Sirt3 Protects Cortical Neurons against Oxidative Stress via Regulating Mitochondrial Ca2+ and Mitochondrial Biogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
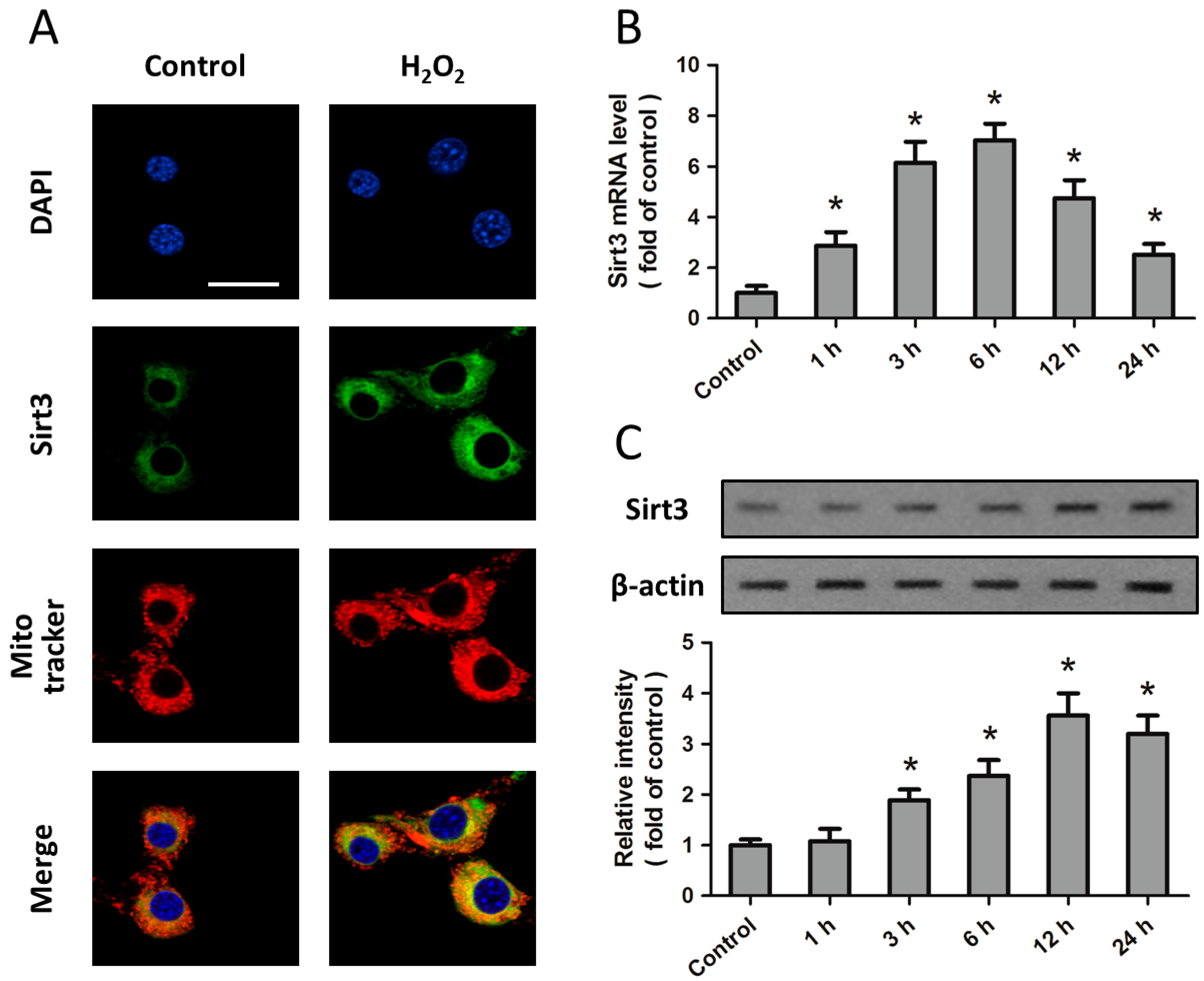
2.1. Expression of Sirt3 after H2O2 Treatment in Cortical Neurons

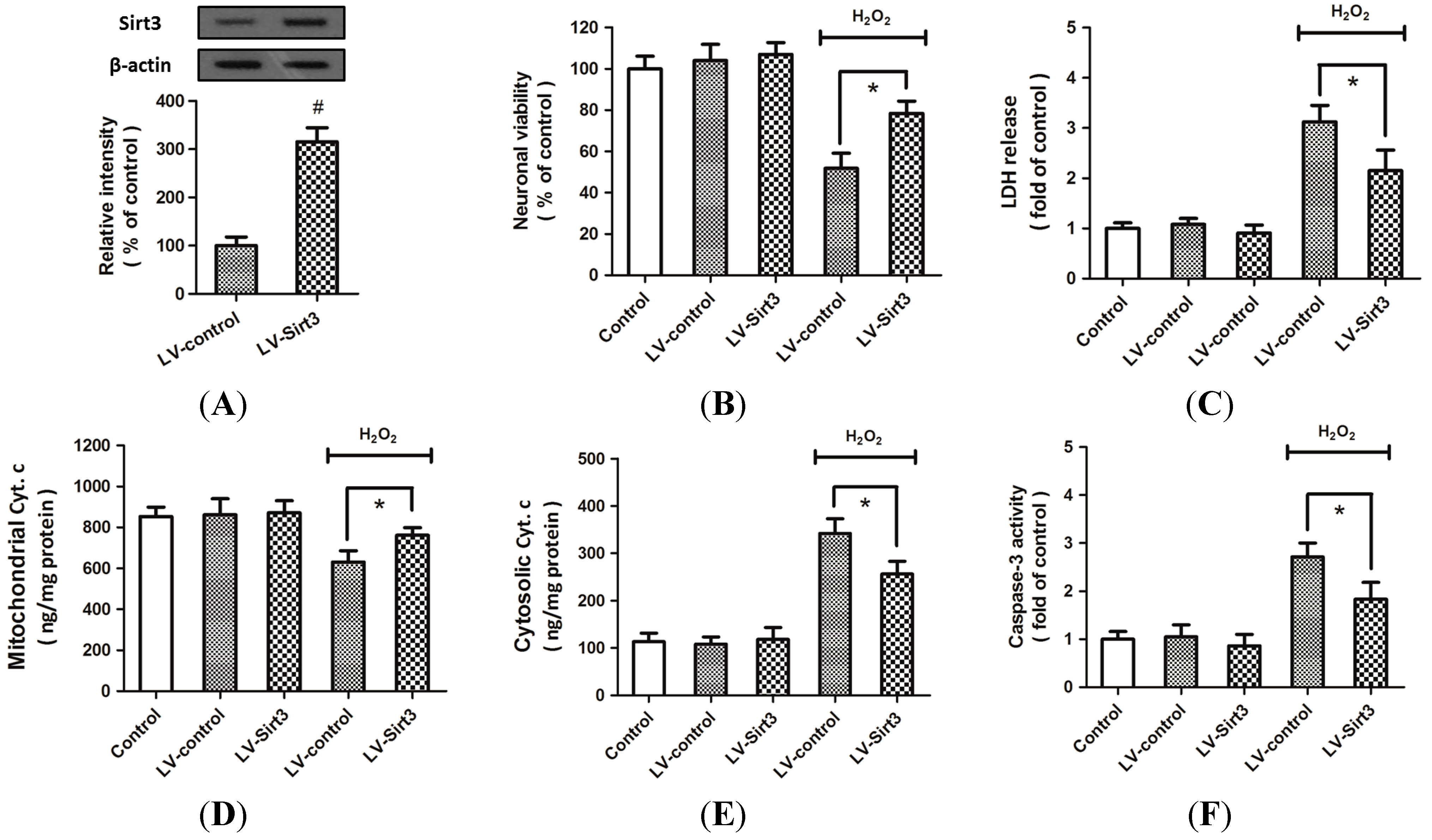
2.2. H2O2-Induced Sirt3 Expression Promotes Neuronal Survival

2.3. Overexpression of Sirt3 Reduces H2O2-Induced Neuronal Injury

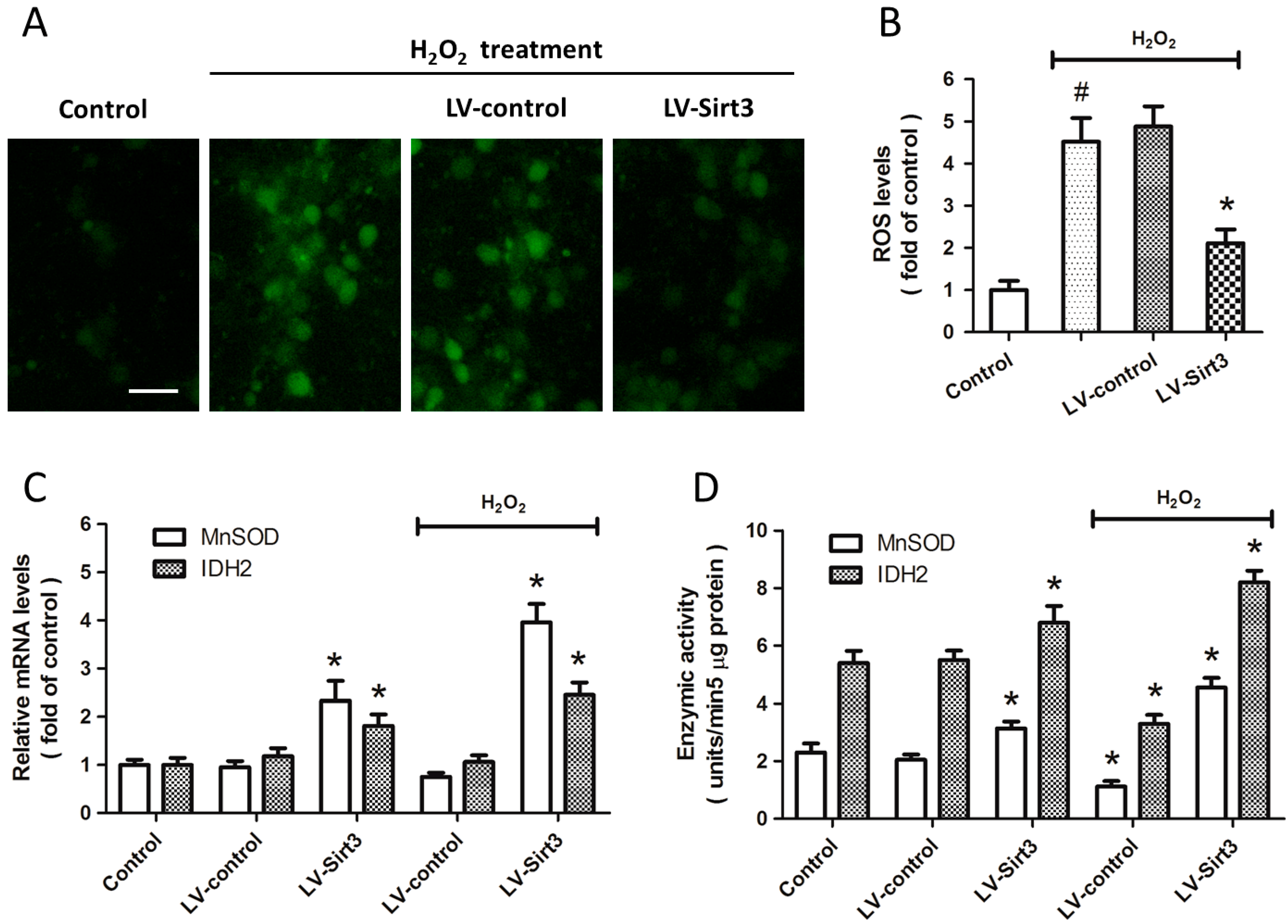
2.4. Overexpression of Sirt3 Inhibits H2O2-Induced Oxidative Stress

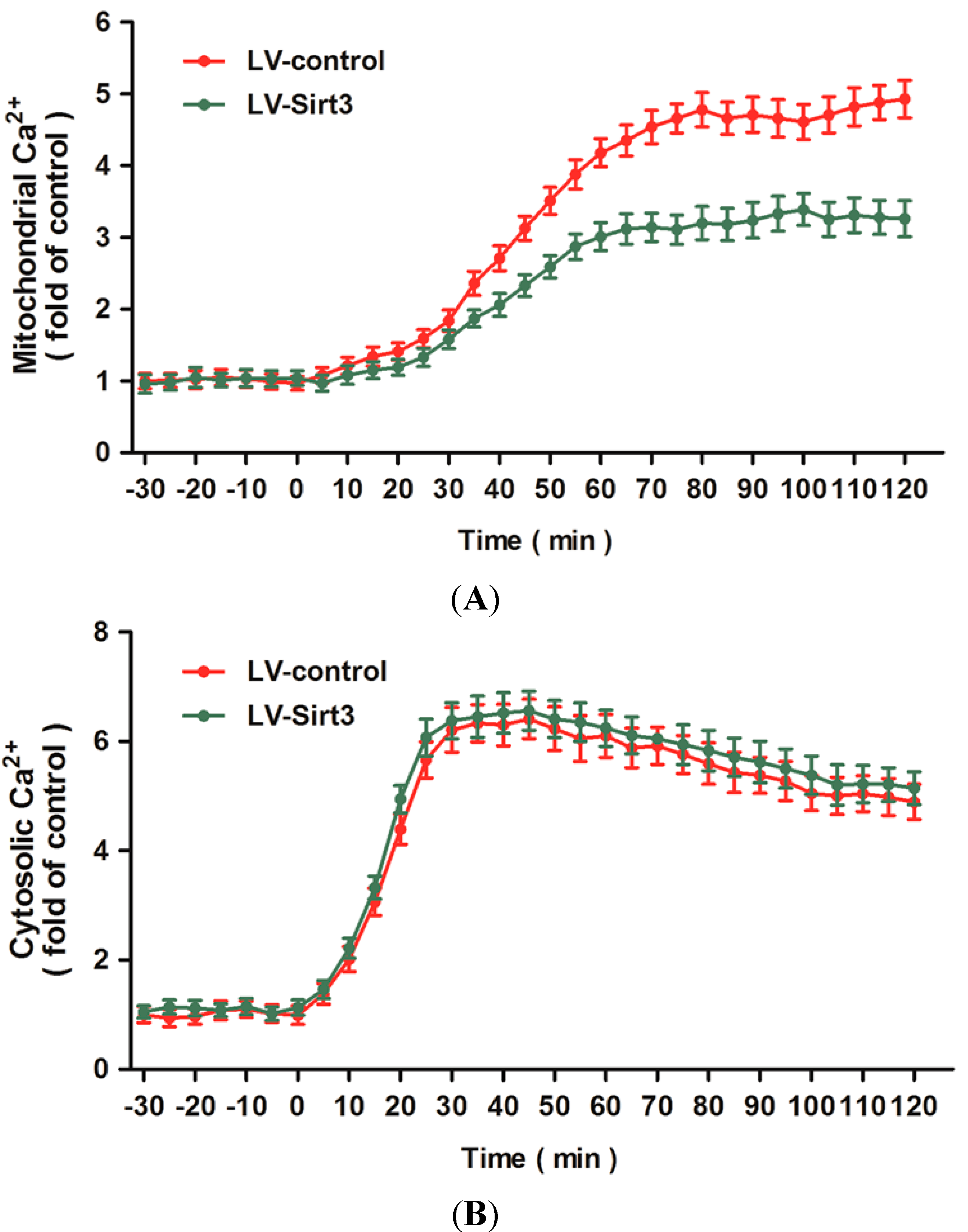
2.5. Overexpression of Sirt3 Blocks H2O2-Induced Mitochondrial Ca2+ Dysregulation

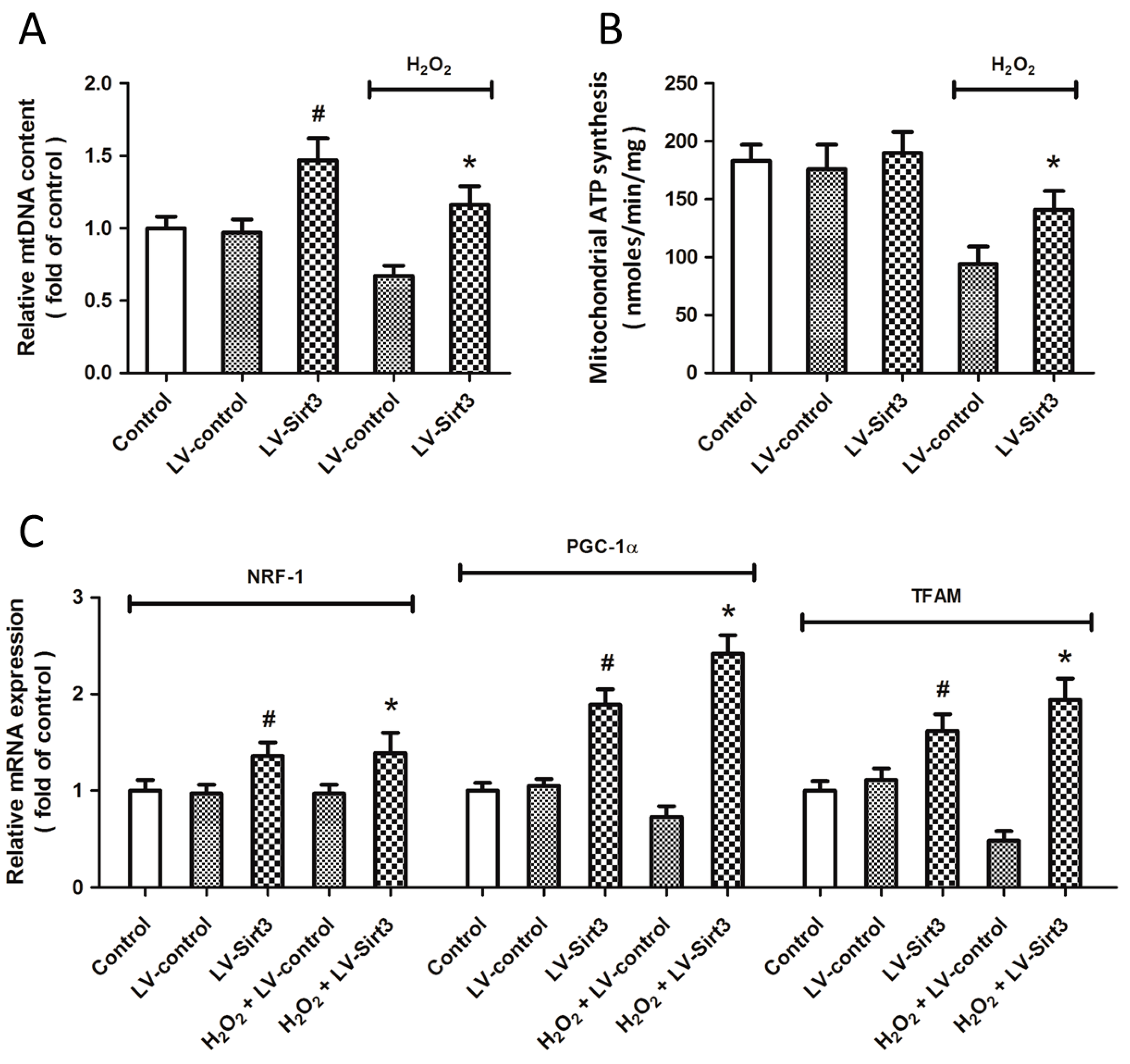
2.6. Overexpression of Sirt3 Promotes Mitochondrial Biogenesis after H2O2 Treatment

3. Discussion
4. Experimental Section
4.1. Primary Culture of Cortical Neurons
4.2. Immunocytochemistry
4.3. Short Interfering RNA (siRNA) and Transfection
4.4. Lentivirus Construction and Transfection
4.5. Neuronal Viability Assay
4.6. Lactate Dehydrogenase (LDH) Release Assay
4.7. Quantification of Cytochrome c Release
4.8. Measurement of Caspase-3 Activity
4.9. Measurement of ROS Generation
4.10. Detection of Antioxidant Enzymes Activities
4.11. Calcium Imaging
4.12. Determination of Mitochondrial DNA Content
4.13. Measurement of ATP Synthesis
4.14. Real-Time RT-PCR
4.15. Western Blot Analysis
4.16. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Avery, S.V. Molecular targets of oxidative stress. Biochem. J. 2011, 434, 201–210. [Google Scholar] [CrossRef]
- Luo, P.; Chen, T.; Zhao, Y.; Xu, H.; Huo, K.; Zhao, M.; Yang, Y.; Fei, Z. Protective effect of Homer 1a against hydrogen peroxide-induced oxidative stress in PC12 cells. Free Radic. Res. 2012, 46, 766–776. [Google Scholar] [CrossRef]
- Krohn, K.; Maier, J.; Paschke, R. Mechanisms of disease: Hydrogen peroxide, DNA damage and mutagenesis in the development of thyroid tumors. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 713–720. [Google Scholar] [CrossRef]
