
Liver Toxicity of Cadmium Telluride Quantum Dots (CdTe QDs) Due to Oxidative Stress in Vitro and in Vivo


Abstract
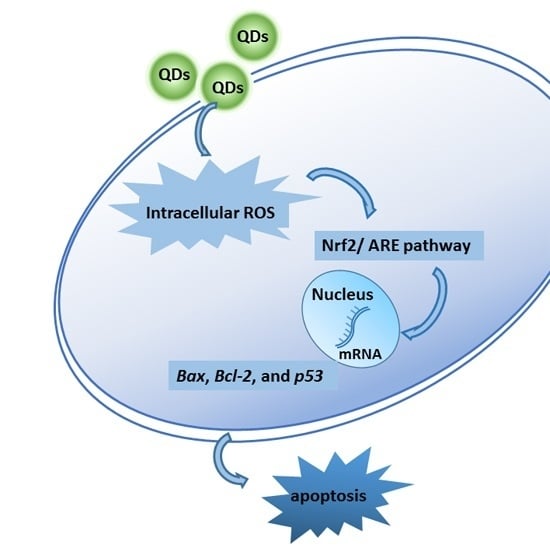
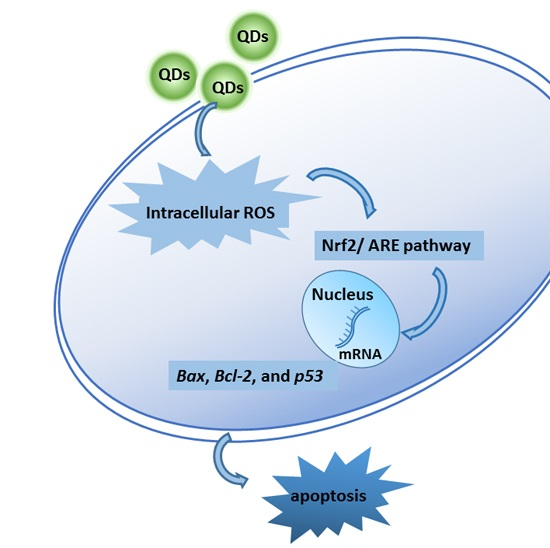
:
1. Introduction
2. Results
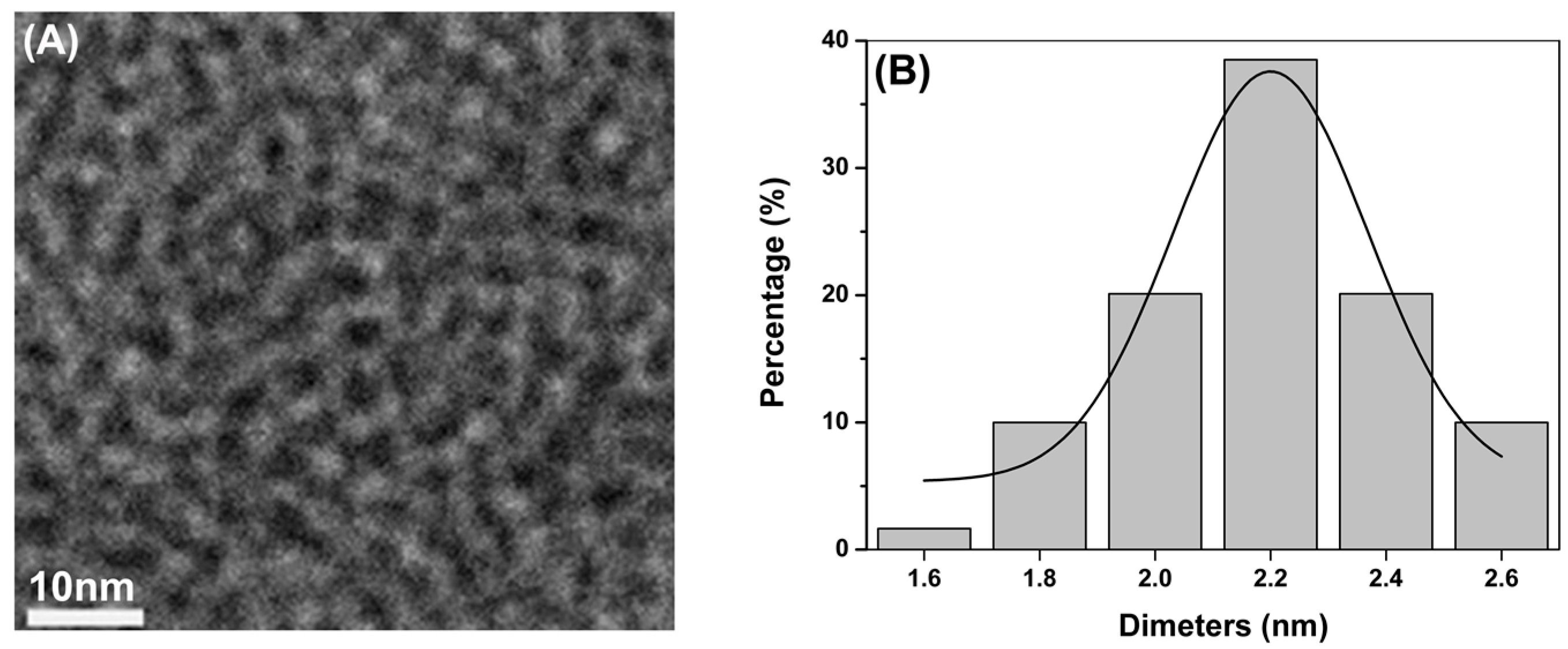
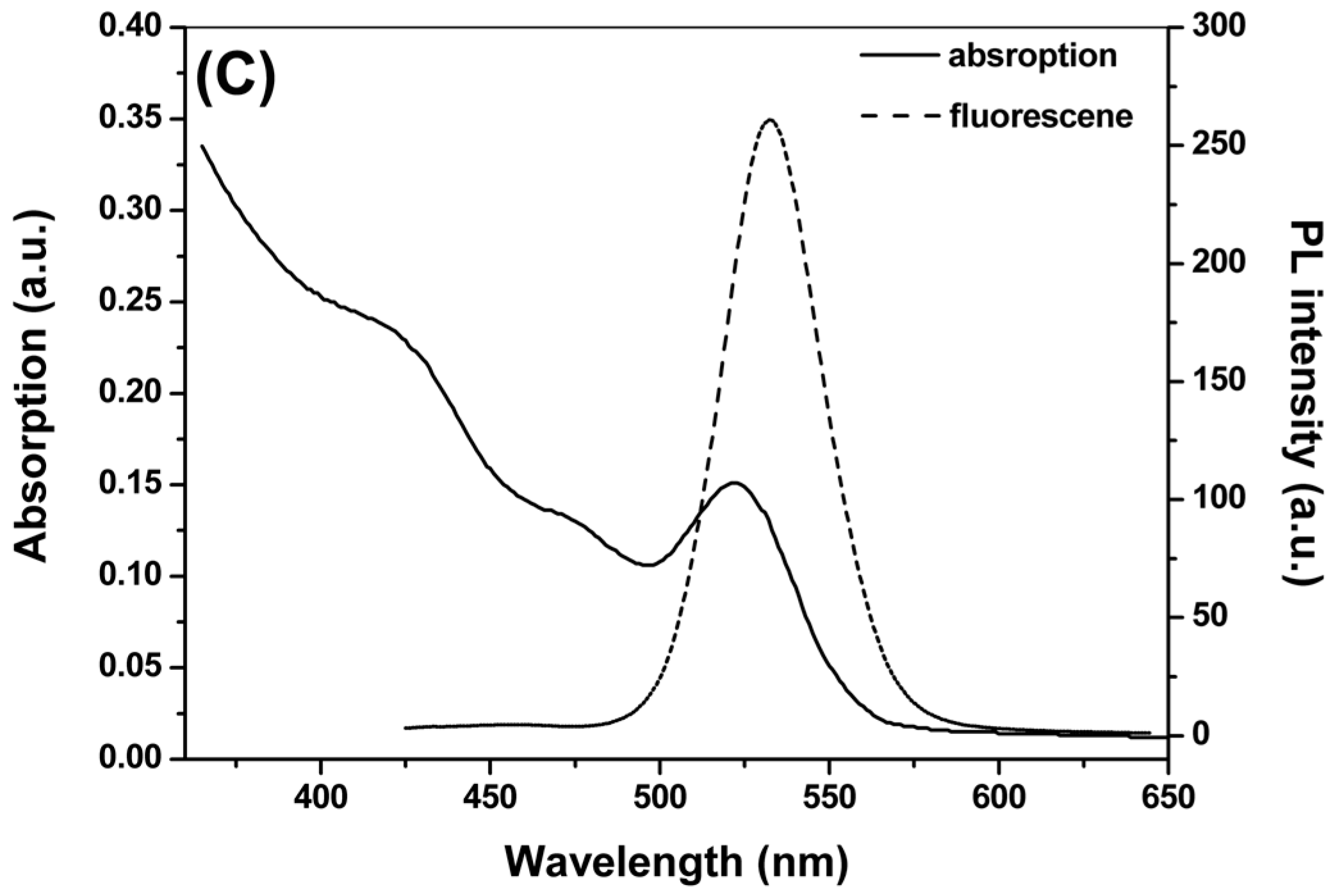
2.1. Synthesis and Characterization of CdTe QDs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Values (Mean ± SD) |
|---|---|
| Hydrodynamic size in distilled water (nm) | 9.82 ± 1.14 |
| Hydrodynamic size in culture medium (nm) | 26.79 ± 1.59 |
| Zeta potential in distilled water (mV) | −26.46 ± 4.75 |
| Zeta potential in culture medium (mV) | −13.86 ± 3.44 |


2.2. Body Weight, Liver Weight, Histopathological Change and Serum Biochemistry Analysis by 28 Day-Repeated Intravenous Administration of CdTe QDs
| Group | Number | Initial Body Weight (g) | Final Body Weight (g) | Liver Weight (g) | Liver/bw Ratio |
|---|---|---|---|---|---|
| control | 6 | 19.80 ± 1.48 | 44.60 ± 2.41 | 2.60 ± 0.38 | 5.81 ± 0.58 |
| 4.12 | 6 | 20.40 ± 1.14 | 43.40 ± 1.82 | 2.46 ± 0.14 | 5.68 ± 0.39 |
| 8.25 | 6 | 20.40 ± 1.14 | 42.80 ± 5.58 | 2.51 ± 0.54 | 5.83 ± 0.65 |
| 16.5 | 6 | 20.20 ± 1.48 | 43.40 ± 2.19 | 2.17 ± 0.20 * | 4.99 ± 0.24 * |
| Parameters | Concentration of CdTe QDs (mg/kg.bw) | |||
|---|---|---|---|---|
| 4.12 | 8.25 | 16.5 | NS | |
| TP (g/L) | 50.60 ± 3.36 | 53.60 ± 3.97 | 53.00 ± 1.73 | 51.25 ± 2.39 |
| ALB (g/L) | 13.60 ± 1.14* | 13.40 ± 0.89 | 13.60 ± 0.89* | 12.00 ± 0.71 |
| TBIL (µmol/L) | 4.52 ± 0.85 | 4.78 ± 1.01 | 3.62 ± 0.41 | 4.55 ± 0.59 |
| DBIL (µmol/L) | 3.52 ± 0.53 | 4.14 ± 0.94 | 3.14 ± 0.49 | 4.06 ± 0.46 |
| ALT (IU/L) | 27.00 ± 10.77 | 58.00 ± 24.82 | 73.60 ± 59.21 | 46.00 ± 9.98 |
| AST (IU/L) | 97.80 ± 8.26 | 121.40 ± 50.19 | 130.80 ± 15.12 | 124.60 ± 21.34 |
| ALP (IU/L) | 100.60 ± 24.69 | 115.60 ± 45.63 | 106.80 ± 18.91 | 89.50 ± 26.37 |
| γ-GT (IU/L) | 1.40 ± 0.89 | 1.80 ± 1.31 | 1.80 ± 0.84 | 1.80 ± 0.45 |
