
Whole Exome Sequencing in Females with Autism Implicates Novel and Candidate Genes

 ,
, 
Abstract
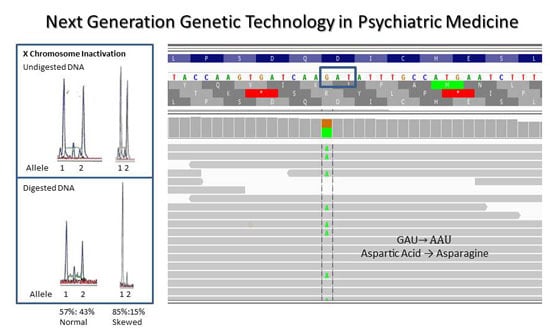
:
1. Introduction
2. Results

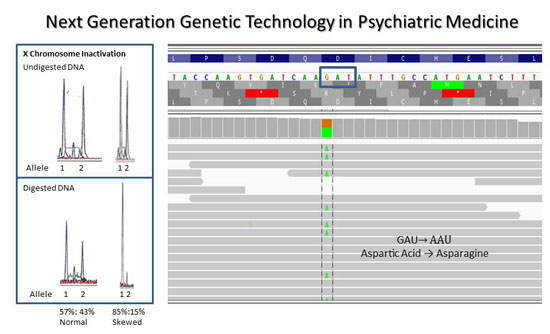{kind=link}
| ID Number XCI | Gene Symbol (Category) * | Chromosome Position (Hg19) | Genomic Variant | Classification Score | GERP2 | PolyPhen2 Score | SIFT Score | Blosum Score/Blosum62 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP Effect | Amino Acid | SNP Codon | SNP Function | VCF Allele Depth | ||||||||
| HI0405 46%:54% | KCNC2 (P) [12] | 12:75444895 | Non-synon | p.F296C | tTt/tGt | Missense | 162, 30 | 0.790 | 0 | 0.984 | 0 | 0.787/−2 |
| ASPM (F) [23] | 1:197112823 | Non-synon | p.R186G | Aga/Gga | Missense | 69, 53 | 0.777 | 0.007 | 0.994 | 0 | 0.787/−2 | |
| TAF3 (F) [24] | 10:8006662 | Non-synon | p.R396G | Cga/Gga | Missense | 69, 60 | 0.740 | 0 | 0.956 | 0 | 0.787/−2 | |
| FLRT2 (F) [25] | 14:86089552 | Non-synon | p.H564R | cAt/cGt | Missense | 28, 29 | 0.736 | 0 | 0.958 | 0.02 | 0.787/0 | |
| SYTL4 (F) [26] | X:99941091 | Non-synon | p.H448D | Cat/Gat | Missense | 36, 37 | 0.710 | 0.006 | 1 | 0.03 | 0.787/−1 | |
| HI0555/58%:42% | SETD2 (A) [12,27,28] | 3:47164293 | Non-synon | p.K610O | aaG/aaT | Missense | 23, 21 | 0.778 | 0 | 0.970 | 0 | 8.522/0 |
| HI0558 52%:48% | IFT122 (F) [29] | 3:129238526 | Non-synon | p.R986H | cGc/cAc | Missense | 58, 61 | 0.795 | 0.008 | 0.989 | 0 | 0.074/0 |
| NUP98 (F) [30] | 11:3756456 | Non-synon | p.R519W | Cgg/Tgg | Missense | 12, 9 | 0.793 | 0.006 | 0.999 | 0 | 24.449/−3 | |
| ZKSCAN1 (F) [31] | 7:99621487 | Non-synon | p.R119C | Cgc/Tgc | Missense | 8, 5 | 0.782 | 0.006 | 1 | 0 | 0.787/−3 | |
| DNAH8 (F) [32] | 6:38834433 | Non-synon | p.M1971V | Atg/Gtg | Missense | 8, 14 | 0.783 | 0.009 | 0.902 | 0 | 4.182/1 | |
| CENPJ (F) [33] | 13:25487103 | Non-synon | p.M20L | Atg/Ttg | Missense | 45, 42 | 0.761 | 0.007 | 0.991 | 0.01 | 4.312/2 | |
| TRIM5 (F) [34] | 11:5878550 | Non-synon | p.R127P | cGc/cCc | Missense | 42, 24 | 0.742 | 0.0089 | 0.971 | 0 | 0.787/−2 | |
| PCDH8 (A) [35] | 13:53421358 | Non-synon | p.A404V | gCg/gTg | Missense | 13, 14 | 0.731 | 0 | 0.924 | 0 | 0.787/0 | |
| CTNNA3 (A) [36] | 10:68381521 | Non-synon | p.M434V | Atg/Gtg | Missense | 30, 17 | 0.723 | 0.009 | 0.942 | 0.01 | 4.183/1 | |
| PPP1R18 (P) [37] | 6:30652604 | Non-synon | p.S397P | Tct/Cct | Missense | 12, 6 | 0.703 | 0.005 | 0.958 | 0 | 0.787/−1 | |
| HI0605 53%:47% | CCDC64 (A) [38] | 12:120428012 | Non-synon | p.A113T | Gcc/Acc | Missense | 13, 24 | 0.794 | 0.006 | 0.96 | 0 | 4.774/−1 |
| DCHS1 (F) [39] | 11:6662745 | Codon Change Plus Codon Insertion | p.L32LW | ctg/ctCTGg | None | 11, 8 | 0.700 | 0 | 0.979 | 0.02 | 0/0 | |
| HI0714 40%:60% | TRAK2 (F) [40] | 2:202264189 | Non-synon | p.R130G | Cga/Gga | Missense | 31, 27 | 0.805 | 0.010 | 1 | 0 | 0.064/−2 |
| MPST (F) [41] | 22:37425405 | Utr_3_Prime | p.Null-1Null | Null | None | 20, 21 | 0.731 | 0.008 | 1 | 0 | 0.787/−2 | |
| FSTL3 (F) [42] | 19:680331 | Non-synon | p.R115H | cGc/cAc | Missense | 7, 3 | 0.729 | 0.010 | 1 | 0.01 | 8.723/0 | |
| HI0751 16%:84% | IST1 (F) [43,44] | 16:71956522 | Non-synon | p.P232R | cCc/cGc | Missense | 63, 50 | 0.754 | 0.008 | 0.972 | 0.01 | 0.787/−2 |
| KIF27 (P) [43,44] | 9:86474259 | Non-synon | p.S921P | Tca/Cca | Missense | 22, 25 | 0.730 | 0.009 | 0.970 | 0.01 | 0.787/−1 | |
| PCDHGA1 (P) [45] | 5:140870234 | Non-synon | p.V475A | gTg/gCg | Missense | 152, 141 | 0.713 | 0 | 0.995 | 0 | 0.787/0 | |
| HI0765 77%:23% | SEMA3F (P) [46] | 3:50225525 | Non-synon | p.R679W | Cgg/Tgg | Missense | 21, 7 | 0.793 | 0.006 | 1 | 0 | 0.787/−3 |
| SCN11A (P) [47] | 3:38968409 | Frame-shift | p.-166SS? | -/TCTT CACT | None | 23, 17 | 0.748 | 0.008 | 0.982 | 0 | 0/0 | |
| GPRASP2 (F) [48] | X:101972234 | Non-synon | p.R812C | Cgt/Tgt | Missense | 69, 51 | 0.706 | 0 | 0.989 | 0.01 | 0.787/−3 | |
| HI0793 37%:64% | ELTD1 (F) [49] | 1:79356850 | Non-synon | p.C687R | Tgt/Cgt | Missense | 50, 25 | 0.807 | 0.008 | 0.998 | 0 | 10.199/−3 |
| DENND3 (F) [50] | 8:142161854 | Non-synon | p.S252L | tCg/tTg | Missense | 40, 20 | 0.806 | 0.006 | 0.992 | 0 | 0.787/−2 | |
| MIDN (F) [51] | 19:1250353 | Non-synon | p.C19G | Tgc/Ggc | Missense | 8, 3 | 0.793 | 0.008 | 0.998 | 0 | 17.850/−3 | |
| RADIL (F) [52] | 7:4917586 | Non-synon | p.P61R | cCt/cGt | Missense | 42, 27 | 0.774 | 0.004 | 1 | 0 | 14.302/−2 | |
| EXOC7 (P) [53,54] | 17:74097856 | Non-synon | p.R71Q | cGg/cAg | Missense | 36, 46 | 0.755 | 0.006 | 0.992 | 0.01 | 0.059/1 | |
| PLCE1 (F) [55] | 10:96064250 | Non-synon | p.H1515Y | Cat/Tat | Missense | 22, 14 | 0.737 | 0.008 | 0.999 | 0.02 | 7.078/2 | |
| KDM5A (F) [56] | 12:416817 | Non-synon | p.A1244T | Gcc/Acc | Missense | 42, 25 | 0.706 | 0 | 0.993 | 0 | 0.157/0 | |
| ARAP2 (F) [57] | 4:36115873 | Non-synon | p.D1358N | Gat/Aat | Missense | 70, 53 | 0.702 | 0.008 | 0.907 | 0.02 | 12.052/1 | |
| HI0855 44%:56% | CHAC1 (F) [58] | 15:41247844 | Frame- shift | p.-177? | -/T | None | 23, 19 | 0.941 | 0.009 | 0.997 | 0 | 0.787/0 |
| MIDN (F) [59] | 19:1250353 | Non-synon | p.C19G | Tgc/Ggc | Missense | 3, 7 | 0.793 | 0.008 | 0.998 | 0 | 17.85/−3 | |