- Stone, J.R. An assessment of proposed mechanisms for sensing hydrogen peroxide in mammalian systems. Arch. Biochem. Biophys. 2004, 422, 119–124. [Google Scholar] [CrossRef]
- Halliwell, B.; Clement, M.V.; Long, L.H. Hydrogen peroxide in the human body. FEBS Lett. 2000, 486, 10–13. [Google Scholar] [CrossRef]
- Armogida, M.; Nistico, R.; Mercuri, N.B. Therapeutic potential of targeting hydrogen peroxide metabolism in the treatment of brain ischaemia. Br. J. Pharmacol. 2012, 166, 1211–1224. [Google Scholar] [CrossRef]
- Melo, A.; Monteiro, L.; Lima, R.M.; Oliveira, D.M.; Cerqueira, M.D.; El-Bacha, R.S. Oxidative stress in neurodegenerative diseases: Mechanisms and therapeutic perspectives. Oxid. Med. Cell. Longev. 2011, 2011, 467180. [Google Scholar]
- Cornelius, C.; Crupi, R.; Calabrese, V.; Graziano, A.; Milone, P.; Pennisi, G.; Radak, Z.; Calabrese, E.J.; Cuzzocrea, S. Traumatic brain injury: Oxidative stress and neuroprotection. Antioxid. Redox Signal. 2013, 19, 836–853. [Google Scholar] [CrossRef]
- Brown, G.C.; Borutaite, V. Regulation of apoptosis by the redox state of cytochrome c. BBA Bioenerg. 2008, 1777, 877–881. [Google Scholar] [CrossRef]
- Green, D.R. Apoptotic pathways: Ten minutes to dead. Cell 2005, 121, 671–674. [Google Scholar] [CrossRef]
- Caroppi, P.; Sinibaldi, F.; Fiorucci, L.; Santucci, R. Apoptosis and human diseases: Mitochondrion damage and lethal role of released cytochrome c as proapoptotic protein. Curr. Med. Chem. 2009, 16, 4058–4065. [Google Scholar] [CrossRef]
- Dias, T.R.; Rato, L.; Martins, A.D.; Simoes, V.L.; Jesus, T.T.; Alves, M.G.; Oliveira, P.F. Insulin deprivation decreases caspase-dependent apoptotic signaling in cultured rat sertoli cells. ISRN Urol. 2013, 2013, 970370. [Google Scholar]
- Simoes, V.L.; Alves, M.G.; Martins, A.D.; Dias, T.R.; Rato, L.; Socorro, S.; Oliveira, P.F. Regulation of apoptotic signaling pathways by 5α-dihydrotestosterone and 17β-estradiol in immature rat Sertoli cells. J. Steroid Biochem. Mol. Biol. 2013, 135, 15–23. [Google Scholar] [CrossRef]
