
Abnormalities in Alternative Splicing of Apoptotic Genes and Cardiovascular Diseases

Abstract
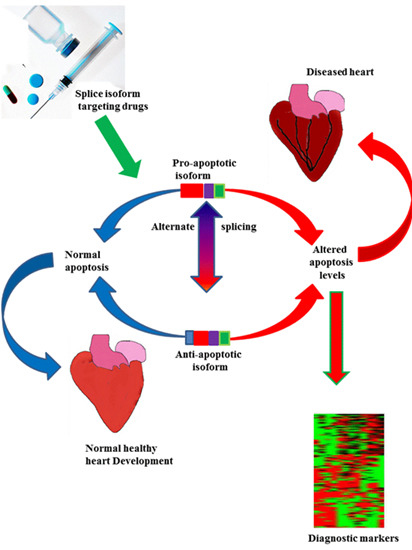
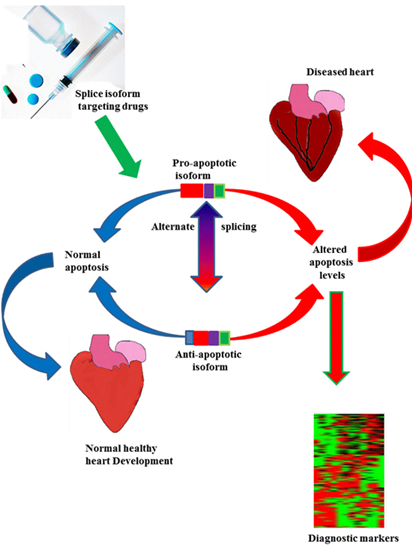
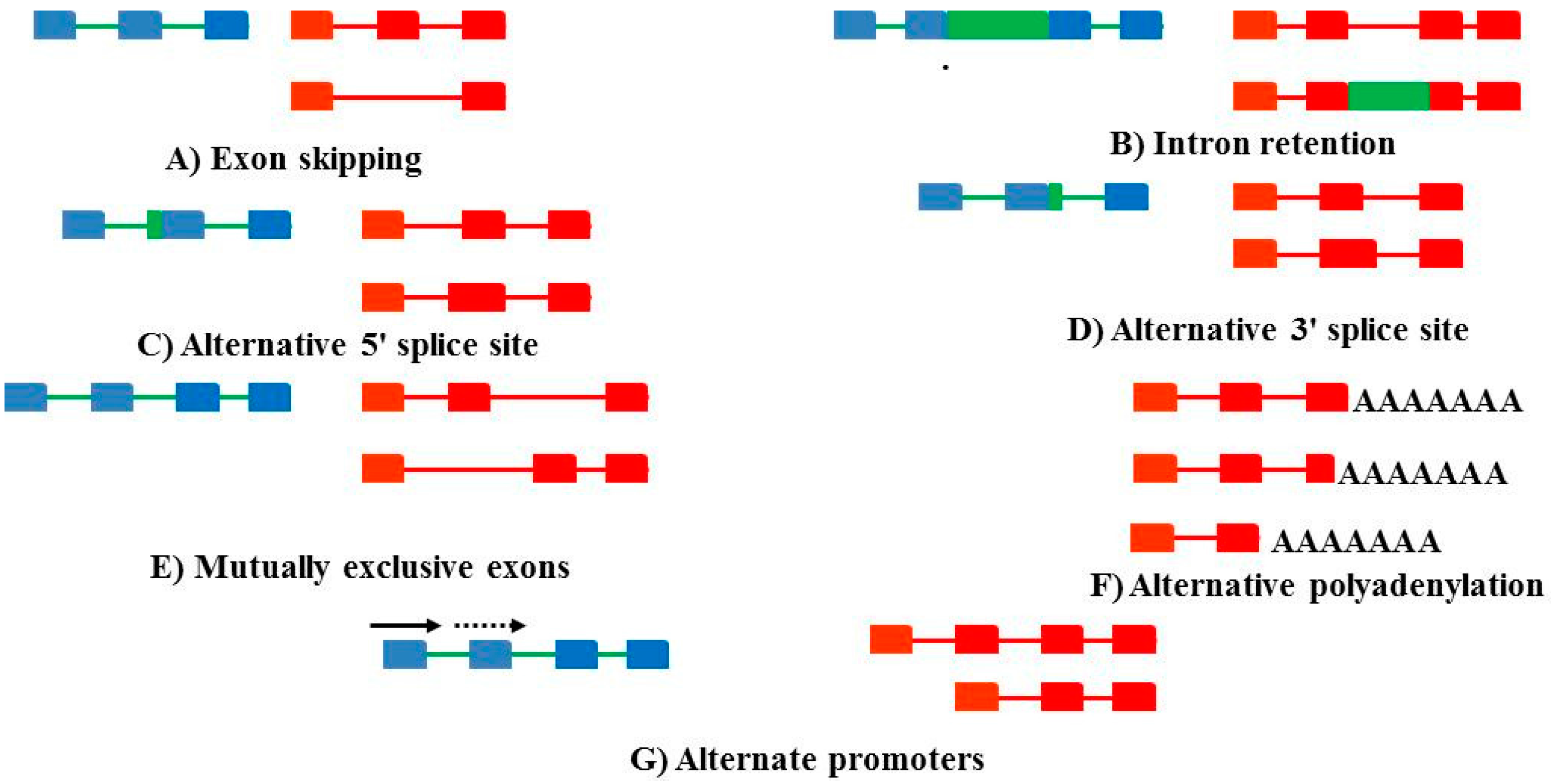
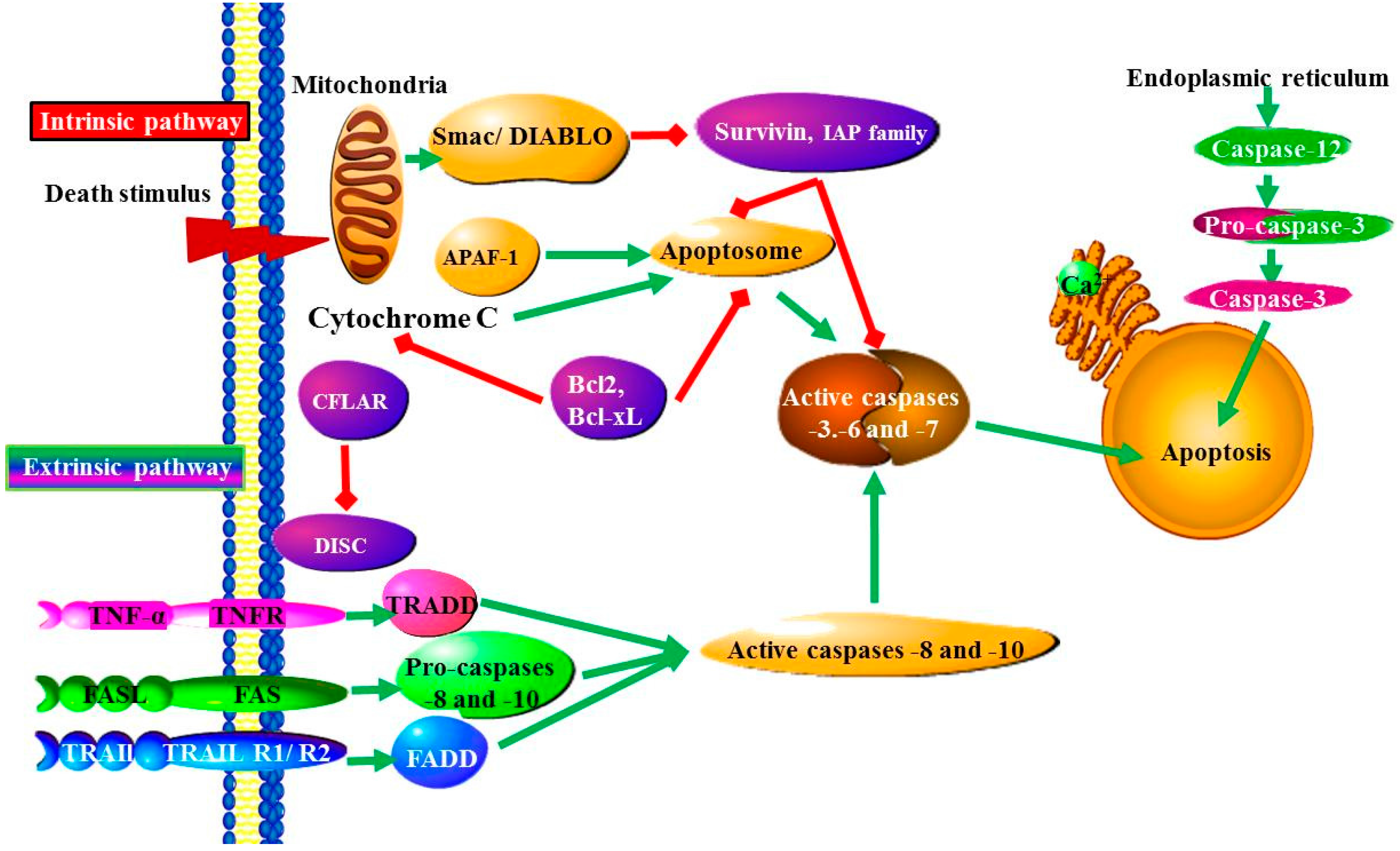
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction


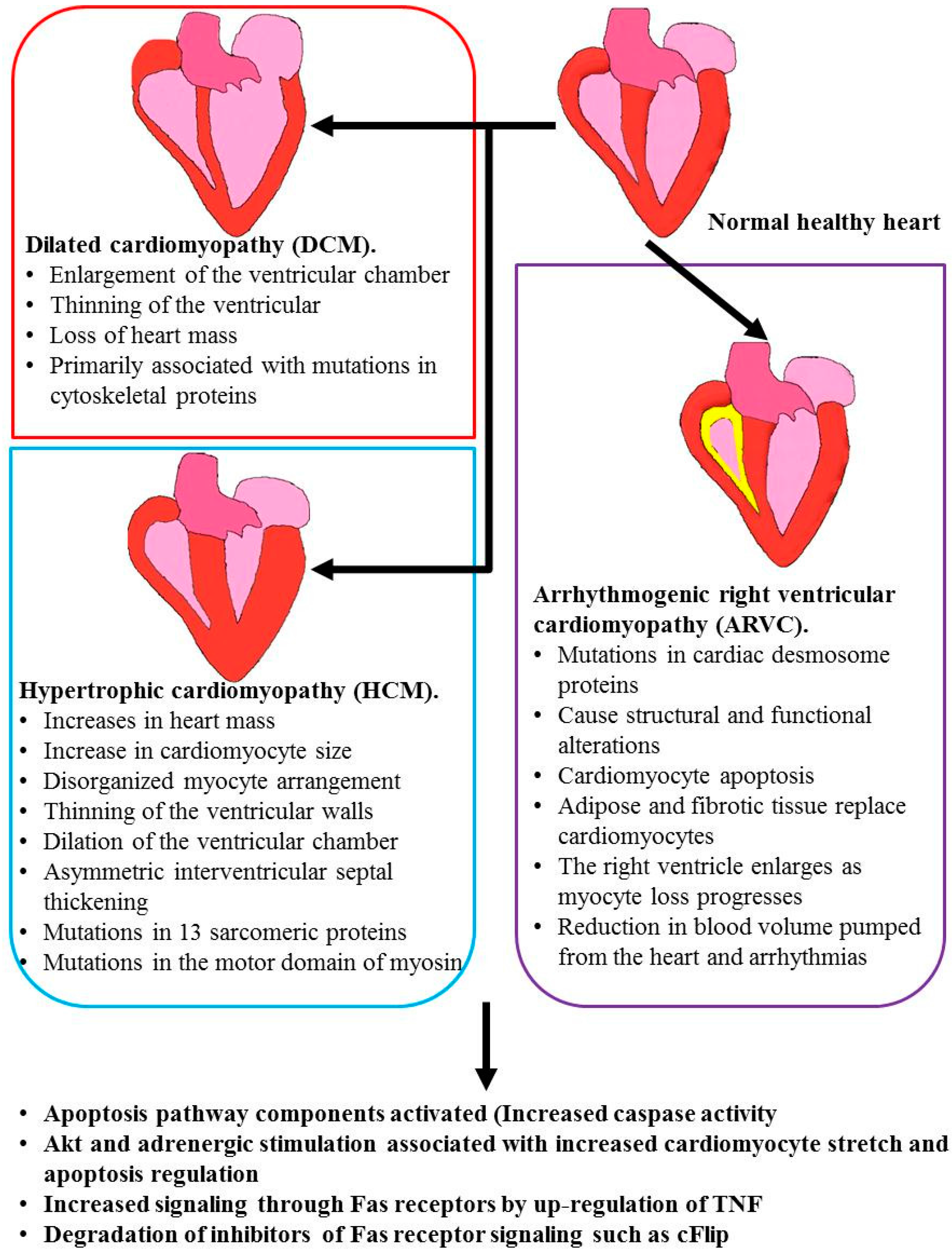
2. Cardiomyopathies that Have Apoptosis Related Causes

2.1. Ischemia/Reperfusion Injury