| CDH6 (P) [60] | 5:31323179 | Non-synon | p.R712G | Aga/Gga | Missense | 76, 81 | 0.749 | 0.003 | 0.988 | 0.01 | 0.787/−2 | |
| HI0868 58%:42% | CHST3 (P) [61] | 10:73767391 | Non-synon | p.Y200C | tAc/tGc | Missense | 43, 30 | 0.759 | 0 | 0.998 | 0.02 | 17.489/−2 |
| FBXO5 (P) [62] | 6:153296090 | Non-synon | p.H256R | cAt/cGt | Missense | 103, 76 | 0.756 | 0.003 | 1 | 0 | 0.787/0 | |
| HI0890/NI | BTAF1 (A) [63] | 10:93719892 | Non-synon | p.Y414F | tAt/tTt | Missense | 29, 16 | 0.765 | 0.005 | 0.997 | 0 | 3/3 |
| HI1143/14%:86% | CLPTM1 (F) [64] | 19:45476442 | Non-synon | p.N94I | aAc/aTc | Missense | 30, 27 | 0.744 | 0.007 | 1 | 0 | 0.787/−3 |
| HI1157 61%:39% | AMIGO1 (F) [65] | 1:110050220 | Non-synon | p.R438W | Cgg/Tgg | Missense | 42, 28 | 0.806 | 0 | 0.994 | 0 | 0.787/−3 |
| PEX5 (P) [66] | 12:7362354 | Non-synon | p.R560C | Cgc/Tgc | Missense | 71, 50 | 0.801 | 0.006 | 1 | 0 | 0.787/−3 | |
| KIF23 (P) [67] | 15:69718474 | Non-synon | p.E266A | gAa/gCa | Missense | 52, 48 | 0.775 | 0.007 | 0.998 | 0 | 0.787/−1 | |
| HI1228 42%:58% | FOXA3 (P) [68] | 19:46376266 | Non-synon | p.G334R | Gga/Aga | Missense | 30, 29 | 0.801 | 0.004 | 0.975 | 0 | 26.000/−2 |
| FRMD4A (F) [69] | 10:13698719 | Non-synon | p.S941L | tCg/tTg | Missense | 6, 8 | 0.775 | 0 | 0.957 | 0 | 0.064/−2 | |
| HI1305 73%:27% | PIR (F) [70] | X:15474053 | Non-synon | p.E132G | gAa/gGa | Missense | 32, 28 | 0.811 | 0.007 | 0.962 | 0 | 0.213/−2 |
| DNAH2 (F) [71] | 17:7637815 | Non-synon | p.I255T | aTa/aCa | Missense | 25, 20 | 0.793 | 0.010 | 0.942 | 0 | 7.006/−1 | |
| HI1375 17%:83% | LHX2 (F) [72] | 9:126777468 | Non-synon | p.A130P | Gct/Cct | Missense | 50, 34 | 0.775 | 0.006 | 0.998 | 0 | 0.787/−1 |
| EPHA4 (P) [73] | 2:222428829 | Non-synon | p.A148T | Gct/Act | Missense | 18, 25 | 0.744 | 0 | 0.977 | 0 | 0.787/0 | |
| HI1402/85%:15% | IL1RAPL1 (A) [12,74] | X:29973282 | Non-synon | p.P478Q | cCa/cAa | Missense | 57, 42 | 0.761 | 0 | 0.999 | 0 | 7.684/−1 |
| HI1422 56%:44% | RTN4R (F) [75] | 22:20230387 | Non-synon | p.S89L | tCg/tTg | Missense | 11, 18 | 0.778 | 0.004 | 0.998 | 0 | 0.064/−4 |
| DHX8 (F) [76] | 17:41606990 | Non-synon | p.R282H | cGc/cAc | Missense | 4, 8 | 0.724 | 0.010 | 1 | 0 | 0.787/0 | |
| HI1433 93%:7% | FAT2 (P) [77,78,79] | 5:150947258 | Non-synon | p.T411I | aCt/aTt | Missense | 32, 29 | 0.757 | 0 | 0.992 | 0 | 6.276/−1 |
| FITM2 (F) [80,81] | 20:42939631 | Non-synon | p.R52H | cGc/cAc | Missense | 6, 5 | 0.750 | 0.008 | 0.999 | 0.02 | 8.723/0 | |
| RALGPS2 (F) [82,83] | 1:179064186 | Non-synon | p.V342M | Gtg/Atg | Missense | 69, 73 | 0.734 | 0.010 | 0.970 | 0 | 0.787/1 | |
| HI1739 31%:69% | ASB3 (F) [84] | 2:53941656 | Non-synon | p.S208N | aGc/aAc | Missense | 53, 30 | 0.773 | 0.007 | 0.980 | 0 | 0.403/1 |
| AKAP6 (F) [85] | 14:33291533 | Non-synon | p.D1504P | gAt/gTt | Missense | 43, 41 | 0.761 | 0 | 0.983 | 0.01 | 0.787/−3 | |
| GRM4 (A) [86] | 6:34100940 | Non-synon | p.D111N | Gac/Aac | Missense | 58, 51 | 0.726 | 0.005 | 0.993 | 0.02 | 0.787/1 | |
| HI1884/48%:52% | TRAF3IP1 (F) [87] | 2:239306221 | Non-synon | p.M537T | aTg/aCg | Missense | 13, 19 | 0.759 | 0.007 | 0.999 | 0.01 | 0.033/−1 |
| HI1954 23%:77% | DDX5 (P) [88] | 17:62500102 | Non-synon | p.S146C | tCt/tGt | Missense | 88, 91 | 0.788 | 0.007 | 0.950 | 0 | 0.787/−1 |
| KIF5A (P) [89] | 12:57974875 | Non-synon | p.R891Q | cGg/cAg | Missense | 25, 19 | 0.767 | 0.005 | 0.989 | 0 | 0.787/1 | |
| HI2126/41%:59% | ANKRD11 (A) [90,91] | 16:89351565 | Non-synon | p.T461R | aCa/aGa | Missense | 20, 11 | 0.767 | 0 | 0.961 | 0 | 0.787/−1 |
| HI2172 61%:39% | TNFRSF21 (F) [92] | 6:47221105 | Non-synon | p.W465R | Tgg/Agg | Missense | 8, 16 | 0.805 | 0.004 | 1 | 0 | 10.610/−3 |
| LRSAM1 (F) [93,94] | 9:130253524 | Non-synon | p.L484M | Ctg/Atg | Missense | 18, 23 | 0.796 | 0.010 | 0.935 | 0 | 0.787/−1 | |
| CYFIP1 (A) [95] | 15:22960872 | Non-synon | p.D716N | Gac/Aac | Missense | 28, 30 | 0.767 | 0.009 | 0.999 | 0 | 0.067/1 | |
| HI2215 60%:40% | FPGT- TNNI3K (F) [96] | 1:74836022 | Non-synon | p.G673V | gGc/gTc | Missense | 34, 53 | 0.802 | 0.009 | 0.998 | 0 | 0.787/−3 |
| MAP1A (P) [96] | 15:43820377 | Non-synon | p.P2235S | Ccc/Tcc | Missense | 65, 67 | 0.793 | 0 | 0.998 | 0 | 0.787/−1 | |
| MAP1A (P) [96] | 15:43819396 | Non-synon | p.Y1908H | Tac/Cac | Missense | 13, 21 | 0.782 | 0.004 | 0.994 | 0 | 0.787/2 | |
| HI2244 32%:68% | KDM5B (P) [97,98,99] | 1:202724501 | Non-synon | p.Y320K | aTa/aAa | Missense | 52, 47 | 0.808 | 0.007 | 0.988 | 0 | 0.787/−4 |
| TRPM2 (P) [97,98,99] | 21: 45826549 | Non-synon | p.I954F | Atc/Ttc | Missense | 17, 16 | 0.786 | 0.008 | 0.997 | 0 | 12.615/−3 | |
| RAI1 (A) [100] | 17:17699284 | Non-synon | p.P959A | Ccc/Gcc | Missense | 37, 26 | 0.763 | 0 | 0.992 | 0 | 0.787/−1 | |
| HI2278 8%:92% | DCHS1 (F) [39] | 11:6662745 | Codon Change Plus Codon Insertion | p.L32LW | Ctg/ctCTGg | None | 12, 11 | 0.700 | 0 | 0.979 | 0.02 | 0/0 |
| HI2843 25%:75% | UBE2E2 (P) [101] | 3:23631311 | Non-synon | p.Y198H | Tac/Cac | Missense | 64, 33 | 0.787 | 0 | 0.994 | 0 | 0.787/2 |