- Chan, P.H. Mitochondria and neuronal death/survival signaling pathways in cerebral ischemia. Neurochem. Res. 2004, 29, 1943–1949. [Google Scholar] [CrossRef]
- Ye, R.; Yang, Q.; Kong, X.; Han, J.; Zhang, X.; Zhang, Y.; Li, P.; Liu, J.; Shi, M.; Xiong, L.; et al. Ginsenoside Rd attenuates early oxidative damage and sequential inflammatory response after transient focal ischemia in rats. Neurochem. Int. 2011, 58, 391–398. [Google Scholar] [CrossRef]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef]
- Bause, A.S.; Haigis, M.C. Sirt3 regulation of mitochondrial oxidative stress. Exp. Gerontol. 2013, 48, 634–639. [Google Scholar] [CrossRef]
- Pillai, V.B.; Sundaresan, N.R.; Kim, G.; Gupta, M.; Rajamohan, S.B.; Pillai, J.B.; Samant, S.; Ravindra, P.V.; Isbatan, A.; Gupta, M.P. Exogenous NAD blocks cardiac hypertrophic response via activation of the Sirt3-LKB1-AMP-activated kinase pathway. J. Biol. Chem. 2010, 285, 3133–3144. [Google Scholar] [CrossRef]
- Lombard, D.B.; Alt, F.W.; Cheng, H.L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 homolog Sirt3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef]
- Kim, S.H.; Lu, H.F.; Alano, C.C. Neuronal Sirt3 protects against excitotoxic injury in mouse cortical neuron culture. PLoS One 2011, 6, e14731. [Google Scholar] [CrossRef]
- Tseng, A.H.; Shieh, S.S.; Wang, D.L. Sirt3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic.Biol. Med. 2013, 63, 222–234. [Google Scholar] [CrossRef]
- Haigis, M.C.; Guarente, L.P. Mammalian sirtuins—Emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006, 20, 2913–2921. [Google Scholar] [CrossRef]
- Finkel, T.; Deng, C.X.; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 460, 587–591. [Google Scholar] [CrossRef]
- Kim, E.J.; Kho, J.H.; Kang, M.R.; Um, S.J. Active regulator of Sirt1 cooperates with Sirt1 and facilitates suppression of p53 activity. Mol. Cell 2007, 28, 277–290. [Google Scholar] [CrossRef]