| LDH (IU/L) | 420.00 ± 88.36 * | 540.40 ± 130.34 * | 539.00 ± 80.77 * | 954.00 ± 247.01 |
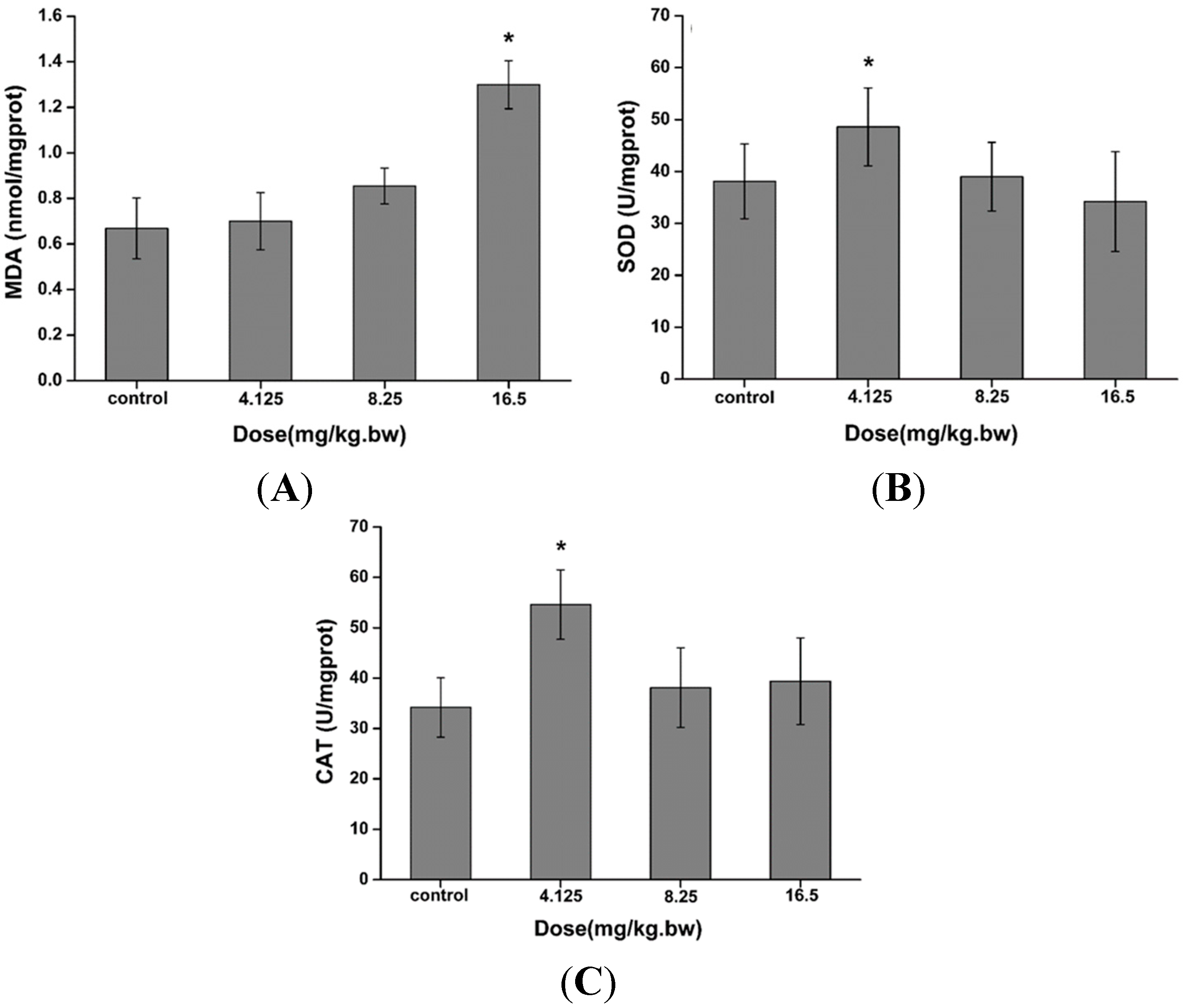
2.3. Induction of Oxidative Stress in the Liver
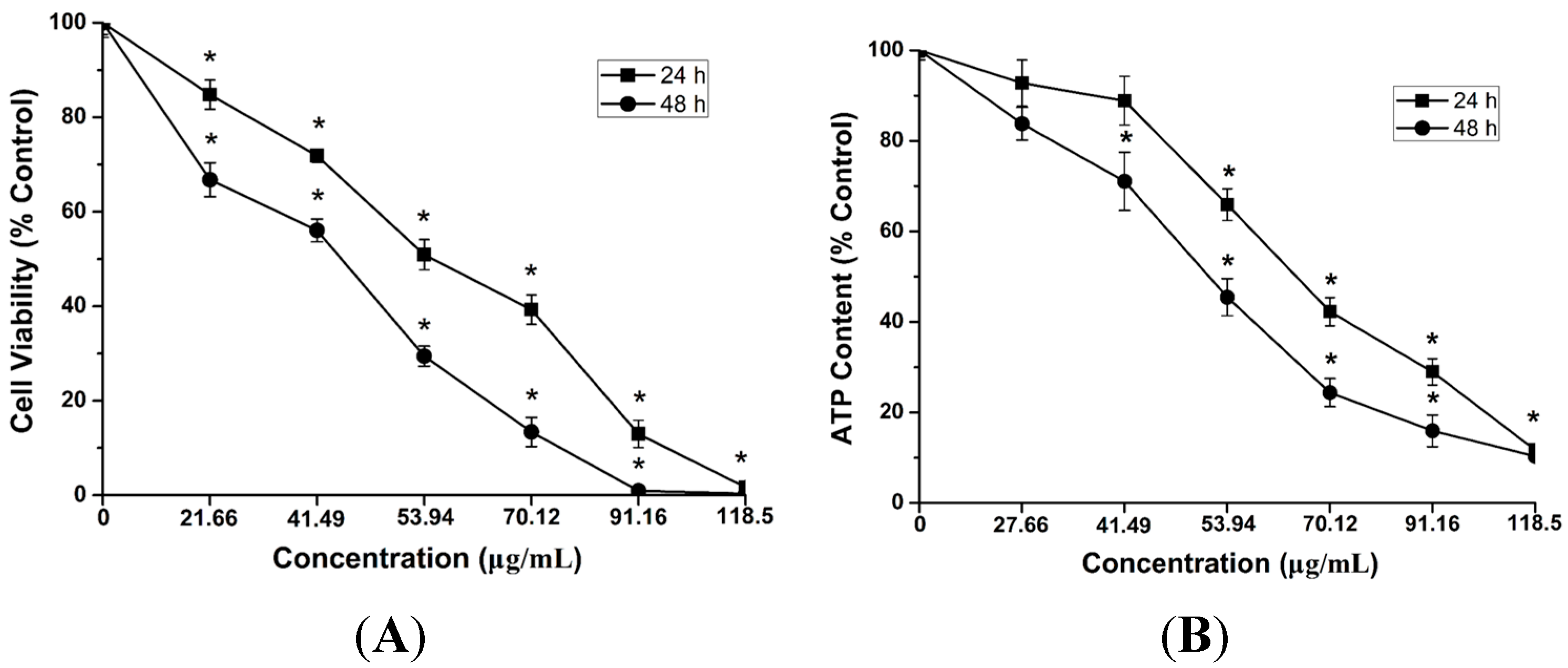
2.4. Cytotoxicity and ATP Synthesis Induced by CdTe QDs


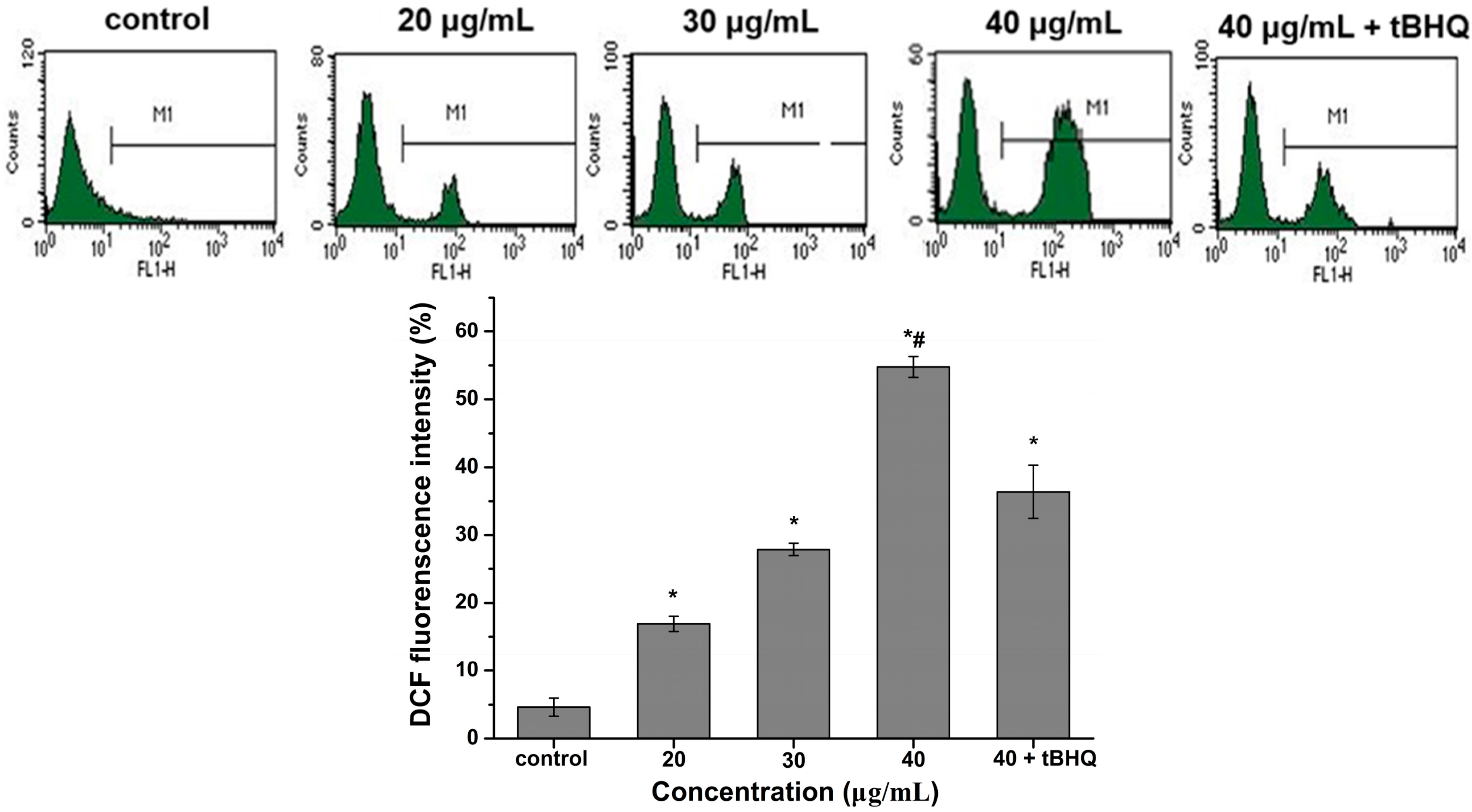
2.5. Reactive Oxygen Species (ROS) Generation by CdTe QDs

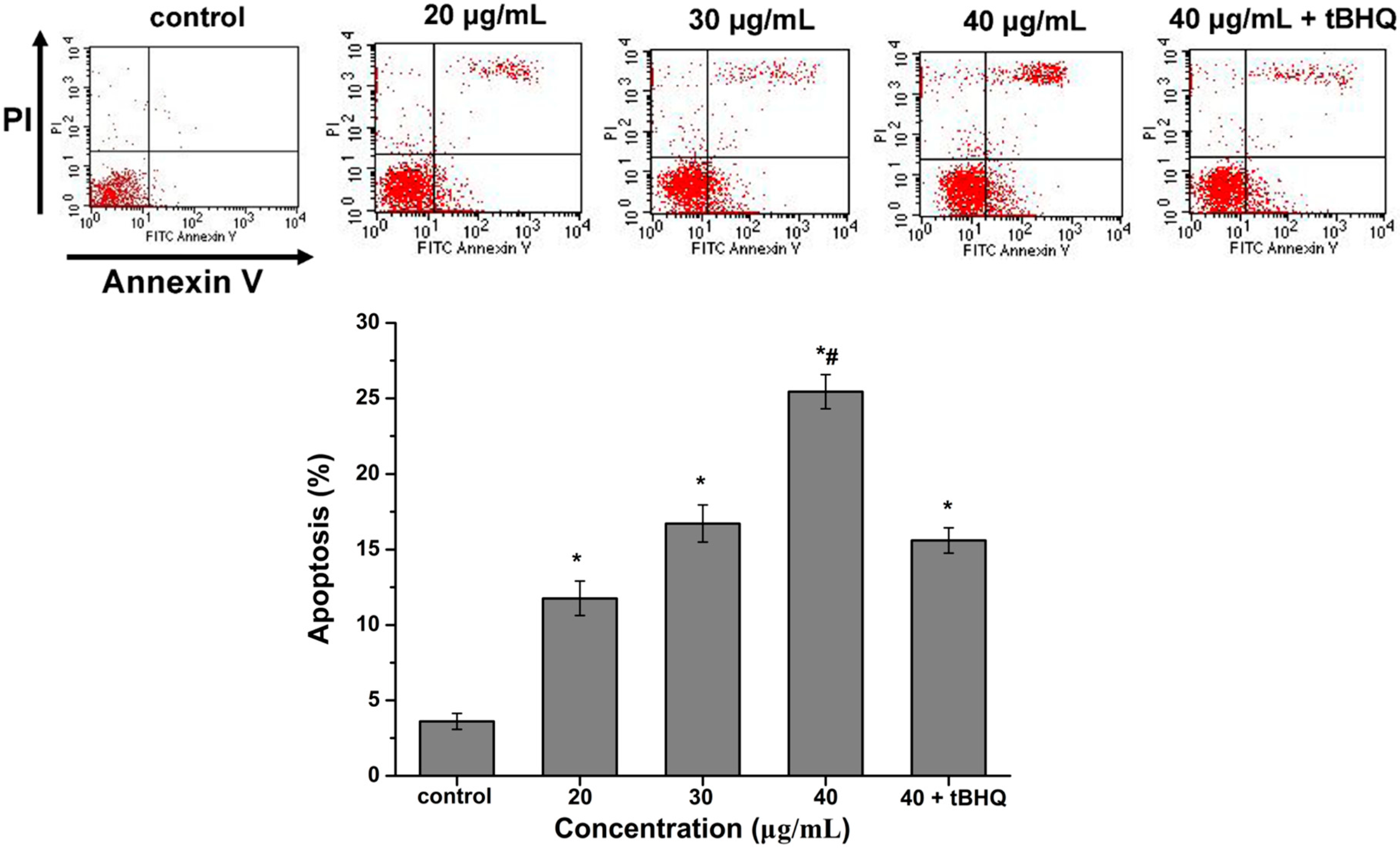
2.6. CdTe QDs Nanoparticles Induced Apoptosis

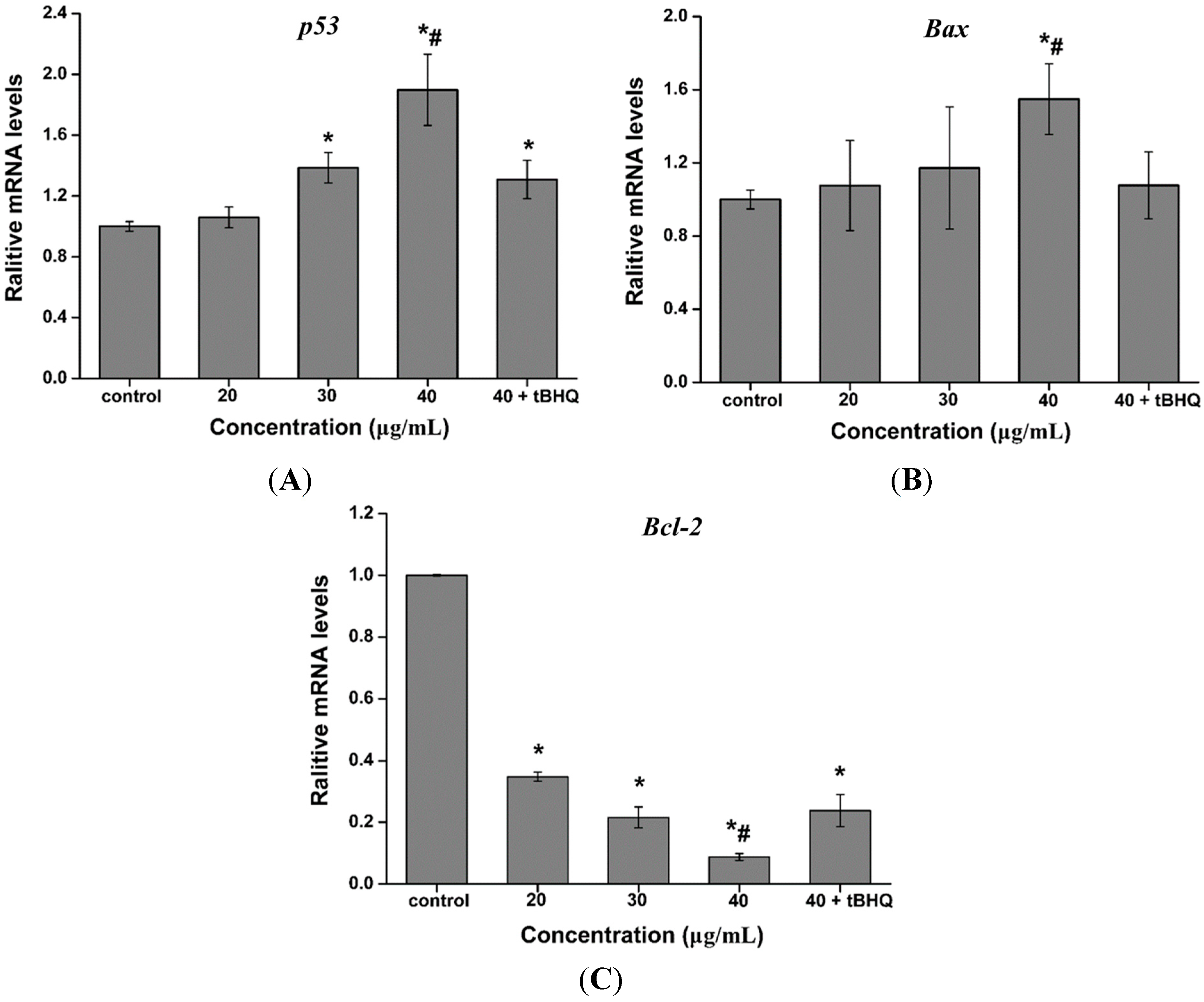
2.7. Effect of CdTe QDs on the Expression of p53, Bax and Bcl-2 mRNA

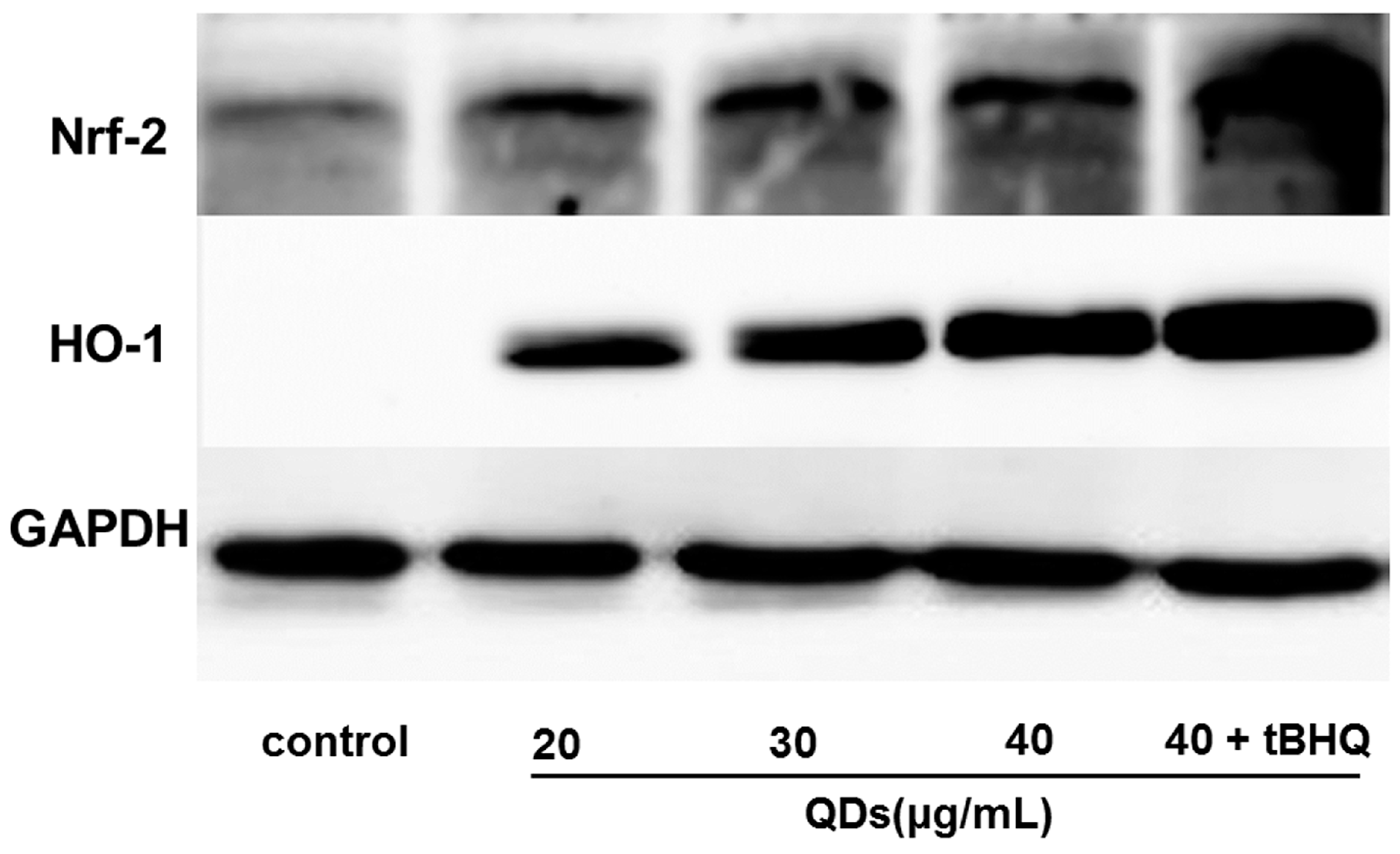
2.8. Effect of CdTe QDs on the Expression of Nrf2 Signal Pathway Proteins

3. Discussion
4. Experimental Section
4.1. Preparation and Characterization of Quantum Dots
4.2. In Vivo Sample Preparation
4.3. Clinical Observations and Pathological Examinations
4.4. Oxidative Stress Markers and Enzyme Activity Assays Analysis
4.5. Cell Culture and Treatments
4.6. Viability Analysis
4.7. Determination of Cellular ATP Levels
4.8. Determination of Intracellular Reactive Oxygen Species (ROS) Generation
4.9. Annexin V-FITC/Propidium Iodide Apoptosis Assay
4.10. Real-Time Quantitative Analysis
| Gene Primer | Sequence (5ʹ-3ʹ) | Product Size (bp) |
|---|---|---|
| Bax | Forward: AGACAGGGGCCTTTTTGCTAC | 137 |
| Reverse: AATTCGCCGGAGACACTCG | ||
| Bcl-2 | Forward: GAGAGCGTCAACAGGGAGATG | 108 |
| Reverse: CCAGCCTCCGTTATCCTGGA | ||
| p53 | Forward: CCCCTGTCATCTTTTGTCCCT | 137 |
| Reverse: AGCTGGCAGAATAGCTTATTGAG | ||
| GAPDH | Forward: AGGTCGGTGTGAACGGATTTG | 123 |
| Reverse: TGTAGACCATGTAGTTGAGGTCA |
4.11. Western Blot Analysis of Nrf2 and HO-1 Activation