| PCDHB15 (P) [45] | 5:140625618 | Non-synon | p.R157W | Cgg/Tgg | Missense | 33, 61 | 0.754 | 0.007 | 0.942 | 0 | 24.449/−3 | |
| SCG2 (F) [102] | 2:224463227 | Non-synon | p.E257D | gaG/gaC | Missense | 70, 64 | 0.750 | 0 | 0.993 | 0.01 | 0.787/2 | |
| HI2879 18%:82% | TRIM9 (F) [103] | 14:51448554 | Non-synon | p.R623Q | cGg/cAg | Missense | 64, 69 | 0.774 | 0 | 1 | 0 | 0.787/1 |
| SOX7 (A) [104] | 8:10583274 | Non-synon | p.Y432H | Tac/Cac | Missense | 25, 33 | 0.766 | 0 | 0.997 | 0 | 0.787/2 | |
| ANKFN1 (P) [105] | 17:54559849 | Non-synon | p.G744R | Ggg/Agg | Missense | 28, 16 | 0.715 | 0 | 0.995 | 0.04 | 26.000/−2 | |
| DCHS1 (F) [39] | 11:6662745 | Codon Change Plus Codon Insertion | p.L32LW | ctg/ctCTGg | None | 11, 12 | 0.700 | 0 | 0.979 | 0.02 | 0/0 | |
| HI2898 85%:15% | MPHOSPH8 (F) [106] | 13:20242552 | Non-synon | p.Y736C | tAc/tGc | Missense | 39, 28 | 0.779 | 0.007 | 0.999 | 0 | 0.787/−2 |
| GABRQ (A) [107] | X:151819020 | Non-synon | p.S292F | tCc/tTc | Missense | 9, 16 | 0.749 | 0.008 | 0.963 | 0 | 0.787/−2 | |
| Gene Variant Category | Number of Subjects (%) | Number of Genes (%) | Range |
|---|---|---|---|
| Known ASD Genes | 11 (37%) | 12 (15%) | 0–2 |
| Paralogues of Known ASD Genes | 16 (53%) | 23 (30%) | 0–2 |
| Neurodevelopmental Function Genes | 23 (77%) | 43 (54%) | 0–7 |
3. Discussion
3.1. Single Gene Variants
3.2. Involvement of Skewness of X Chromosome Inactivation and Putative Disease Causing Genes
3.3. Other Autosomal Putative Disease Causing Genes
4. Experimental Section
4.1. Samples from Females with Autism
4.2. Whole Exome Sequencing
4.2.1. Exome Sequencing Methods
4.2.2. Data Analysis
4.2.3. Gene Filtration/Selection Parameters
4.2.4. Clinical Relevance to ASD
4.3. X Chromosome Inactivation in Females with Autism
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 4th ed.; American Psychiatric Association Press: Washington, DC, USA, 2000. [Google Scholar]
- Johnson, C.P.; Myers, S.M. The Council on Children With Disabilities. Identification and evaluation of children with autism spectrum disorders. Pediatrics 2007, 120, 1183–1215. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.; Risi, S.; Lambrecht, L.; Cook, E.H., Jr.; Leventhal, B.L.; DiLavore, P.C.; Pickles, A.; Rutter, M. The autism diagnostic observation schedule-generic: A standard measure of social and communication deficits associated with the spectrum of autism. J. Autism Dev. Disord. 2000, 30, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Le Couteur, A.; Lord, C.; Ruter, M. Autism Diagnostic Interview-Reviewed (ADI-R); Western Psychological Services: Los Angeles, CA, USA, 2003. [Google Scholar]
- Constantino, J.N.; Davis, S.A.; Todd, R.D.; Schindler, M.K.; Gross, M.M.; Brophy, S.L.; Metzger, L.M.; Shoushtari, C.S.; Splinter, R.; Reich, W. Validation of a brief quantitative measure of autistic traits: Comparison of the social responsiveness scale with the autism diagnostic interview-revised. J. Autism Dev. Disord. 2003, 33, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Developmental Disabilities Monitoring Network Surveillance Year 2010 Principal Investigators; Centers for Disease Control and Prevention (CDC). Prevalence of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill. Summ. 2014, 63, 1–21. [Google Scholar]
- Rapin, I.; Dunn, M. The neurology of autism: Many unanswered questions. Eur. J. Neurol. 1995, 2, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, D.H.; Levitt, P. Autism spectrum disorders: Developmental disconnection syndromes. Curr. Opin. Neurobio. 2007, 17, 103–111. [Google Scholar] [CrossRef]
- Butler, M.G.; Dasouki, M.J.; Zhou, X.P.; Talebizadeh, Z.; Brown, M.; Takahashi, T.N.; Miles, J.H.; Wang, C.H.; Stratton, R.; Pilarski, R.; et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 2005, 42, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Herman, G.E.; Henninger, N.; Ratliff-Schaub, K.; Pastore, M.; Fitzgerald, S.; McBride, K.L. Genetic testing in autism: How much is enough? Genet. Med. 2007, 9, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.L.; Hovanes, K.; Dasouki, M.; Manzardo, A.M.; Butler, M.G. Chromosomal microarray analysis of consecutive individuals with autism spectrum disorders or learning disability presenting for genetic services. Gene 2014, 535, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Butler, MG; Rafi, S.K.; Manzardo, AM. Clinically relevant candidate and known genes for autism spectrum disorders (ASD) with representation on high resolution chromosome ideograms. OA Autism 2014, 2, 5–28. [Google Scholar]
- Lyon, M.F. X-chromosome inactivation and human genetic disease. Acta Paediatr. 2002, 91, 107–112. [Google Scholar] [CrossRef]
- Plenge, R.M.; Stevenson, R.A.; Lubs, H.A.; Schwartz, C.E.; Willard, H.F. Skewed X-chromosome inactivation is a common feature of X-linked mental retardation disorders. Am. J. Hum. Genet. 2002, 71, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Talebizadeh, Z.; Bittel, D.C.; Veatch, O.J.; Kibiryeva, N.; Butler, M.G. Brief report: Non-random X chromosome inactivation in females with autism. J. Autism Dev. Disord. 2005, 35, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Buyse, I.M.; Fang, P.; Hoon, K.T.; Amir, R.E.; Zoghbi, H.Y.; Roa, B.B. Diagnostic testing for Rett syndrome by DHPLC and direct sequencing analysis of the MECP2 gene: Identification of several novel mutations and polymorphisms. Am. J. Hum. Genet. 2000, 67, 1428–1436. [Google Scholar] [CrossRef] [PubMed]