- Shindler, K.S.; Ventura, E.; Rex, T.S.; Elliott, P.; Rostami, A. Sirt1 activation confers neuroprotection in experimental optic neuritis. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3602–3609. [Google Scholar] [CrossRef]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting FOXO3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar]
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef]
- Park, S.H.; Ozden, O.; Jiang, H.; Cha, Y.I.; Pennington, J.D.; Aykin-Burns, N.; Spitz, D.R.; Gius, D.; Kim, H.S. Sirt3, Mitochondrial ROS, ageing, and carcinogenesis. Int. J. Mol. Sci. 2011, 12, 6226–6239. [Google Scholar] [CrossRef]
- Verdin, E.; Hirschey, M.D.; Finley, L.W.; Haigis, M.C. Sirtuin regulation of mitochondria: Energy production, apoptosis, and signaling. Trends Biochem. Sci. 2010, 35, 669–675. [Google Scholar] [CrossRef]
- Guarente, L. Mitochondria—A nexus for aging, calorie restriction, and sirtuins? Cell 2008, 132, 171–176. [Google Scholar] [CrossRef]
- Smith, K.T.; Workman, J.L. Introducing the acetylome. Nat. biotechnol. 2009, 27, 917–919. [Google Scholar] [CrossRef]
- Marfe, G.; Tafani, M.; Indelicato, M.; Sinibaldi-Salimei, P.; Reali, V.; Pucci, B.; Fini, M.; Russo, M.A. Kaempferol induces apoptosis in two different cell lines via Akt inactivation, Bax and Sirt3 activation, and mitochondrial dysfunction. J. Cell. Biochem. 2009, 106, 643–650. [Google Scholar] [CrossRef]
- Allison, S.J.; Milner, J. Sirt3 is pro-apoptotic and participates in distinct basal apoptotic pathways. Cell Cycle 2007, 6, 2669–2677. [Google Scholar] [CrossRef]
- Yang, H.; Yang, T.; Baur, J.A.; Perez, E.; Matsui, T.; Carmona, J.J.; Lamming, D.W.; Souza-Pinto, N.C.; Bohr, V.A.; Rosenzweig, A.; et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 2007, 130, 1095–1107. [Google Scholar] [CrossRef]
- Sundaresan, N.R.; Samant, S.A.; Pillai, V.B.; Rajamohan, S.B.; Gupta, M.P. Sirt3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol. Cell. Biol. 2008, 28, 6384–6401. [Google Scholar] [CrossRef]
- Kincaid, B.; Bossy-Wetzel, E. Forever young: Sirt3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front. Aging Neurosci. 2013, 5, 48. [Google Scholar]