4.12. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ho, C.-C.; Luo, Y.-H.; Chuang, T.-H.; Yang, C.-S.; Ling, Y.-C.; Lin, P. Quantum dots induced monocyte chemotactic protein-1 expression via myd88-dependent toll-like receptor signaling pathways in macrophages. Toxicology 2013, 308, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hauck, T.S.; Anderson, R.E.; Fischer, H.C.; Newbigging, S.; Chan, W.C.W. In vivo quantum-dot toxicity assessment. Small 2010, 6, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Tang, M. Toxicity of quantum dots on respiratory system. Inhal. Toxicol. 2014, 26, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.M.; Im, H.-Y.; Seo, J.-E.; Hasan, M.; Woo, K.; Kwon, O.-S. Acute toxicity and tissue distribution of CdSe/CdS-MPA quantum dots after repeated intraperitoneal injection to mice. J. Appl. Toxicol. 2013, 33, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; He, Y.; Su, Y.; Li, X.; Huang, Q.; Wang, H.; Zhang, X.; Tai, R.; Fan, C. The cytotoxicity of cadmium-based quantum dots. Biomaterials 2012, 33, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Hollingsworth, J.A.; Hu, B.; Steinbrück, A.; Stark, P.C.; Rios Valdez, C.; Vuyisich, M.; Stewart, M.H.; Atha, D.H.; Nelson, B.C.; et al. Functionalization-dependent induction of cellular survival pathways by CdSe quantum dots in primary normal human bronchial epithelial cells. ACS Nano 2013, 7, 8397–8411. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.H.; Shiao, N.H.; Lu, P.Z. CdSe quantum dots induce apoptosis in human neuroblastoma cells via mitochondrial-dependent pathways and inhibition of survival signals. Toxicol. Lett. 2006, 167, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Q.; Tang, M. Research advances on apoptosis caused by quantum dots. Biol. Trace Elem. Res. 2014, 161, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Shingo, K.; Kyoko, I.; Takeshi, Y.; Takenori, T.; Yutaka, Y.; Hiroyuki, Y.; Narisato, K.; Akiyoshi, H.; Noriyoshi, M.; Kenji, Y.; et al. Organ distribution of quantum dots after intraperitoneal administration, with special reference to area-specific distribution in the brain. Nanotechnology 2010, 21, 335103. [Google Scholar]