- Maier, E.M.; Kammerer, S.; Muntau, A.C.; Wichers, M.; Braun, A.; Roscher, A.A. Symptoms in carriers of adrenoleukodystrophy relate to skewed X inactivation. Ann. Neurol. 2002, 52, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Theodoro, M.F.; Bittel, D.C.; Kuipers, P.J.; Driscoll, D.J.; Talebizadeh, Z. X-chromosome inactivation patterns in females with Prader-Willi syndrome. Am. J. Med. Genet. A 2007, 143, 469–475. [Google Scholar] [CrossRef]
- Butler, M.G.; Sturich, J.; Myers, S.E.; Gold, J.A.; Kimonis, V.; Driscoll, D.J. Is gestation in Prader-Willi syndrome affected by the genetic subtype? J. Assist. Reprod. Genet. 2009, 26, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, S.B.; Lai, L.W.; Erickson, R.P.; Magnuson, L.; Thomas, E.; Gendron, R.; Herrmann, J. Trisomy 15 with loss of the paternal 15 as a cause of Prader-Willi syndrome due to maternal disomy. Am. J. Hum. Genet. 1992, 51, 701–708. [Google Scholar] [PubMed]
- Allen, R.C.; Zoghbi, H.Y.; Moseley, A.B.; Rosenblatt, H.M.; Belmont, J.W. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with x chromosome inactivation. Am. J. Hum. Genet. 1992, 51, 1229–1239. [Google Scholar] [PubMed]
- Bittel, D.C.; Theodoro, M.F.; Kibiryeva, N.; Fischer, W.; Talebizadeh, Z.; Butler, M.G. Comparison of X-chromosome inactivation patterns in multiple tissues from human females. J. Med. Genet. 2008, 45, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.; Roberts, E.; Mochida, G.H.; Hampshire, D.J.; Scott, S.; Askham, J.M.; Springell, K.; Mahadevan, M.; Crow, Y.J.; Markham, A.F.; et al. ASPM is a major determinant of cerebral cortical size. Nat. Genet. 2002, 32, 316–320. [Google Scholar] [CrossRef]
- Vermeulen, M.; Mulder, K.W.; Denissov, S.; Pim Pijnappel, W.W. M.; van Schaik, F.M.A.; Varier, R.A.; Baltissen, M.P.A.; Stunnenberg, H.G.; Mann, M.; Timmers, H.T.M. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 2007, 131, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Lacy, S.E.; Bonnemann, C.G.; Buzney, E.A.; Kunkel, L.M. Identification of FLRT1, FLRT2, and FLRT3: A novel family of transmembrane leucine-rich repeat proteins. Genomics 1999, 62, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Takeuchi, T.; Yokota, H.; Izumi, T. Novel rabphilin-3-like protein associates with insulin-containing granules in pancreatic beta cells. J. Biol. Chem. 1999, 274, 28542–28548. [Google Scholar] [CrossRef] [PubMed]
- Faber, P.W.; Barnes, G.T.; Srinidhi, J.; Chen, J.; Gusella, J.F.; MacDonald, M.E. Huntingtin interacts with a family of WW domain proteins. Hum. Mol. Genet. 1998, 7, 1463–1474. [Google Scholar] [CrossRef] [PubMed]
- Nagase, T.; Kikuno, R.; Hattori, A.; Kondo, Y.; Okumura, K.; Ohara, O. Prediction of the coding sequences of unidentified human genes. XIX. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 2000, 7, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Claudio, J.O.; Liew, C.C.; Ma, J.; Heng, H.H.Q.; Stewart, A.K.; Hawley, R.G. Cloning and expression analysis of a novel WD repeat gene, WDR3, mapping to 1p12-p13. Genomics 1999, 59, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Asakawa, H.; Hiraoka, Y.; Haraguchi, T. Nucleoporin Nup98: A gatekeeper in the eukaryotic kingdoms. Genes Cells 2010, 15, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Tommerup, N.; Vissing, H. Isolation and fine mapping of 16 novel human zinc finger-encoding cDNAs identify putative candidate genes for developmental and malignant disorders. Genomics 1995, 27, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Chapelin, C.; Duriez, N.; Magnino, F.; Goossens, M.; Escudier, E.; Amselem, S. Isolation of several human axonemal dynein heavy chain genes: Genomic structure of the catalytic site, phylogenetic analysis and chromosomal assignment. FEBS Lett. 1997, 412, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.; Roberts, E.; Springell, K.; Lizarraga, S.B.; Scott, S.; Higgins, J.; Hampshire, D.J.; Morrison, E.E.; Leal, G.F.; Silva, E.O.; et al. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat. Genet. 2005, 37, 353–355. [Google Scholar] [CrossRef]
- Pertel, T.; Hausmann, S.; Morger, D.; Zuger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef]
- Strehl, S.; Glatt, K.; Liu, Q.M.; Glatt, H.; Lalande, M. Characterization of two novel protocadherins (PCDH8 and PCDH9) localized on human chromosome 13 and mouse chromosome 14. Genomics 1998, 53, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, A.; Arai, H.; Asada, T.; Imagawa, M.; Matsubara, E.; Shoji, M.; Higuchi, S.; Urakami, K.; Kakita, A.; Takahashi, H.; et al. Genetic association of CTNNA3 with late-onset Alzheimer’s disease in females. Hum. Mol. Genet. 2007, 16, 2854–2869. [Google Scholar] [CrossRef]