- Rato, L.; Duarte, A.I.; Tomas, G.D.; Santos, M.S.; Moreira, P.I.; Socorro, S.; Cavaco, J.E.; Alves, M.G.; Oliveira, P.F. Pre-diabetes alters testicular PGC1-α/Sirt3 axis modulating mitochondrial bioenergetics and oxidative stress. BBA Bioenerg. 2014, 1837, 335–344. [Google Scholar] [CrossRef]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a new target of PGC-1α, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS One 2010, 5, e11707. [Google Scholar] [CrossRef]
- Hafner, A.V.; Dai, J.; Gomes, A.P.; Xiao, C.Y.; Palmeira, C.M.; Rosenzweig, A.; Sinclair, D.A. Regulation of the mPTP by Sirt3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging 2010, 2, 914–923. [Google Scholar]
- Shulga, N.; Wilson-Smith, R.; Pastorino, J.G. Sirtuin-3 deacetylation of cyclophilin D induces dissociation of hexokinase II from the mitochondria. J. Cell Sci. 2010, 123, 894–902. [Google Scholar] [CrossRef]
- Schild, L.; Huppelsberg, J.; Kahlert, S.; Keilhoff, G.; Reiser, G. Brain mitochondria are primed by moderate Ca2+ rise upon hypoxia/reoxygenation for functional breakdown and morphological disintegration. J. Biol. Chem. 2003, 278, 25454–25460. [Google Scholar] [CrossRef]
- Starkov, A.A.; Chinopoulos, C.; Fiskum, G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium 2004, 36, 257–264. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Tanaka, K.; Iijima, T.; Mishima, T.; Suga, K.; Akagawa, K.; Iwao, Y. Ca2+ buffering capacity of mitochondria after oxygen-glucose deprivation in hippocampal neurons. Neurochem. Res. 2009, 34, 221–226. [Google Scholar] [CrossRef]
- Martinez-Sanchez, M.; Striggow, F.; Schroder, U.H.; Kahlert, S.; Reymann, K.G.; Reiser, G. Na+ and Ca2+ homeostasis pathways, cell death and protection after oxygen-glucose-deprivation in organotypic hippocampal slice cultures. Neuroscience 2004, 128, 729–740. [Google Scholar] [CrossRef]
- Peng, T.I.; Jou, M.J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188. [Google Scholar] [CrossRef]