2.2. Hypertrophic Cardiomyopathy (HCM) and Dilated Cardiomyopathy (DCM)
2.3. Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC)
2.4. Hypoplastic Left Heart Syndrome (HLHS)
3. The Impact of Alternative Splicing on Cardiac Disease
3.1. Hypertrophic Cardiomyopathy
3.2. Arrhythmogenic Right Ventricular Cardiomyopathy
4. Alternative Splicing and Apoptotic Genes
4.1. Serine-Arginine (SR)-Rich Proteins and Their Specific Kinases
4.2. Caspases
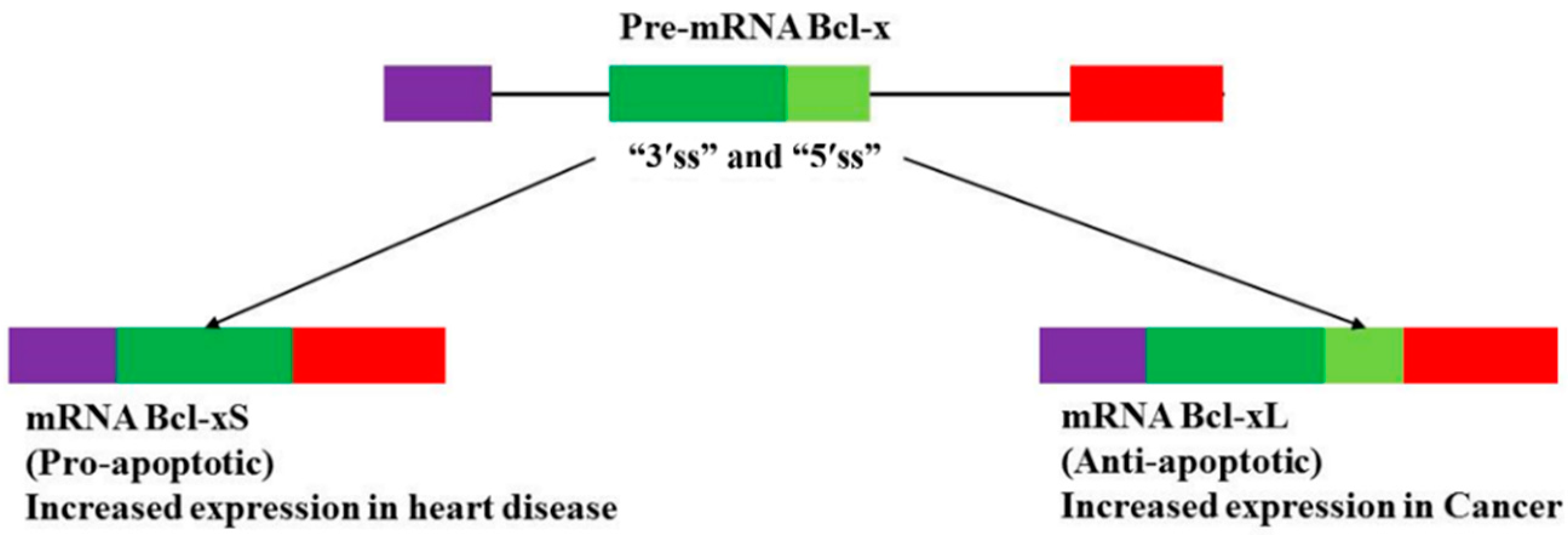
