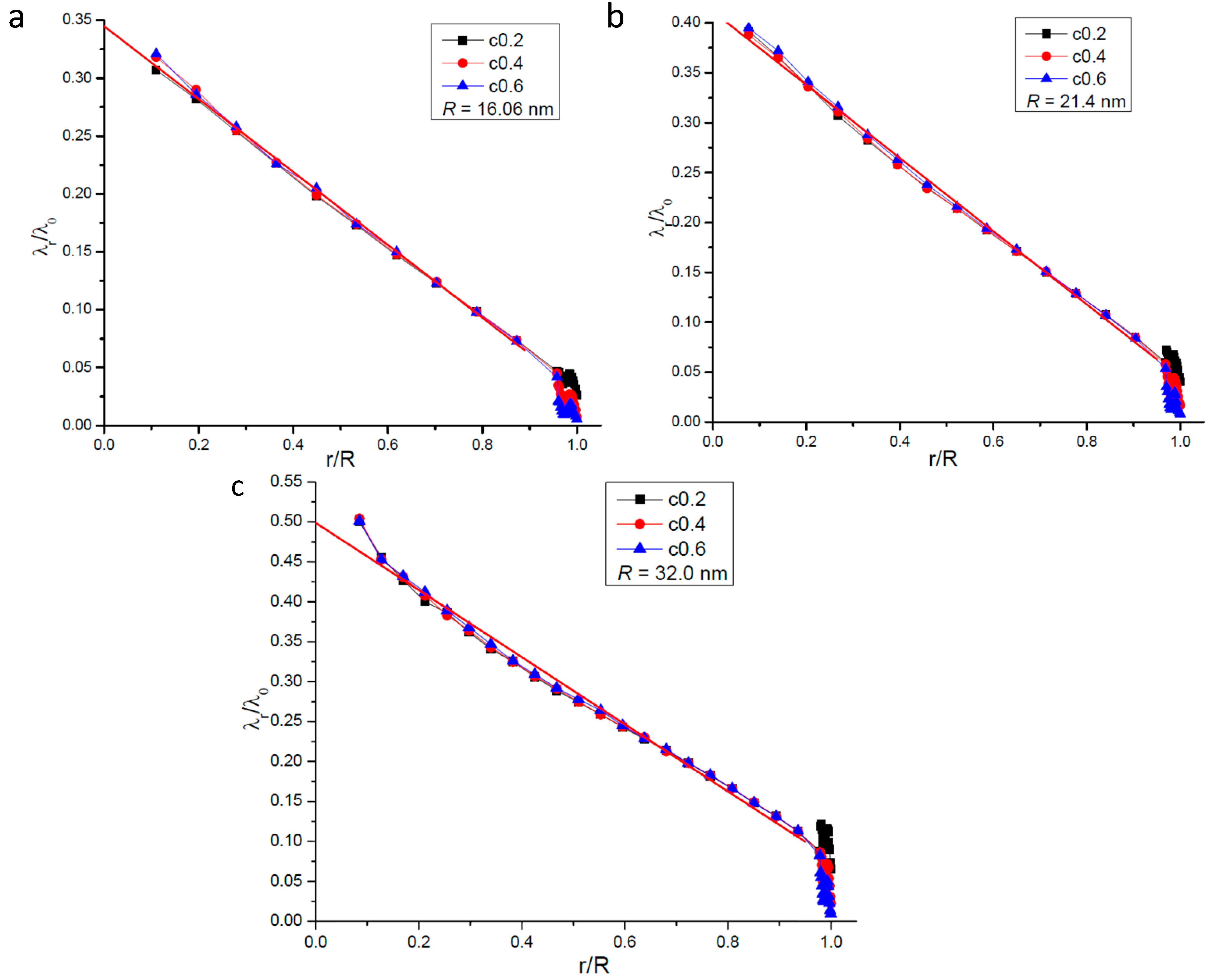
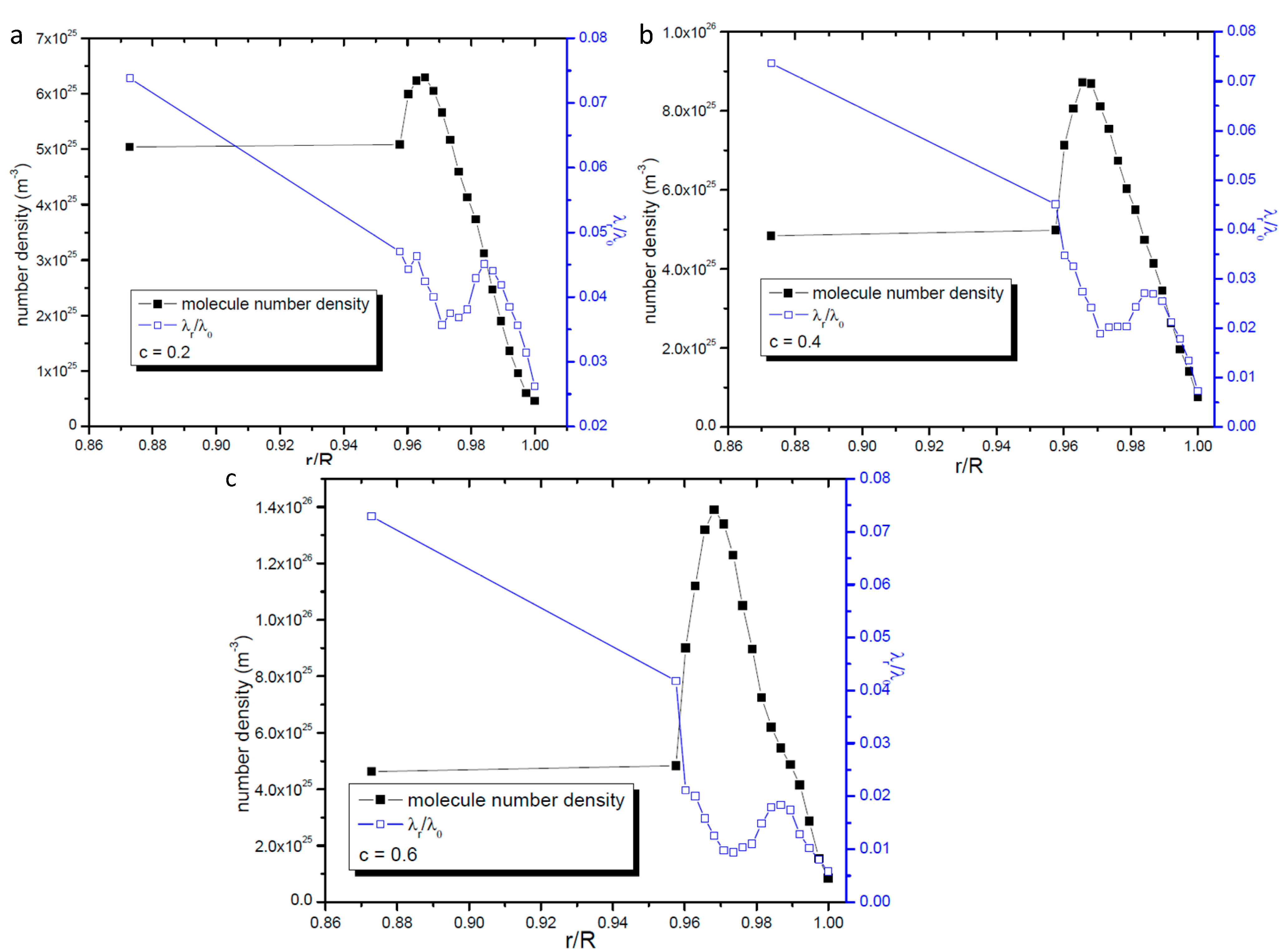
2.3. Profile of the Radial Gas Molecular Mean Free Path


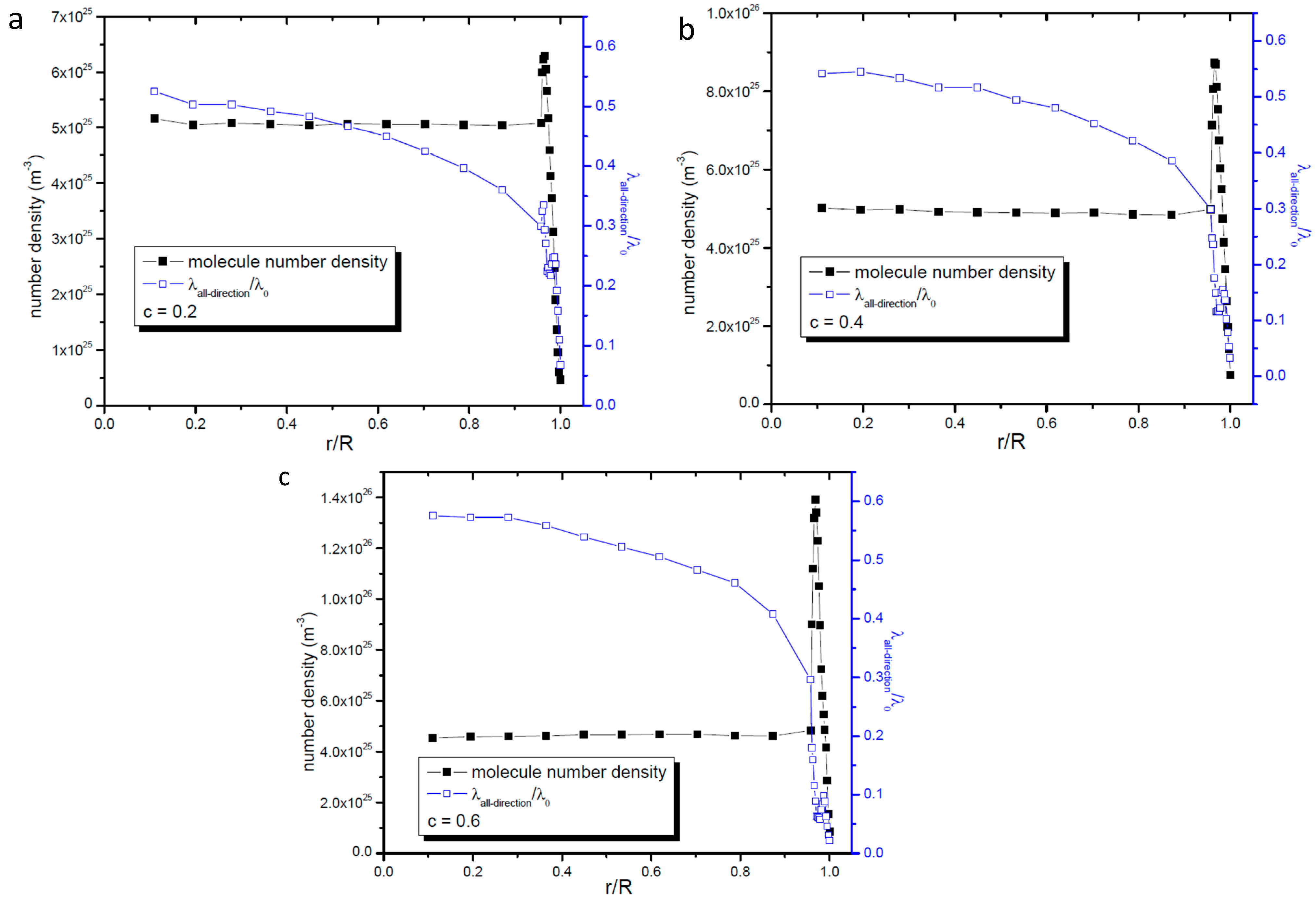
2.4. Relationship between Gas Number Density and Molecular Mean Free Path in Nanopores




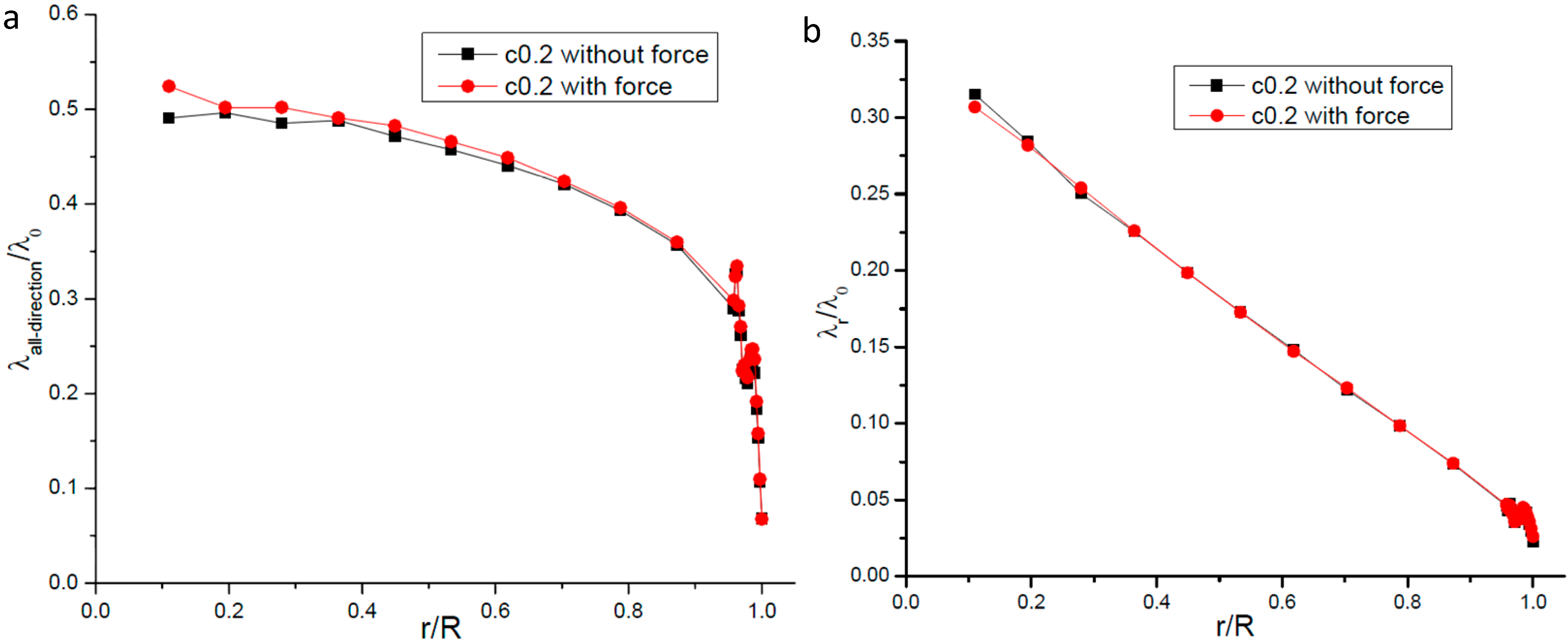
2.5. The Body Force’s Effects on the Gas Molecular Mean Free Path

3. Numerical Experimental Section
3.1. Simulation Model







3.2. Boundary Conditions
3.3. Method of Gas Molecular Mean Free Path Calculation
3.4. Other Simulation Details
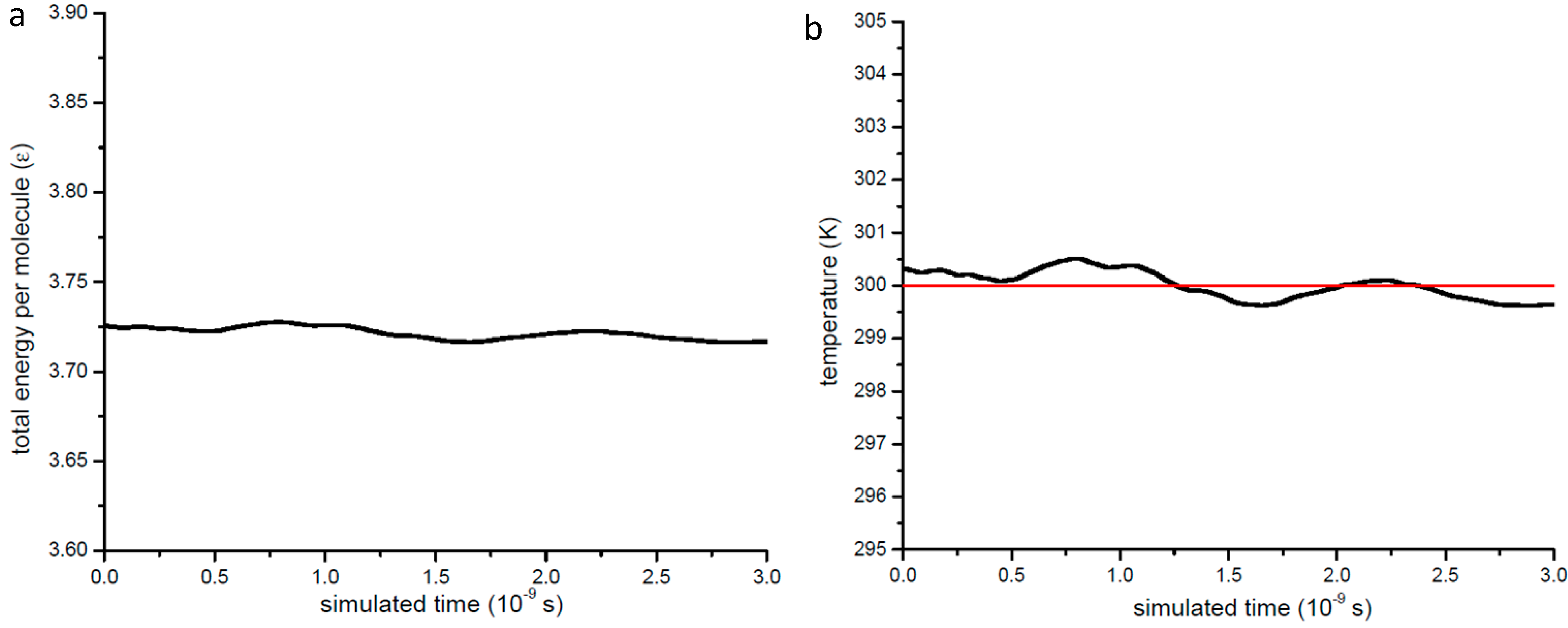
3.5. Certification of the Methodology