- Zong, W.X.; Li, C.; Hatzivassiliou, G.; Lindsten, T.; Yu, Q.C.; Yuan, J.; Thompson, C.B. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J. Cell Biol. 2003, 162, 59–69. [Google Scholar] [CrossRef]
- Pinton, P.; Ferrari, D.; Rapizzi, E.; di Virgilio, F.; Pozzan, T.; Rizzuto, R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: Significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001, 20, 2690–2701. [Google Scholar] [CrossRef]
- Scorrano, L.; Korsmeyer, S.J. Mechanisms of cytochrome c release by proapoptotic BCL-2 family members. Biochem. Biophys. Res. Commun. 2003, 304, 437–444. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Chalmers, S. The integration of mitochondrial calcium transport and storage. J. Bioenerg. Biomembr. 2004, 36, 277–281. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef]
- Fukumori, R.; Takarada, T.; Nakazato, R.; Fujikawa, K.; Kou, M.; Hinoi, E.; Yoneda, Y. Selective inhibition by ethanol of mitochondrial calcium influx mediated by uncoupling protein-2 in relation to N-methyl-d-aspartate cytotoxicity in cultured neurons. PLoS One 2013, 8, e69718. [Google Scholar]
- Vassalle, M.; Lin, C.I. Calcium overload and cardiac function. J. Biomed. Sci. 2004, 11, 542–565. [Google Scholar] [CrossRef]
- Tian, Y.; Li, B.; Shi, W.Z.; Chang, M.Z.; Zhang, G.J.; Di, Z.L.; Liu, Y. Dynamin-related protein 1 inhibitors protect against ischemic toxicity through attenuating mitochondrial Ca2+ uptake from endoplasmic reticulum store in PC12 cells. Int. J. Mol. Sci. 2014, 15, 3172–3185. [Google Scholar] [CrossRef]
- Cheng, A.; Hou, Y.; Mattson, M.P. Mitochondria and neuroplasticity. ASN Neuro 2010, 2, e00045. [Google Scholar]
- Seo, A.Y.; Joseph, A.M.; Dutta, D.; Hwang, J.C.; Aris, J.P.; Leeuwenburgh, C. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J. Cell Sci. 2010, 123, 2533–2542. [Google Scholar] [CrossRef]
- McLeod, C.J.; Pagel, I.; Sack, M.N. The mitochondrial biogenesis regulatory program in cardiac adaptation to ischemia—A putative target for therapeutic intervention. Trends Cardiovasc. Med. 2005, 15, 118–123. [Google Scholar] [CrossRef]