- Kao, S.C.; Chen, C.Y.; Wang, S.L.; Yang, J.J.; Hung, W.C.; Chen, Y.C.; Lai, N.S.; Liu, H.T.; Huang, H.; Chen, H.C.; et al. Identification of phostensin, a PP1 F-actin cytoskeleton targeting subunit. Biochem. Biophys. Res. Commun. 2007, 356, 594–598. [Google Scholar] [CrossRef] [PubMed]
- Leidinger, P.; Backes, C.; Deutscher, S.; Schmitt, K.; Mueller, S.C.; Frese, K.; Haas, J.; Ruprecht, K.; Paul, F.; Stahler, C.; et al. A blood based 12-miRNA signature of Alzheimer disease patients. Genome Biol. 2013, 14. [Google Scholar] [CrossRef]
- Cappello, S.; Gray, M.J.; Badouel, C.; Lange, S.; Einsiedler, M.; Srour, M.; Chitayat, D.; Hamdan, F.F.; Jenkins, Z.A.; Morgan, T.; et al. Mutations in genes encoding the cadherin receptor-ligand pair DCHS1 and FAT4 disrupt cerebral cortical development. Nat. Genet. 2013, 45, 1300–1308. [Google Scholar]
- Grishin, A.; Li, H.; Levitan, E.S.; Zaks-Makhina, E. Identification of gamma-aminobutyric acid receptor-interacting factor 1 (TRAK2) as a trafficking factor for the K+ channel Kir2.1. J. Biol. Chem. 2006, 281, 30104–30111. [Google Scholar] [CrossRef] [PubMed]
- Billaut-Laden, I.; Rat, E.; Allorge, D.; Crunelle-Thibaut, A.; Cauffiez, C.; Chevalier, D.; Lo-Guidice, J.M.; Broly, F. Evidence for a functional genetic polymorphism of the human mercaptopyruvate sulfurtransferase (MPST), a cyanide detoxification enzyme. Toxicol. Lett. 2006, 165, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Hayette, S.; Gadoux, M.; Martel, S.; Bertrand, S.; Tigaud, I.; Magaud, J.P.; Rimokh, R. FLRG (follistatin-related gene), a new target of chromosomal rearrangement in malignant blood disorders. Oncogene 1998, 16, 2949–2954. [Google Scholar] [CrossRef] [PubMed]
- Bajorek, M.; Morita, E.; Skalicky, J.J.; Morham, S.G.; Babst, M.; Sundquist, W.I. Biochemical analyses of human IST1 and its function in cytokinesis. Mol. Biol. Cell. 2009, 20, 1360–1373. [Google Scholar] [CrossRef] [PubMed]
- Klejnot, M.; Kozielski, F. Structural insights into human Kif7, a kinesin involved in hedgehog signalling. Acta Crystallogr. 2012, 68, 154–159. [Google Scholar]
- Wu, Q.; Maniatis, T. A striking organization of a large family of human neural cadherin-like cell adhesion genes. Cell 1999, 97, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.S.; Rubio, M.E.; Clem, R.L.; Johnson, D.; Case, L.; Tessier-Lavigne, M.; Huganir, R.L.; Ginty, D.D.; Kolodkin, A.L. Secreted semaphorins control spine distribution and morphogenesis in the postnatal CNS. Nature 2009, 462, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Wen, J.; Yang, W.; Wang, C.; Gao, L.; Zheng, L.H.; Wang, T.; Ran, K.; Li, Y.; Li, X.; et al. Gain-of-function mutations in SCN11a cause familial episodic pain. Am. J. Hum. Genet. 2013, 93, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Piton, A.; Gauthier, J.; Hamdan, F.F.; Lafreniere, R.G.; Yang, Y.; Henrion, E.; Laurent, S.; Noreau, A.; Thibodeau, P.; Karemera, L.; et al. Systematic resequencing of X-chromosome synaptic genes in autism spectrum disorder and schizophrenia. Mol. Psychiatry 2011, 16, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Pergadia, M.L.; Saccone, S.F.; Lynskey, M.T.; Wang, J.C.; Martin, N.G.; Statham, D.; Henders, A.; Campbell, M.; Garcia, R.; et al. An autosomal linkage scan for cannabis use disorders in the nicotine addiction genetics project. Arch. Gen. Psychiatry 2008, 65, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, S.; Gerondopoulos, A.; Linford, A.; Rigden, D.J.; Barr, F.A. Family-wide characterization of the DENN domain Rab GDP-GTP exchange factors. J. Cell Biol. 2010, 191, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Le Meur, N.; Martin, C.; Saugier-Veber, P.; Joly, G.; Lemoine, F.; Moirot, H.; Rossi, A.; Bachy, B.; Cabot, A.; Joly, P.; et al. Complete germline deletion of the STK11 gene in a family with Peutz-Jeghers syndrome. Eur. J. Hum. Genet. 2004, 12, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Smolen, G.A.; Schott, B.J.; Stewart, R.A.; Diederichs, S.; Muir, B.; Provencher, H.L.; Look, A.T.; Sgroi, D.C.; Peterson, R.T.; Haber, D.A. A Rap GTPase interactor, RADIL, mediates migration of neural crest precursors. Genes Dev. 2007, 21, 2131–2136. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Zhang, J.; Zhang, Y.; Hsu, S.C.; Zhou, D.; Guo, W. Exo70 interacts with the Arp2/3 complex and regulates cell migration. Nat. Cell Biol. 2006, 8, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Oeffner, F.; Moch, C.; Neundorf, A.; Hofmann, J.; Koch, M.; Grzeschik, K.H. Novel interaction partners of Bardet-Biedl syndrome proteins. Cell Motil. Cytoskelet. 2008, 65, 143–155. [Google Scholar] [CrossRef]
- Lopez, I.; Mak, E.C.; Ding, J.; Hamm, H.E.; Lomasney, J.W. A novel bifunctional phospholipase C that is regulated by Galpha 12 and stimulates the Ras/mitogen-activated protein kinase pathway. J. Biol. Chem. 2001, 276, 2758–2765. [Google Scholar] [CrossRef] [PubMed]
- Liefke, R.; Oswald, F.; Alvarado, C.; Ferres-Marco, D.; Mittler, G.; Rodriguez, P.; Dominguez, M.; Borggrefe, T. Histone demethylase KDM5A is an integral part of the core Notch-RBP-J repressor complex. Genes Dev. 2010, 24, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.W.; Jian, X.; Yoon, H.Y.; Randazzo, P.A. ARAP2 signals through Arf6 and Rac1 to control focal adhesion morphology. J. Biol. Chem. 2013, 288, 5849–5860. [Google Scholar] [CrossRef] [PubMed]