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hummer, G.; Rasaiah, J.C.; Noworyta, J.P. Water conduction through the hydrophobic channel of a carbon nanotube. Nature 2001, 414, 188–190. [Google Scholar] [CrossRef]
- Holt, J.K.; Park, H.G.; Wang, Y.M. Fast mass transport through sub-2-nanometer carbon nanotubes. Science 2006, 312, 1034–1037. [Google Scholar] [CrossRef]
- Cao, B.Y.; Sun, J.; Chen, M.; Guo, Z.Y. Molecular momentum transport at fluid-solid interfaces in MEMS/NEMS: A review. Int. J. Mol. Sci. 2009, 10, 4638–4706. [Google Scholar] [CrossRef]
- Travis, K.P.; Todd, B.D.; Evans, D.J. Departure from Navier-Stokes hydrodynamics in confined liquids. Phys. Rev. E 1997, 55, 4288–4295. [Google Scholar] [CrossRef]
- Song, X.; Chen, J.K. A comparative study on poiseuille flow of simple fluids through cylindrical and slit-like nanochannels. Int. J. Heat Mass Transf. 2008, 51, 1770–1779. [Google Scholar]
- Murat, B.; Beskok, A. Molecular dynamics simulations of shear-driven gas flows in nano-channels. Microfluid. Nanofluid. 2011, 11, 611–622. [Google Scholar]
- Stops, D.W. The mean free path of gas molecules in the transition regime. J. Phys. D Appl. Phys. 1970, 3, 685–696. [Google Scholar]
- Dongari, N.; Zhang, Y.H.; Reese, J.M. Molecular free path distribution in rarefied gases. J. Phys. D Appl. Phys. 2011, 44. [Google Scholar] [CrossRef]
- Dongari, N.; Zhang, Y.H.; Reese, J.M. Modeling of Knudsen layer effects in micro/nanoscale gas flows. J. Fluids Eng. 2011, 7. [Google Scholar] [CrossRef] [Green Version]