- Ayrton, A.; Morgan, P. Role of transport proteins in drug absorption, distribution and excretion. Xenobiotica 2001, 31, 469–497. [Google Scholar] [CrossRef] [PubMed]
- Kmieć, Z. Introduction—Morphology of the liver lobule. In Cooperation of Liver Cells in Health and Disease; Springer Berlin Heidelberg: New York, NY, USA, 2001; Volume 161, pp. 1–6. [Google Scholar]
- Kermanizadeh, A.; Løhr, M.; Roursgaard, M.; Messner, S.; Gunness, P.; Kelm, J.M.; Møller, P.; Stone, V.; Loft, S. Hepatic toxicology following single and multiple exposure of engineered nanomaterials utilising a novel primary human 3D liver microtissue model. Part. Fibre Toxicol. 2014, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, S.; Wang, L.; Qu, C.; Zhang, C.; Hong, L.; Yuan, L.; Huang, Z.; Wang, Z.; Liu, S.; et al. CdSe quantum dot (QD)-induced morphological and functional impairments to liver in mice. PLoS ONE 2011, 6, e24406. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.C.; Willmore, W.G.; Tayabali, A.F. Cadmium telluride quantum dots cause oxidative stress leading to extrinsic and intrinsic apoptosis in hepatocellular carcinoma HepG2 cells. Toxicology 2013, 306, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.C.; Rippstein, P.; Tayabali, A.F.; Willmore, W.G. Mitochondrial toxicity of cadmium telluride quantum dot nanoparticles in mammalian hepatocytes. Toxicol. Sci. 2015, 146, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Yaghini, E.; Pirker, K.F.; Kay, C.W.M.; Seifalian, A.M.; MacRobert, A.J. Quantification of reactive oxygen species generation by photoexcitation of pegylated quantum dots. Small 2014, 10, 5106–5115. [Google Scholar] [CrossRef] [PubMed]
- Diesen, D.L.; Kuo, P.C. Nitric oxide and redox regulation in the liver: Part I. General considerations and redox biology in hepatitis. J. Surg. Res. 2010, 162, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Glinka, Y.Y.; Youdim, M.B. Inhibition of mitochondrial complexes I and IV by 6-hydroxydopamine. Eur. J. Pharmacol. 1995, 292, 329–332. [Google Scholar] [CrossRef]