- Ljubicic, V.; Joseph, A.M.; Saleem, A.; Uguccioni, G.; Collu-Marchese, M.; Lai, R.Y.; Nguyen, L.M.; Hood, D.A. Transcriptional and post-transcriptional regulation of mitochondrial biogenesis in skeletal muscle: Effects of exercise and aging. BBA Gen. Subj. 2010, 1800, 223–234. [Google Scholar] [CrossRef]
- Vina, J.; Gomez-Cabrera, M.C.; Borras, C.; Froio, T.; Sanchis-Gomar, F.; Martinez-Bello, V.E.; Pallardo, F.V. Mitochondrial biogenesis in exercise and in ageing. Adv. Drug Deliv. Rev. 2009, 61, 1369–1374. [Google Scholar] [CrossRef]
- Nisoli, E.; Carruba, M.O. Nitric oxide and mitochondrial biogenesis. J. Cell Sci. 2006, 119, 2855–2862. [Google Scholar] [CrossRef]
- Weir, H.J.; Murray, T.K.; Kehoe, P.G.; Love, S.; Verdin, E.M.; O’Neill, M.J.; Lane, J.D.; Balthasar, N. CNS Sirt3 expression is altered by reactive oxygen species and in Alzheimer’s disease. PLoS One 2012, 7, e48225. [Google Scholar] [CrossRef]
- Sano, R.; Annunziata, I.; Patterson, A.; Moshiach, S.; Gomero, E.; Opferman, J.; Forte, M.; d’Azzo, A. GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca2+-dependent mitochondrial apoptosis. Mol. Cell 2009, 36, 500–511. [Google Scholar] [CrossRef]
- Kaufman, R.J.; Malhotra, J.D. Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics. BBA Mol. Cell Res. 2014, 1843, 2233–2239. [Google Scholar]
- Baumeister, P.; Dong, D.; Fu, Y.; Lee, A.S. Transcriptional induction of GRP78/BiP by histone deacetylase inhibitors and resistance to histone deacetylase inhibitor-induced apoptosis. Mol. Cancer Ther. 2009, 8, 1086–1094. [Google Scholar] [CrossRef]
- Ageberg, M.; Rydstrom, K.; Relander, T.; Drott, K. The histone deacetylase inhibitor valproic acid sensitizes diffuse large B-cell lymphoma cell lines to CHOP-induced cell death. Am. J. Transl. Res. 2013, 5, 170–183. [Google Scholar]
- Redmond, L.; Kashani, A.H.; Ghosh, A. Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron 2002, 34, 999–1010. [Google Scholar] [CrossRef]