4.3. The Bcl-2 Family

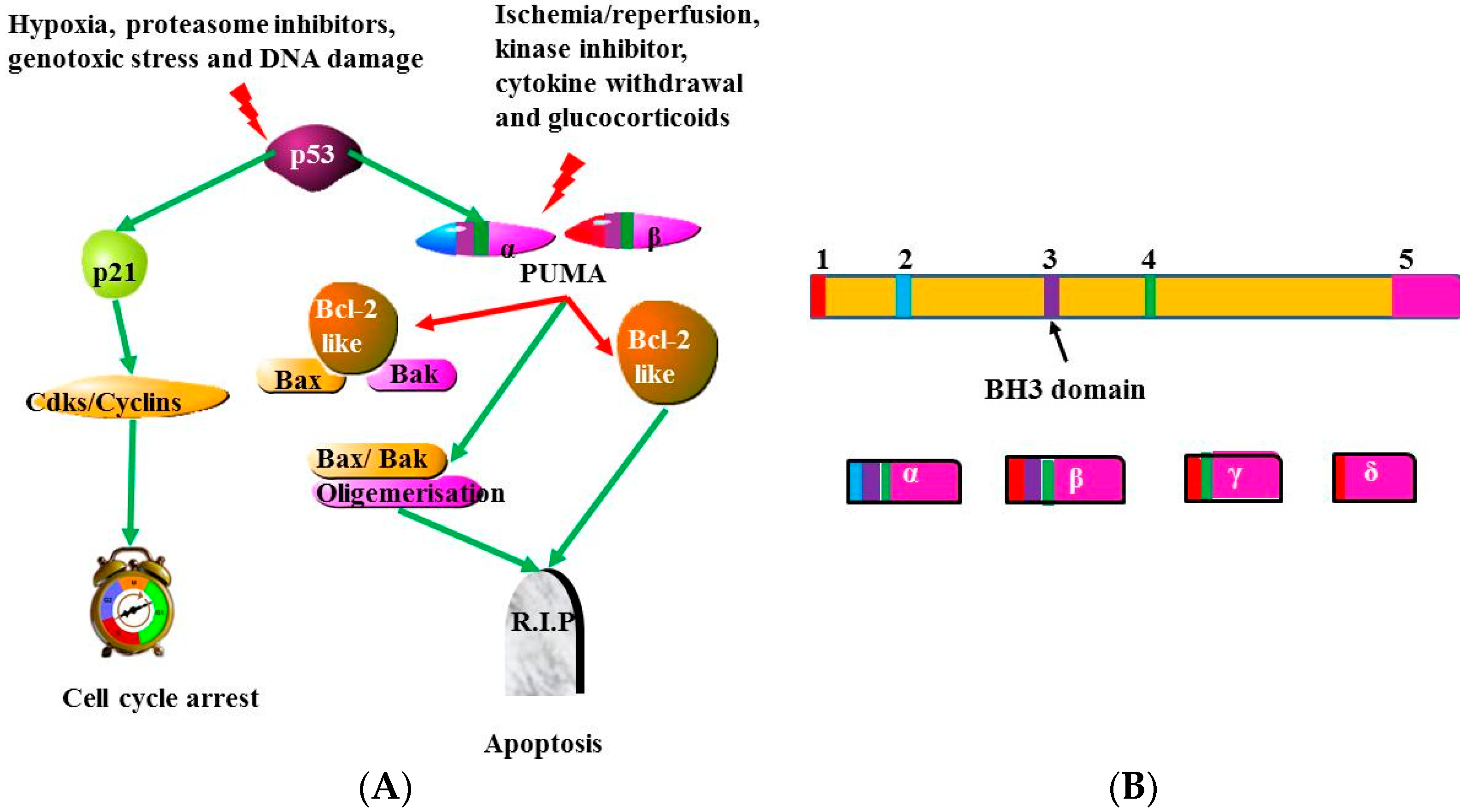
4.4. p53 and p53-Upregulated Modulator of Apoptosis (PUMA)

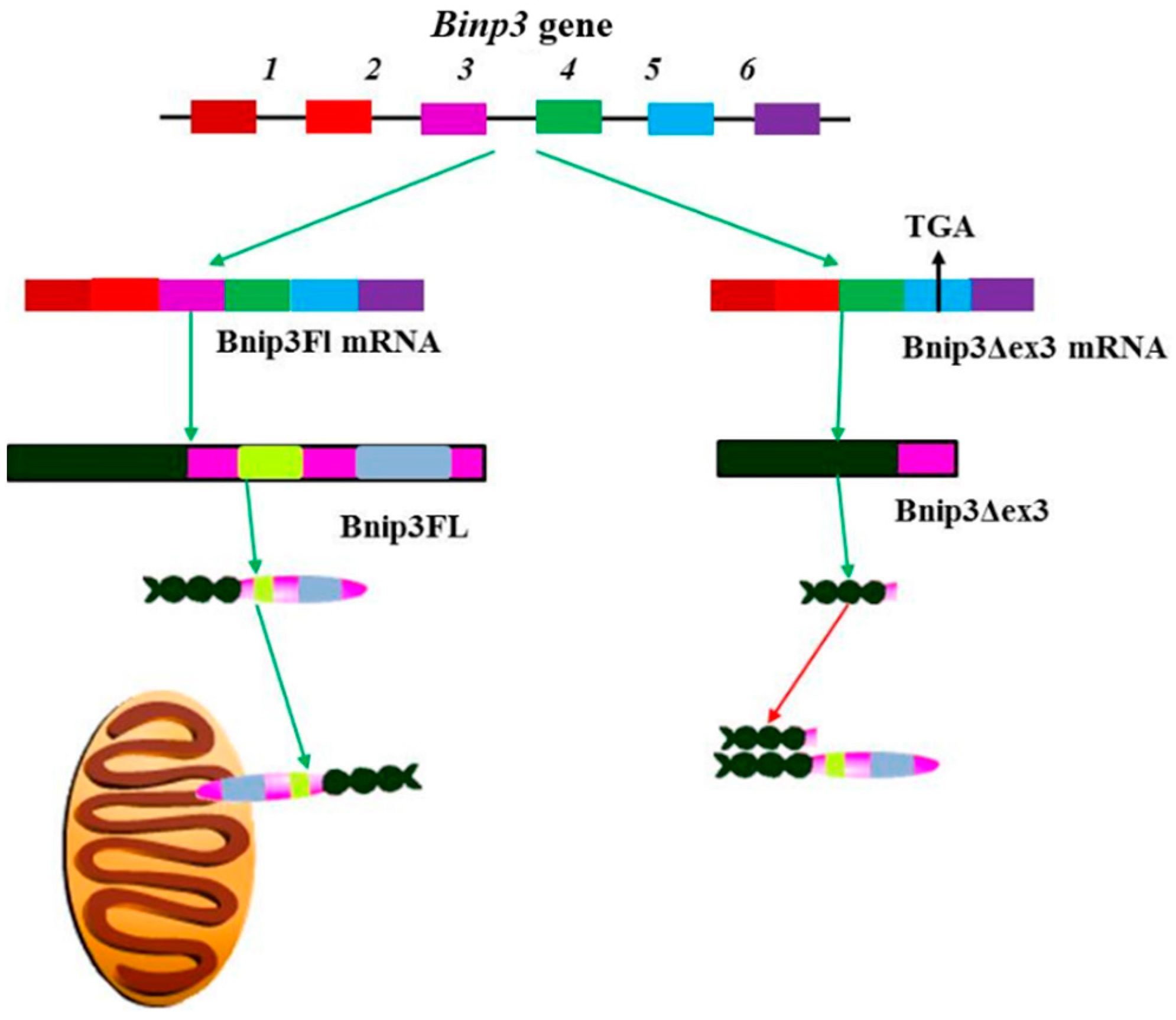
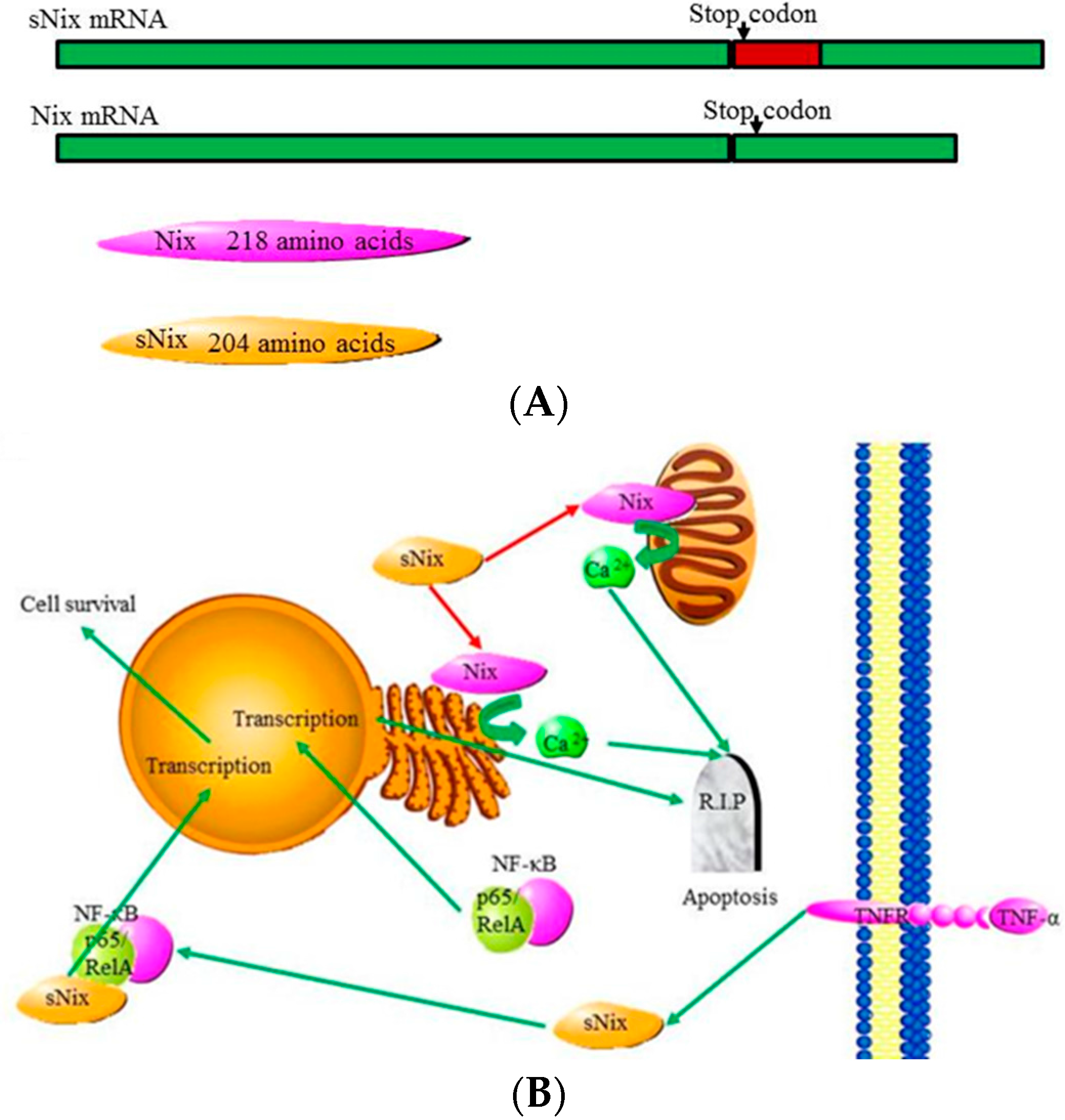
4.5. Bnip3 and Nix


5. Novel Therapeutic Strategies for Targeting Diseases Associated with Abnormal Splicing

6. Conclusion
Conflicts of Interest
Abbreviations
References
