
GHRH, PRP-PACAP and GHRHR Target Sequencing via an Ion Torrent Personal Genome Machine Reveals an Association with Growth in Orange-Spotted Grouper (Epinephelus coioides)


Abstract
:1. Introduction
2. Results
2.1. The Phenotype and Genotype



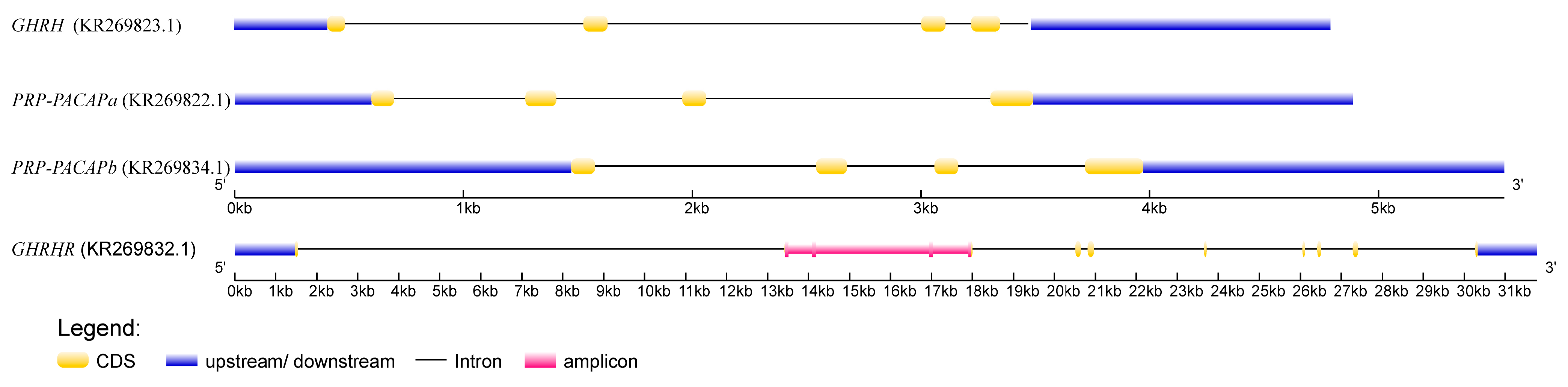{kind=link}
{kind=link}
{kind=link}
| Source | Sum of Squares | Degrees of Freedom | Mean Square | F | p-Value |
|---|---|---|---|---|---|
| Mean coverage depth | |||||
| Individual | 1,543,230 | 158 | 9767.28 | 6.786137 | 1.8 × 10−59 |
| Fragment | 76,483 | 3 | 25,494.44 | 17.71309 | 6.5 × 10−11 |
| Error | 682,227 | 474 | 1439.30 | ||
| Total | 2,301,941 | 635 | |||
| Sensitivity | |||||
| Individual | 7.7933 | 158 | 0.0493 | 2.794009 | 1.0 × 10−17 |
| Fragment | 0.3277 | 3 | 0.1092 | 6.187525 | 4.0 × 10−4 |
| Error | 8.3679 | 474 | 0.0177 | ||
| Total | 16.4889 | 635 | |||
| Region | Number (bp) | Number of Polymorphic Sites | Nucleotide Diversity | Tajima’s D c | Fu’s Fs | dN/dS d | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| π | θw | Fs | p | Stat | p | |||||
| GHRH (KR269823.1) | Synonymous | 101.66 | 2 | 0.00165 | 0.00310 | −0.59938 | ||||
| Silent b | 4431.66 | 112 | 0.00362 | 0.00399 | −0.27736 | |||||
| Non-synonymous | 324.34 | 5 | 0.00210 | 0.00243 | −0.24643 | |||||
| Total gene a | 4756 | 117 | 0.00352 | 0.00388 | −0.28353 | −126.856 | 0.000 | 0.250 | 0.803 | |
| PRP-PACAPa (KR269822.1) | Synonymous | 127.00 | 0 | 0.00000 | 0.00000 | NA | ||||
| Silent b | 4457.00 | 81 | 0.00318 | 0.00287 | 0.00267 | |||||
| Non-synonymous | 395.00 | 1 | 0.00006 | 0.00040 | 0.00267 | |||||
| Total gene a | 4852 | 82 | 0.00292 | 0.00267 | 0.00267 | −134.514 | 0.000 | 1.029 | 0.305 | |
| PRP-PACAPb (KR269834.1) | Synonymous | 134.82 | 0 | 0.00000 | 0.00000 | NA | ||||
| Silent b | 5084.82 | 53 | 0.00107 | 0.00164 | −1.01201 | |||||
| Non-synonymous | 465.18 | 1 | 0.00016 | 0.00034 | −0.50730 | |||||
| Total gene a | 5550 | 54 | 0.00099 | 0.00154 | −1.02345 | −73.275 | 0.000 | 0.000 | 1.000 | |
| GHRHR (KR269832.1) | Synonymous | 82.19 | 2 | 0.00266 | 0.00384 | −0.39137 | ||||
| Silent b | 4009.19 | 79 | 0.00285 | 0.00311 | −0.24244 | |||||
| Non-synonymous | 265.81 | 6 | 0.00311 | 0.00356 | −0.24054 | |||||
| Total gene a | 4280 | 85 | 0.00287 | 0.00313 | −0.25251 | −46.197 | 0.000 | 0.202 | 0.840 | |
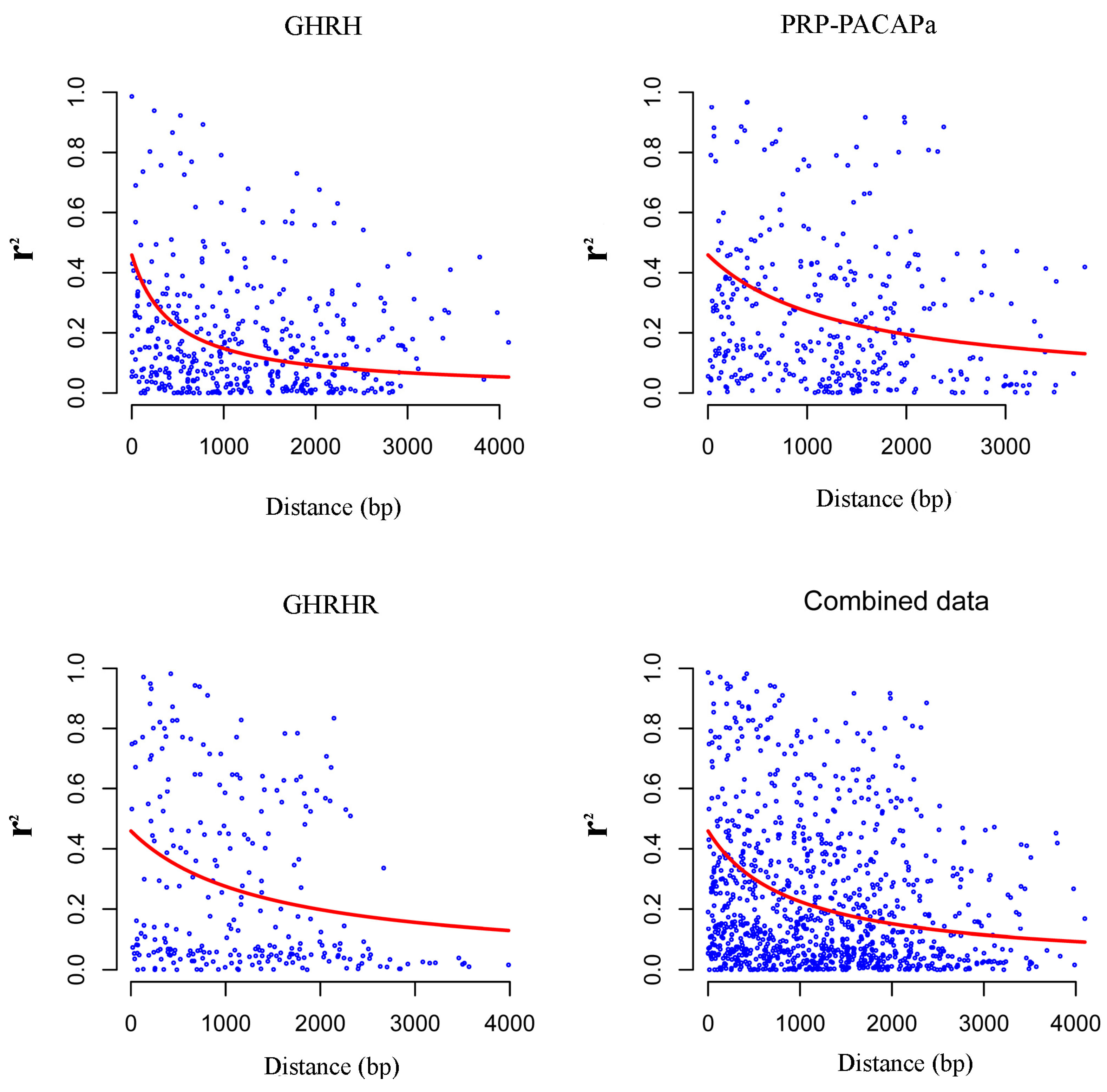
2.2. Nucleotide Diversity and Linkage Disequilibrium

2.3. The Relationship between Growth Hormone-Releasing Hormone (GHRH), PACAP-Related Peptide/Pituitary Adenylate Cyclase Activating Polypeptide (PRP-PACAP), Growth Hormone-Releasing Hormone (GHRHR) and Growth Traits