- Chen, T.; Yang, Y.F.; Luo, P.; Liu, W.; Dai, S.H.; Zheng, X.R.; Fei, Z.; Jiang, X.F. Homer1 knockdown protects dopamine neurons through regulating calcium homeostasis in an in vitro model of Parkinson’s disease. Cell. Signal. 2013, 25, 2863–2870. [Google Scholar] [CrossRef]
- Chen, H.; Hu, C.J.; He, Y.Y.; Yang, D.I.; Xu, J.; Hsu, C.Y. Reduction and restoration of mitochondrial dna content after focal cerebral ischemia/reperfusion. Stroke 2001, 32, 2382–2387. [Google Scholar] [CrossRef]
- Parone, P.A.; Da Cruz, S.; Han, J.S.; McAlonis-Downes, M.; Vetto, A.P.; Lee, S.K.; Tseng, E.; Cleveland, D.W. Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J. Neurosci. 2013, 33, 4657–4671. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dai, S.-H.; Chen, T.; Wang, Y.-H.; Zhu, J.; Luo, P.; Rao, W.; Yang, Y.-F.; Fei, Z.; Jiang, X.-F. Sirt3 Protects Cortical Neurons against Oxidative Stress via Regulating Mitochondrial Ca2+ and Mitochondrial Biogenesis. Int. J. Mol. Sci. 2014, 15, 14591-14609. https://doi.org/10.3390/ijms150814591
Dai S-H, Chen T, Wang Y-H, Zhu J, Luo P, Rao W, Yang Y-F, Fei Z, Jiang X-F. Sirt3 Protects Cortical Neurons against Oxidative Stress via Regulating Mitochondrial Ca2+ and Mitochondrial Biogenesis. International Journal of Molecular Sciences. 2014; 15(8):14591-14609. https://doi.org/10.3390/ijms150814591
Chicago/Turabian StyleDai, Shu-Hui, Tao Chen, Yu-Hai Wang, Jie Zhu, Peng Luo, Wei Rao, Yue-Fan Yang, Zhou Fei, and Xiao-Fan Jiang. 2014. "Sirt3 Protects Cortical Neurons against Oxidative Stress via Regulating Mitochondrial Ca2+ and Mitochondrial Biogenesis" International Journal of Molecular Sciences 15, no. 8: 14591-14609. https://doi.org/10.3390/ijms150814591
APA StyleDai, S. -H., Chen, T., Wang, Y. -H., Zhu, J., Luo, P., Rao, W., Yang, Y. -F., Fei, Z., & Jiang, X. -F. (2014). Sirt3 Protects Cortical Neurons against Oxidative Stress via Regulating Mitochondrial Ca2+ and Mitochondrial Biogenesis. International Journal of Molecular Sciences, 15(8), 14591-14609. https://doi.org/10.3390/ijms150814591