- Ozaki, M.; Deshpande, S.S.; Angkeow, P.; Bellan, J.; Lowenstein, C.J.; Dinauer, M.C.; Goldschmidt-Clermont, P.J.; Irani, K. Inhibition of the Rac1 GTPase protects against nonlethal ischemia/reperfusion-induced necrosis and apoptosis in vivo. FASEB J. 2000, 14, 418–429. [Google Scholar] [PubMed]
- Finkel, T. Oxygen radicals and signaling. Curr. Opin. Cell Biol. 1998, 10, 248–253. [Google Scholar] [CrossRef]
- Delgado-Buenrostro, N.L.; Medina-Reyes, E.I.; Lastres-Becker, I.; Freyre-Fonseca, V.; Ji, Z.; Hernández-Pando, R.; Marquina, B.; Pedraza-Chaverri, J.; Espada, S.; Cuadrado, A.; et al. Nrf2 protects the lung against inflammation induced by titanium dioxide nanoparticles: A positive regulator role of Nrf2 on cytokine release. Environ. Toxicol. 2015, 30, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, H.R. Estimating LD50 using the probit technique: A basic computer program. Drug Chem. Toxicol. 1983, 6, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.C.; Snyder, S.H. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl. Acad. Sci. USA 1999, 96, 13978–13982. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Tang, M.; Zhang, T.; Hu, Y.; Zhang, S.; Kong, L.; Xue, Y. Determination of a threshold dose to reduce or eliminate CdTe-induced toxicity in L929 cells by controlling the exposure dose. PLoS ONE 2013, 8, e59359. [Google Scholar] [CrossRef] [PubMed]
- Novelli, E.L.B.; Hernandes, R.T.; Novelli Filho, J.L.V.B.; Barbosa, L.L. Differential/combined effect of water contamination with cadmium and nickel on tissues of rats. Environ. Pollut. 1998, 103, 295–300. [Google Scholar] [CrossRef]
- Das, K.K.; Das, S.N.; DasGupta, S. The influence of ascorbic acid on nickel-induced hepatic lipid peroxidation in rats. J. Basic Clin. Physiol. Pharmacol. 2001, 12, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Kovochich, M.; Brant, J.; Hotze, M.; Sempf, J.; Oberley, T.; Sioutas, C.; Yeh, J.I.; Wiesner, M.R.; Nel, A.E. Comparison of the abilities of ambient and manufactured nanoparticles to induce cellular toxicity according to an oxidative stress paradigm. Nano Lett. 2006, 6, 1794–1807. [Google Scholar] [CrossRef] [PubMed]