| Marker | Gene | Mutation Type | Location | Site | MAF | Trait | p | Q | R2 (%) | 2a 1 | d 2 | d/a 1,2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KR269823.1:g.475A>C | GHRH | Synonymous | Exon1 | +72 | 0.24 | SL | 1.41 × 10−6 | 0.0002 | 16.1 | 3.4989 | −0.8404 | −0.4804 |
| MBD | 3.73 × 10−5 | 0.0048 | 12.4 | 0.8379 | −0.2173 | −0.5186 | ||||||
| BWT | 6.10 × 10−7 | 0.0001 | 17.0 | 54.7356 | −12.8061 | −0.4679 | ||||||
| CPD | 3.65 × 10−4 | 0.0467 | 9.8 | 0.2052 | −0.0199 | −0.1938 | ||||||
| TL | 1.76 × 10−5 | 0.0023 | 13.3 | 3.8517 | −0.9712 | −0.5043 | ||||||
| KR269823.1:g.2143T>C | GHRH | Untranslated | Intron2 | +1740 | 0.14 | SL | 6.34 × 10−4 | 0.0406 | 9.2 | 1.6475 | −0.1210 | −0.1469 |
| BWT | 7.44 × 10−4 | 0.0476 | 9.0 | 17.9322 | 3.5977 | 0.4013 |
| Marker | Trait | p Value | R2 | Haplotype | Frequency |
|---|---|---|---|---|---|
| KR269823.1:g.2143T>C | SL | 0.0389 | 8.96% | SNPs15–18 | |
| BWT | 0.0294 | 12.34% | T_A_C_G | 0.780 | |
| T_G_T_A | 0.070 | ||||
| C_A_C_G | 0.128 |
3. Discussion
3.1. Target Sequencing
3.2. Nucleotide Diversity and Linkage Disequilibrium
3.3. Association Mapping of Growth
4. Experimental Section
4.1. The Sample and Phenotype Records
4.2. Candidate Gene Sequencing and SNP Determination
| ID | Gene | Forward 5′ to 3′ | Reverse 5′ to 3′ | Amplicon Length 3 | GenBank Accession Number or Predicted (mRNA) |
|---|---|---|---|---|---|
| KR269823.1 | GHRH 1 | TGTGGGAGTGACTGGGAGC | GAGACACGCACAATACCAG | 4756 | GU966634.1 |
| KR269822.1 | PRP-PACAPa 1 | GATGCACTCCAATGGGAC | GAGTCGTCTGCACAGATG | 4852 | predicted |
| KR269834.1 | PRP-PACAPb 1 | GACTGCTCCTTCTTGGTTAA | TTTGTCTCCTGCTCTTCCT | 5550 | AY869693.1 |
| KR269832.1 | GHRHR 2 | GCTTTGTCGAGCCTGCACC | CAACACAGCAACAACCAGC | 4280 | GU966635.1 |
4.3. Nucleotide Diversity and Linkage Disequilibrium
4.4. Associations between the Single-Nucleotide Polymorphisms (SNPs) and Phenotype
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Heemstra, P.C.; Randall, J.E. FAO Species Catalogue vol. 16 Groupers of the World (Family Serranidae, Subfamily Epinephelinae): An Annotated and Illustrated Catalogue of the Grouper, Rockcod, Hind, Coral Grouper, and Lyretail Species Known to Date; Food and Agriculture Organization of the United Nations: Rome, Italy, 1993. [Google Scholar]
- Pierre, S.; Gaillard, S.; Prevot-D’Alvise, N.; Aubert, J.; Rostaing-Capaillon, O.; Leung-Tack, D.; Grillasca, J. Grouper aquaculture: Asian success and Mediterranean trials. Aquat. Conserv. 2008, 18, 297–308. [Google Scholar] [CrossRef]