- Chi, Z.; Zhang, J.; Tokunaga, A.; Harraz, M.M.; Byrne, S.T.; Dolinko, A.; Xu, J.; Blackshaw, S.; Gaiano, N.; Dawson, T.M.; et al. Botch promotes neurogenesis by antagonizing Notch. Dev. Cell 2012, 22, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, M.; Suemori, H.; Noguchi, S.; Ji, Z.S.; Tsunoo, H. Novel nucleolar protein, midnolin, is expressed in the mesencephalon during mouse development. Gene 2000, 254, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Sano, K.; Tanihara, H. Diversity of the cadherin family: Evidence for eight new cadherins in nervous tissue. Cell Regul. 1991, 2, 261–270. [Google Scholar] [PubMed]
- Unger, S.; Lausch, E.; Rossi, A.; Megarbane, A.; Sillence, D.; Alcausin, M.; Aytes, A.; Mendoza-Londono, R.; Nampoothiri, S.; Afroze, B.; et al. Phenotypic features of carbohydrate sulfotransferase 3 (CHST3) deficiency in 24 patients: Congenital dislocations and vertebral changes as principal diagnostic features. Am. J. Med. Genet. A 2010, 152A, 2543–2549. [Google Scholar] [CrossRef]
- Reimann, J.D.; Freed, E.; Hsu, J.Y.; Kramer, E.R.; Peters, J.M.; Jackson, P.K. Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell 2001, 105, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Van Der Knaap, J.A.; van Den Boom, V.; Kuipers, J.; van Eijk, M.J.; van Der Vliet, P.C.; Timmers, H.T. The gene for human TATA-binding-protein-associated factor (TAFII) 170: Structure, promoter and chromosomal localization. Biochem. J. 2000, 345, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Yoshiura, K.; Machida, J.; Daack-Hirsch, S.; Patil, S.R.; Ashworth, L.K.; Hecht, J.T.; Murray, J.C. Characterization of a novel gene disrupted by a balanced chromosomal translocation t(2;19)(q11.2;q13.3) in a family with cleft lip and palate. Genomics 1998, 54, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Kuja-Panula, J.; Kiiltomaki, M.; Yamashiro, T.; Rouhiainen, A.; Rauvala, H. AMIGO, a transmembrane protein implicated in axon tract development, defines a novel protein family with leucine-rich repeats. J. Cell Biol. 2003, 160, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Dammai, V.; Subramani, S. The human peroxisomal targeting signal receptor, Pex5p, is translocated into the peroxisomal matrix and recycled to the cytosol. Cell 2001, 105, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Iwamori, T.; Iwamori, N.; Ma, L.; Edson, M.A.; Greenbaum, M.P.; Matzuk, M.M. TEX14 interacts with CEP55 to block cell abscission. Mol. Cell. Biol. 2010, 30, 2280–2292. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Kaestner, K.H. The Foxa family of transcription factors in development and metabolism. Cell Mol. Life Sci. 2006, 63, 2317–2328. [Google Scholar] [CrossRef] [PubMed]
- Yoon, D.; Kim, Y.J.; Cui, W.Y.; Van der Vaart, A.; Cho, Y.S.; Lee, J.Y.; Ma, J.Z.; Payne, T.J.; Li, M.D.; Park, T. Large-scale genome-wide association study of asian population reveals genetic factors in FRMD4A and other loci influencing smoking initiation and nicotine dependence. Hum. Genet. 2012, 131, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- Licciulli, S.; Cambiaghi, V.; Scafetta, G.; Gruszka, A.M.; Alcalay, M. Pirin downregulation is a feature of AML and leads to impairment of terminal myeloid differentiation. Leukemia 2010, 24, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Pazour, G.J.; Agrin, N.; Walker, B.L.; Witman, G.B. Identification of predicted human outer dynein arm genes: Candidates for primary ciliary dyskinesia genes. J. Med. Genet. 2006, 43, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.J.; O’Leary, D.D. Role for Lhx2 in corticogenesis through regulation of progenitor differentiation. Mol. Cell. Neurosci. 2013, 56, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Leighton, P.A.; Mitchell, K.J.; Goodrich, L.V.; Lu, X.; Pinson, K.; Scherz, P.; Skarnes, W.C.; Tessier-Lavigne, M. Defining brain wiring patterns and mechanisms through gene trapping in mice. Nature 2001, 410, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Piton, A.; Michaud, J.L.; Peng, H.; Aradhya, S.; Gauthier, J.; Mottron, L.; Champagne, N.; Lafreniere, R.G.; Hamdan, F.F.; team, S.D.; et al. Mutations in the calcium-related gene IL1RAPL1 are associated with autism. Hum. Mol. Genet. 2008, 17, 3965–3974. [Google Scholar] [CrossRef] [PubMed]
- Sinibaldi, L.; De Luca, A.; Bellacchio, E.; Conti, E.; Pasini, A.; Paloscia, C.; Spalletta, G.; Caltagirone, C.; Pizzuti, A.; Dallapiccola, B. Mutations of the Nogo-66 receptor (RTN4R) gene in schizophrenia. Hum. Mutat. 2004, 24, 534–535. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Ohno, M.; Shimura, Y. Identification of a putative RNA helicase (HRH1), a human homolog of yeast Prp22. Mol. Cell. Biol. 1994, 14, 7611–7620. [Google Scholar] [PubMed]
- Katoh, Y.; Katoh, M. Comparative integromics on FAT1, FAT2, FAT3 and FAT4. Int. J. Mol. Med. 2006, 18, 523–528. [Google Scholar] [PubMed]