- Pujalte, I.; Passagne, I.; Brouillaud, B.; Treguer, M.; Durand, E.; Ohayon-Courtes, C.; L’Azou, B. Cytotoxicity and oxidative stress induced by different metallic nanoparticles on human kidney cells. Part. Fibre Toxicol. 2011, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Shukla, R.K.; Kumar, A.; Vallabani, N.V.S.; Pandey, A.K.; Dhawan, A. Titanium dioxide nanoparticle-induced oxidative stress triggers DNA damage and hepatic injury in mice. Nanomedicine 2013, 9, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, F.; Zeng, Z.; Yang, H.; Jiang, H. The protective effect of bafilomycin A1 against cobalt nanoparticle-induced cytotoxicity and aseptic inflammation in macrophages in vitro. Biol. Trace Elem. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.K.; AbdElgawad, H.; Giblen, T.; Zinta, G.; de Rop, M.; Asard, H.; Blust, R.; de Boeck, G. Anti-oxidative defences are modulated differentially in three freshwater teleosts in response to ammonia-induced oxidative stress. PLoS ONE 2014, 9, e95319. [Google Scholar] [CrossRef] [PubMed]
- Hanaa Ali Hassan Mostafa Abd, E.-A. Lipid peroxidation end-products as a key of oxidative stress: Effect of antioxidant on their production and transfer of free radicals. INTECH 2012. [Google Scholar] [CrossRef]
- Chelikani, P.; Fita, I.; Loewen, P.C. Diversity of structures and properties among catalases. Cell. Mol. Life Sci. CMLS 2004, 61, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Glutathione homeostasis and functions: Potential targets for medical interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef] [PubMed]