- You, X.; Shu, L.; Li, S.; Chen, J.; Luo, J.; Lu, J.; Mu, Q.; Bai, J.; Xia, Q.; Chen, Q.; et al. Construction of high-density genetic linkage maps for orange-spotted grouper Epinephelus coioides using multiplexed shotgun genotyping. BMC Genet. 2013, 14, 113. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Huang, H.; Meng, Z.; Zhang, Y.; Luo, J.; Chen, G.; Lin, H. Single Nucleotide polymorphisms in the Leptin-a gene and associations with growth traits in the orange-spotted grouper (Epinephelus coioides). Int. J. Mol. Sci. 2013, 14, 8625–8637. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wei, Y.; Meng, Z.; Zhang, Y.; Liu, X.; Guo, L.; Luo, J.; Chen, G.; Lin, H. Polymorphisms of Leptin-b gene associated with growth traits in orange-spotted grouper (Epinephelus coioides). Int. J. Mol. Sci. 2014, 15, 11996–12006. [Google Scholar] [CrossRef] [PubMed]
- Gjedrem, T.; Robinson, N.; Rye, M. The importance of selective breeding in aquaculture to meet future demands for animal protein: A review. Aquaculture 2012, 350, 117–129. [Google Scholar] [CrossRef]
- Yu, J.M.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Hindorff, L.A.; MacArthur, J.; Morales, J.; Junkins, H.A.; Hall, P.N.; Klemm, A.K.; Manolio, T.A. A Catalog of Published Genome-Wide Association Studies. Available online: http://www.genome.gov/gwastudies (accessed on 17 March 2015).
- Wang, J.; Wang, H.; Chi, R.; Guo, J.; Wu, Y. Progresses in research of genome-wide association studies in livestock and poultry. Sci. Agric. Sin. 2013, 46, 819–829. [Google Scholar]
- Zhu, C.; Gore, M.; Buckler, E.S.; Yu, J. Status and prospects of association mapping in plants. Plant Genome 2008, 1, 5–20. [Google Scholar] [CrossRef]
- Caniato, F.F.; Hamblin, M.T.; Guimaraes, C.T.; Zhang, Z.; Schaffert, R.E.; Kochian, L.V.; Magalhaes, J.V. Association mapping provides insights into the origin and the fine structure of the sorghum aluminum tolerance locus, AltSB. PLoS ONE 2014, 9, e87438. [Google Scholar] [CrossRef] [PubMed]
- Thavamanikumar, S.; Tibbits, J.; McManus, L.; Ades, P.; Stackpole, D.; Hadjigol, S.; Vaillancourt, R.; Zhu, P.; Bossinger, G. Candidate gene-based association mapping of growth and wood quality traits in Eucalyptus globulus Labill. BMC Proc. 2011, 5, O15. [Google Scholar] [CrossRef]