- Neale, B.M.; Kou, Y.; Liu, L.; Ma'ayan, A.; Samocha, K.E.; Sabo, A.; Lin, C.F.; Stevens, C.; Wang, L.S.; Makarov, V.; et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, 485, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Kenny, E.M.; Cormican, P.; Furlong, S.; Heron, E.; Kenny, G.; Fahey, C.; Kelleher, E.; Ennis, S.; Tropea, D.; Anney, R.; et al. Excess of rare novel loss-of-function variants in synaptic genes in schizophrenia and autism spectrum disorders. Mol. Psychiatry 2014, 19, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Kadereit, B.; Kumar, P.; Wang, W.J.; Miranda, D.; Snapp, E.L.; Severina, N.; Torregroza, I.; Evans, T.; Silver, D.L. Evolutionarily conserved gene family important for fat storage. Proc. Natl. Acad. Sci. USA 2008, 105, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.W.; Herrera-Abreu, M.T.; Rohn, J.L.; Racine, V.; Tajadura, V.; Suryavanshi, N.; Bechtel, S.; Wiemann, S.; Baum, B.; Ridley, A.J. Identification and characterization of a set of conserved and new regulators of cytoskeletal organization, cell morphology and migration. BMC Biol. 2011, 9, 54. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Arias-Vasquez, A.; Sleegers, K.; Aulchenko, Y.S.; Kayser, M.; Sanchez-Juan, P.; Feng, B.J.; Bertoli-Avella, A.M.; van Swieten, J.; Axenovich, T.I.; et al. A genomewide screen for late-onset Alzheimer disease in a genetically isolated Dutch population. Am. J. Hum. Genet. 2007, 81, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Smith, F.D.; Stark, C.; Wells, C.D.; Fawcett, J.P.; Kulkarni, S.; Metalnikov, P.; O’Donnell, P.; Taylor, P.; Taylor, L.; et al. Proteomic, functional, and domain-based analysis of in vivo 14–3-3 binding proteins involved in cytoskeletal regulation and cellular organization. Curr. Biol. 2004, 14, 1436–1450. [Google Scholar] [CrossRef] [PubMed]
- Kile, B.T.; Viney, E.M.; Willson, T.A.; Brodnicki, T.C.; Cancilla, M.R.; Herlihy, A.S.; Croker, B.A.; Baca, M.; Nicola, N.A.; Hilton, D.J.; et al. Cloning and characterization of the genes encoding the ankyrin repeat and SOCS box-containing proteins asb-1, asb-2, asb-3 and asb-4. Gene 2000, 258, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Dodge-Kafka, K.L.; Soughayer, J.; Pare, G.C.; Michel, J.J.C.; Langeberg, L.K.; Kapiloff, M.S.; Scott, J.D. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 2005, 437, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wright, R.A.; Rockey, P.K.; Burgett, S.G.; Arnold, J.S.; Rosteck, P.R., Jr.; Johnson, B.G.; Schoepp, D.D.; Belagaje, R.M. Group III human metabotropic glutamate receptors 4, 7 and 8: Molecular cloning, functional expression, and comparison of pharmacological properties in RGT cells. Brain Res. Mol. Brain Res. 1998, 53, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Berbari, N.F.; Kin, N.W.; Sharma, N.; Michaud, E.J.; Kesterson, R.A.; Yoder, B.K. Mutations in Traf3ip1 reveal defects in ciliogenesis, embryonic development, and altered cell size regulation. Dev. Biol. 2011, 360, 66–76. [Google Scholar] [PubMed]
- Davis, B.N.; Hilyard, A.C.; Lagna, G.; Hata, A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature 2008, 454, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Goizet, C.; Boukhris, A.; Mundwiller, E.; Tallaksen, C.; Forlani, S.; Toutain, A.; Carriere, N.; Paquis, V.; Depienne, C.; Durr, A.; et al. Complicated forms of autosomal dominant hereditary spastic paraplegia are frequent in SPG10. Hum. Mutat. 2008, 30, E376–E385. [Google Scholar] [CrossRef]
- Youngs, E.L.; Hellings, J.A.; Butler, M.G. ANKRD11 gene deletion in a 17-year-old male. Clin. Dysmorphol. 2011, 20, 170–171. [Google Scholar] [CrossRef] [PubMed]
- Sirmaci, A.; Spiliopoulos, M.; Brancati, F.; Powell, E.; Duman, D.; Abrams, A.; Bademci, G.; Agolini, E.; Guo, S.; Konuk, B.; et al. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am. J. Hum. Genet. 2011, 89, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Mi, S.; Lee, X.; Hu, Y.; Ji, B.; Shao, Z.; Yang, W.; Huang, G.; Walus, L.; Rhodes, K.; Gong, B.J.; et al. Death receptor 6 negatively regulates oligodendrocyte survival, maturation and myelination. Nat. Med. 2011, 17, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Guernsey, D.L.; Jiang, H.; Bedard, K.; Evans, S.C.; Ferguson, M.; Matsuoka, M.; Macgillivray, C.; Nightingale, M.; Perry, S.; Rideout, A.L.; et al. Mutation in the gene encoding ubiquitin ligase LRSAM1 in patients with Charcot-Marie-Tooth disease. PLoS Genet. 2010, 6. [Google Scholar] [CrossRef]