- Chibli, H.; Carlini, L.; Park, S.; Dimitrijevic, N.M.; Nadeau, J.L. Cytotoxicity of InP/ZnS quantum dots related to reactive oxygen species generation. Nanoscale 2011, 3, 2552–2559. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, D.; Carnuccio, R.; Maiuri, M.C. Nanomaterials toxicity and cell death modalities. J. Drug Deliv. 2012, 2012, 167896. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Garantziotis, S.; Rodrigues-Lima, F.; Dupret, J.-M.; Baeza-Squiban, A.; Boland, S. Intracellular signal modulation by nanomaterials. In Nanomaterial; Capco, D.G., Chen, Y., Eds.; Springer Netherlands: Dordrecht, The Netherlands, 2014; Volume 811, pp. 111–134. [Google Scholar]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Gopinath, P.; Gogoi, S.K.; Sanpui, P.; Paul, A.; Chattopadhyay, A.; Ghosh, S.S. Signaling gene cascade in silver nanoparticle induced apoptosis. Colloids Surf. B 2010, 77, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.A.; Alhadlaq, H.A.; Ahmad, J.; Al-Khedhairy, A.A.; Musarrat, J.; Ahamed, M. Copper oxide nanoparticles induced mitochondria mediated apoptosis in human hepatocarcinoma cells. PLoS ONE 2013, 8, e69534. [Google Scholar] [CrossRef] [PubMed]
- Senapati, V.A.; Jain, A.K.; Gupta, G.S.; Pandey, A.K.; Dhawan, A. Chromium oxide nanoparticle-induced genotoxicity and p53-dependent apoptosis in human lung alveolar cells. J. Appl. Toxicol. 2015, 35, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.G.; Choi, E.J. Apoptotic signaling pathways: Caspases and stress-activated protein kinases. J. Biochem. Mol. Biol. 2002, 35, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Eom, H.-J.; Choi, J. Oxidative stress of CeO2 nanoparticles via p38-Nrf-2 signaling pathway in human bronchial epithelial cell, Beas-2B. Toxicol. Lett. 2009, 187, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Dong, J.; Sulik, K.K.; Chen, S.-Y. Induction of the Nrf2-driven antioxidant response by tert-butylhydroquinone prevents ethanol-induced apoptosis in cranial neural crest cells. Biochem. Pharmacol. 