- Nordborg, M.; Borevitz, J.O.; Bergelson, J.; Berry, C.C.; Chory, J.; Hagenblad, J.; Kreitman, M.; Maloof, J.N.; Noyes, T.; Oefner, P.J.; et al. The extent of linkage disequilibrium in Arabidopsis thaliana. Nat. Genet. 2002, 30, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Campbell, N.R.; LaPatra, S.E.; Overturf, K.; Towner, R.; Narum, S.R. Association mapping of disease resistance traits in rainbow trout using restriction site associated DNA sequencing. G3 (Bethesda) 2014, 4, 2473–2481. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.H.; Lin, G.; He, X.; Liu, P.; Liu, F.; Sun, F.; Tu, R.; Yue, G.H. Whole genome scanning and association mapping identified a significant association between growth and a SNP in the IFABP-a gene of the Asian seabass. BMC Genom. 2013, 14, 295. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Yan, A.; Lin, H.; Li, W. Molecular characterization of the GHRH/GHRHR and its effect on GH synthesis and release in orange-spotted grouper (Epinephelus coioides). Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2012, 163, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Siu, F.; Tam, J.; Lau, I.; Wong, A.; Lin, M.; Vaudry, H.; Chow, B. Discovery of growth hormone-releasing hormones and receptors in nonmammalian vertebrates. Proc. Natl. Acad. Sci. USA 2007, 104, 2133–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, J.K.V.; Lee, L.T.O.; Cheng, C.H.K.; Chow, B.K.C. Discovery of a new reproductive hormone in teleosts: Pituitary adenylate cyclase-activating polypeptide-related peptide (PRP). Gen. Comp. Endocrinol. 2011, 173, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, D.; Alonzeau, J.; Bill, B.R.; Ekker, S.C.; Waschek, J.A. Expression analysis of PAC1-R and PACAP genes in zebrafish embryos. J. Mol. Neurosci. 2011, 43, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Dybus, A.; Kmiec, M.; Sobek, Z.; Pietrzyk, W.; Wisniewski, B. Associations between polymorphisms of growth hormone releasing hormone (GHRH) and pituitary transcription factor 1 (PIT1) genes and production traits of Limousine cattle. Arch. Tierz. 2003, 46, 527–534. [Google Scholar]
- Liu, Y.; Lan, X.; Qu, Y.; Li, Z.; Chen, Z.; Lei, C.; Fang, X.; Chen, H. Effects of genetic variability of the dairy goat growth hormone releasing hormone receptor (GHRHR) gene on growth traits. Mol. Biol. Rep. 2011, 38, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.J.; Boulding, E.G. Associations between single nucleotide polymorphisms in candidate genes and growth rate in Arctic charr (Salvelinus alpinus L.). Heredity (Edinb) 2003, 91, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Paul, J.S.; Albrechtsen, A.; Song, Y.S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011, 12, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.L.; Sousa, S.; Justino, A.; Kay, T.; Fernandes, S.; Cirnes, L.; Schmitt, F.; Machado, J.C. Nonoptical massive parallel DNA sequencing of BRCA1 and BRCA2 genes in a diagnostic setting. Hum. Mutat. 