- Weterman, M.A.; Sorrentino, V.; Kasher, P.R.; Jakobs, M.E.; van Engelen, B.G.; Fluiter, K.; de Wissel, M.B.; Sizarov, A.; Nurnberg, G.; Nurnberg, P.; et al. A frameshift mutation in LRSAM1 is responsible for a dominant hereditary polyneuropathy. Hum. Mol. Genet. 2012, 21, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Burnside, R.D.; Pasion, R.; Mikhail, F.M.; Carroll, A.J.; Robin, N.H.; Youngs, E.L.; Gadi, I.K.; Keitges, E.; Jaswaney, V.L.; Papenhausen, P.R.; et al. Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: A susceptibility region for neurological dysfunction including developmental and language delay. Hum. Genet. 2011, 130, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Pastuszak, I.; Ketchum, C.; Hermanson, G.; Sjoberg, E.J.; Drake, R.; Elbein, A.D. GDP-l-fucose pyrophosphorylase. Purification, cDNA cloning, and properties of the enzyme. J. Biol. Chem. 1998, 273, 30165–30174. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Li, P.P.; Cooke, R.G.; Parikh, S.V.; Wang, K.; Kennedy, J.L.; Warsh, J.J. TRPM2 variants and bipolar disorder risk: Confirmation in a family-based association study. Bipolar Disord. 2009, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.T.; Richard, S. SUMOylation negatively modulates target gene occupancy of the KDM5B, a histone lysine demethylase. Epigenetics 2013, 8, 1162–1175. [Google Scholar] [CrossRef] [PubMed]
- Catchpole, S.; Spencer-Dene, B.; Hall, D.; Santangelo, S.; Rosewell, I.; Guenatri, M.; Beatson, R.; Scibetta, A.G.; Burchell, J.M.; Taylor-Papadimitriou, J. PLU-1/JARID1B/KDM5B is required for embryonic survival and contributes to cell proliferation in the mammary gland and in ER+ breast cancer cells. Int. J. Oncol. 2011, 38, 1267–1277. [Google Scholar] [PubMed]
- Carmona-Mora, P.; Walz, K. Retinoic acid induced 1, RAI1: A dosage sensitive gene related to neurobehavioral alterations including autistic behavior. Curr. Genomics 2010, 11, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Van Wijk, S.J.; Timmers, H.T. The family of ubiquitin-conjugating enzymes (E2s): Deciding between life and death of proteins. FASEB J. 2010, 24, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Kirchmair, R.; Hogue-Angeletti, R.; Gutierrez, J.; Fischer-Colbrie, R.; Winkler, H. Secretoneurin—A neuropeptide generated in brain, adrenal medulla and other endocrine tissues by proteolytic processing of secretogranin II (chromogranin C). Neuroscience 1993, 53, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Tanji, K.; Kamitani, T.; Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. TRIM9, a novel brain-specific E3 ubiquitin ligase, is repressed in the brain of Parkinson's disease and dementia with Lewy bodies. Neurobiol. Dis. 2010, 38, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Takash, W.; Canizares, J.; Bonneaud, N.; Poulat, F.; Mattei, M.G.; Jay, P.; Berta, P. SOX7 transcription factor: Sequence, chromosomal localisation, expression, transactivation and interference with Wnt signalling. Nucleic Acids Res. 2001, 29, 4274–4283. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Lynskey, M.T.; Hinrichs, A.; Grucza, R.; Saccone, S.F.; Krueger, R.; Neuman, R.; Howells, W.; Fisher, S.; Fox, L.; et al. A genome-wide association study of DSM-IV cannabis dependence. Addict. Biol. 2011, 16, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Kokura, K.; Sun, L.; Bedford, M.T.; Fang, J. Methyl-H3K9-binding protein MPP8 mediates E-cadherin gene silencing and promotes tumour cell motility and invasion. EMBO J. 2010, 29, 3673–3687. [Google Scholar] [CrossRef] [PubMed]
- Bonnert, T.P.; McKernan, R.M.; Farrar, S.; le Bourdelles, B.; Heavens, R.P.; Smith, D.W.; Hewson, L.; Rigby, M.R.; Sirinathsinghji, D.J.; Brown, N.; et al. Theta, a novel γ-aminobutyric acid type a receptor subunit. Proc. Natl. Acad. Sci. USA 1999, 96, 9891–9896. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Kuroda, S.; Fukata, M.; Nakamura, T.; Nagase, T.; Nomura, N.; Matsuura, Y.; Yoshida-Kubomura, N.; Iwamatsu, A.; Kaibuchi, K. P140Sra-1 (specifically rac1-associated protein) is a novel specific target for rac1 small GTPase. J. Biol. Chem. 1998, 273, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Schenck, A.; Bardoni, B.; Moro, A.; Bagni, C.; Mandel, J.L. A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with fmrp-related proteins FXR1P and FXR2P. Proc. Natl. Acad. Sci. USA 2001, 98, 8844–8849. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Muzny, D.M.; Xia, F.; Niu, Z.; Person, R.; Ding, Y.; Ward, P.; Braxton, A.; Wang, M.; Buhay, C.; et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 2014, 312, 1870–1879. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butler, M.G.; Rafi, S.K.; Hossain, W.; Stephan, D.A.; Manzardo, A.M. Whole Exome Sequencing in Females with Autism Implicates Novel and Candidate Genes. Int. J. Mol. Sci. 2015, 16, 1312-1335. https://doi.org/10.3390/ijms16011312
Butler MG, Rafi SK, Hossain W, Stephan DA, Manzardo AM. Whole Exome Sequencing in Females with Autism Implicates Novel and Candidate Genes. International Journal of Molecular Sciences. 2015; 16(1):1312-1335. https://doi.org/10.3390/ijms16011312
Chicago/Turabian StyleButler, Merlin G., Syed K. Rafi, Waheeda Hossain, Dietrich A. Stephan, and Ann M. Manzardo. 2015. "Whole Exome Sequencing in Females with Autism Implicates Novel and Candidate Genes" International Journal of Molecular Sciences 16, no. 1: 1312-1335. https://doi.org/10.3390/ijms16011312
APA StyleButler, M. G., Rafi, S. K., Hossain, W., Stephan, D. A., & Manzardo, A. M. (2015). Whole Exome Sequencing in Females with Autism Implicates Novel and Candidate Genes. International Journal of Molecular Sciences, 16(1), 1312-1335. https://doi.org/10.3390/ijms16011312