- Dongari, N.; Barbert, R.W.; Emerson, D.R.; Stefanov, S.K.; Zhang, Y.; Reese, J.M. The effect of Knudsen layers on rarefied cylindrical Couette gas flows. Microfluid. Nanofluid. 2013, 14, 31–43. [Google Scholar] [CrossRef]
- Rapaport, D.C. The Art of Molecular Dynamics Simulation; Cambridge University Press: Cambridge, UK, 2004; p. 15. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, Q.; Cai, Z. Study on the Characteristics of Gas Molecular Mean Free Pathin Nanopores by Molecular Dynamics Simulations. Int. J. Mol. Sci. 2014, 15, 12714-12730. https://doi.org/10.3390/ijms150712714
Liu Q, Cai Z. Study on the Characteristics of Gas Molecular Mean Free Pathin Nanopores by Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2014; 15(7):12714-12730. https://doi.org/10.3390/ijms150712714
Chicago/Turabian StyleLiu, Qixin, and Zhiyong Cai. 2014. "Study on the Characteristics of Gas Molecular Mean Free Pathin Nanopores by Molecular Dynamics Simulations" International Journal of Molecular Sciences 15, no. 7: 12714-12730. https://doi.org/10.3390/ijms150712714
APA StyleLiu, Q., & Cai, Z. (2014). Study on the Characteristics of Gas Molecular Mean Free Pathin Nanopores by Molecular Dynamics Simulations. International Journal of Molecular Sciences, 15(7), 12714-12730. https://doi.org/10.3390/ijms150712714