2013, 34, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Adlakha, N.; Ritturaj Kushwaha, H.; Rajagopal, R.; Yazdani, S.S. Draft genome sequence of the Paenibacillus sp. strain ICGEB2008 (MTCC 5639) isolated from the gut of Helicoverpa armigera. Genome Announc. 2013, 1, e12–e26. [Google Scholar]
- Mamanova, L.; Coffey, A.J.; Scott, C.E.; Kozarewa, I.; Turner, E.H.; Kumar, A.; Howard, E.; Shendure, J.; Turner, D.J. Target-enrichment strategies for next-generation sequencing. Nat. Methods 2010, 7, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.W.; Pearson, J.V.; Szelinger, S.; Sekar, A.; Redman, M.; Corneveaux, J.J.; Pawlowski, T.L.; Laub, T.; Nunn, G.; Stephan, D.A.; et al. Identification of genetic variants using bar-coded multiplexed sequencing. Nat. Methods 2008, 5, 887–893. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, M.; McPartlin, J.; Arthure, K.; Riedel, S.; McMillan, N.D. Comparison of the TLDA with the Nanodrop and the reference Qubit system. J. Phys. Conf. Ser. 2011. [Google Scholar] [CrossRef]
- Wang, L.; Meng, Z.; Liu, X.; Zhang, Y.; Lin, H. Genetic diversity and differentiation of the orange-spotted grouper (Epinephelus coioides) between and within cultured stocks and wild populations inferred from microsatellite DNA analysis. Int. J. Mol. Sci. 2011, 12, 4378–4394. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.D.; Jin, W.; Hao, X.D.; Tang, N.L.; Zhang, Y.P. Evidence for positive selection on the Osteogenin (BMP3) gene in human populations. PLoS ONE 2010, 5, e10959. [Google Scholar] [CrossRef] [PubMed]
- Templeton, A.R. Population Genetics and Microevolutionary Theory; Wiley-Liss: Hoboken, NJ, USA, 2006; p. 705. [Google Scholar]
- Yu, J.; Buckler, E.S. Genetic association mapping and genome organization of maize. Curr. Opin. Biotechnol. 2006, 17, 155–160. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, B.; de la Rosa, D.A.; Rajendran, V.M. Multiple mineralocorticoid response elements localized in different introns regulate intermediate conductance K+ (KCNN4) channel expression in the rat distal colon. PLoS ONE 2014, 9, e98695. [Google Scholar] [CrossRef] [PubMed]
- Froese, R.; Pauly, D. FishBase, version (08/2015); FishBase: Paris, France, 2015; Available online: http://www.fishbase.org/Identification/Morphometrics/centimeters/Index.php (accessed on 24 October 2015).