2010, 80, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, J.; Shen, C.; Zhang, X.; Sun, S.; Cho, M.; Sun, C.; Song, Z. Tert-butylhydroquinone (tBHQ) protects hepatocytes against lipotoxicity via inducing autophagy independently of Nrf2 activation. Biochim. Biophys. Acta 2014, 1841, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.W.; Zhang, Y.; Ge, C.W.; Jin, Y.L.; Hu, S.L.; Gu, N. Temperature-dependent photoluminescence of highly luminescent water-soluble CdTe quantum dots. Chin. Chem. Lett. 2009, 20, 977–980. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Zhang, T.; Tang, M.; Kong, L.; Li, H.; Zhang, T.; Xue, Y.; Pu, Y. Surface modification of multiwall carbon nanotubes determines the pro-inflammatory outcome in macrophage. J. Hazard. Mater. 2015, 284, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative Ct method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Hu, Y.; Tang, M.; Kong, L.; Ying, J.; Wu, T.; Xue, Y.; Pu, Y. Liver Toxicity of Cadmium Telluride Quantum Dots (CdTe QDs) Due to Oxidative Stress in Vitro and in Vivo. Int. J. Mol. Sci. 2015, 16, 23279-23299. https://doi.org/10.3390/ijms161023279
Zhang T, Hu Y, Tang M, Kong L, Ying J, Wu T, Xue Y, Pu Y. Liver Toxicity of Cadmium Telluride Quantum Dots (CdTe QDs) Due to Oxidative Stress in Vitro and in Vivo. International Journal of Molecular Sciences. 2015; 16(10):23279-23299. https://doi.org/10.3390/ijms161023279
Chicago/Turabian StyleZhang, Ting, Yuanyuan Hu, Meng Tang, Lu Kong, Jiali Ying, Tianshu Wu, Yuying Xue, and Yuepu Pu. 2015. "Liver Toxicity of Cadmium Telluride Quantum Dots (CdTe QDs) Due to Oxidative Stress in Vitro and in Vivo" International Journal of Molecular Sciences 16, no. 10: 23279-23299. https://doi.org/10.3390/ijms161023279
APA StyleZhang, T., Hu, Y., Tang, M., Kong, L., Ying, J., Wu, T., Xue, Y., & Pu, Y. (2015). Liver Toxicity of Cadmium Telluride Quantum Dots (CdTe QDs) Due to Oxidative Stress in Vitro and in Vivo. International Journal of Molecular Sciences, 16(10), 23279-23299. https://doi.org/10.3390/ijms161023279