- Green, M.R.; Sambrook, J. Molecular Cloning: A Laboratory Manua; Cold Spring Horbor laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Patricia Arrieta, M.; Parres, F.; Lopez, J.; Jimenez, A. Development of a novel pyrolysis-gas chromatography/mass spectrometry method for the analysis of poly(lactic acid) thermal degradation products. J. Anal. Appl. Pyrol. 2013, 101, 150–155. [Google Scholar] [CrossRef]
- Stephens, M.; Smith, N.J.; Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef] [PubMed]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Watterson, G.A. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 1975, 7, 256–276. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [PubMed]
- Nei, M.; Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986, 3, 418–426. [Google Scholar] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Remington, D.L.; Thornsberry, J.M.; Matsuoka, Y.; Wilson, L.M.; Whitt, S.R.; Doeblay, J.; Kresovich, S.; Goodman, M.M.; Buckler, E.S. Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. USA 2001, 98, 11479–11484. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Hardy, O.J.; Vekemans, X. SPAGeDi: A versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2002, 2, 618–620. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Chang, M.; Du, Q.; Xu, B.; Zhang, D. Single-nucleotide polymorphisms in PtoCesA7 and their association with growth and wood properties in Populus tomentosa. Mol. Genet. Genom. 2014, 289, 439–455. [Google Scholar] [CrossRef] [PubMed]
- Zaykin, D.V.; Westfall, P.H.; Young, S.S.; Karnoub, M.A.; Wagner, M.J.; Ehm, M.G. Testing association of statistically inferred haplotypes with discrete and continuous traits in samples of unrelated individuals. Hum. Hered. 2002, 53, 79–91. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, L.; Xia, J.; Yang, S.; Li, M.; You, X.; Meng, Z.; Lin, H. GHRH, PRP-PACAP and GHRHR Target Sequencing via an Ion Torrent Personal Genome Machine Reveals an Association with Growth in Orange-Spotted Grouper (Epinephelus coioides). Int. J. Mol. Sci. 2015, 16, 26137-26150. https://doi.org/10.3390/ijms161125940
Guo L, Xia J, Yang S, Li M, You X, Meng Z, Lin H. GHRH, PRP-PACAP and GHRHR Target Sequencing via an Ion Torrent Personal Genome Machine Reveals an Association with Growth in Orange-Spotted Grouper (Epinephelus coioides). International Journal of Molecular Sciences. 2015; 16(11):26137-26150. https://doi.org/10.3390/ijms161125940
Chicago/Turabian StyleGuo, Liang, Junhong Xia, Sen Yang, Mingming Li, Xinxin You, Zining Meng, and Haoran Lin. 2015. "GHRH, PRP-PACAP and GHRHR Target Sequencing via an Ion Torrent Personal Genome Machine Reveals an Association with Growth in Orange-Spotted Grouper (Epinephelus coioides)" International Journal of Molecular Sciences 16, no. 11: 26137-26150. https://doi.org/10.3390/ijms161125940
APA StyleGuo, L., Xia, J., Yang, S., Li, M., You, X., Meng, Z., & Lin, H. (2015). GHRH, PRP-PACAP and GHRHR Target Sequencing via an Ion Torrent Personal Genome Machine Reveals an Association with Growth in Orange-Spotted Grouper (Epinephelus coioides). International Journal of Molecular Sciences, 16(11), 26137-26150. https://doi.org/10.3390/ijms161125940