- Chen, L.; Tovar-Corona, J.M.; Urrutia, A.O. Alternative splicing: A potential source of functional innovation in the eukaryotic genome. Int. J. Evolut. Biol. 2012, 2012, 10. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.; Jearawiriyapaisarn, N.; Kole, R. Therapeutic potential of splice-switching oligonucleotides. Oligonucleotides 2009, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Revil, T.; Gaffney, D.; Dias, C.; Majewski, J.; Jerome-Majewska, L.A. Alternative splicing is frequent during early embryonic development in mouse. BMC Genom. 2010, 11, 399. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Fujibuchi, W.; Unno, M. Splice variants in apoptotic pathway. Exp. Oncol. 2012, 34, 212–217. [Google Scholar] [PubMed]
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.W.J.; Valcárcel, J. Alternative pre-mRNA splicing: The logic of combinatorial control. Trends Biochem. Sci. 2000, 25, 381–388. [Google Scholar] [CrossRef]
- Schwerk, C.; Schulze-Osthoff, K. Regulation of apoptosis by alternative pre-mRNA splicing. Mol. Cell 2005, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Akgul, C.; Moulding, D.A.; Edwards, S.W. Alternative splicing of Bcl-2-related genes: Functional consequences and potential therapeutic applications. Cell. Mol. Life Sci. CMLS 2004, 61, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Shin, N.-H.; Sun, Y.; Wang, K.K.W. Molecular cloning and characterization of a novel caspase-3 variant that attenuates apoptosis induced by proteasome inhibition. Biochem. Biophys. Res. Commun. 2001, 283, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, N.; Kamachi, M.; Aratake, K.; Izumi, Y.; Ida, H.; Tanaka, F.; Tamai, M.; Arima, K.; Nakamura, H.; Origuchi, T.; et al. Regulation of alternative splicing of caspase-2 through an intracellular signaling pathway in response to pro-apoptotic stimuli. J. Lab. Clin. Med. 2005, 145, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Antonsson, B. Bax and other pro-apoptotic bcl-2 family “Killer-proteins” and their victim the mitochondrion. Cell Tissue Res. 2001, 306, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Srivastava, R.K. Involvement of Bcl-2 family members, phosphatidylinositol 3′-kinase/Akt and mitochondrial p53 in curcumin (diferulolylmethane)-induced apoptosis in prostate cancer. Int. J. Oncol. 2007, 30, 905–918. [Google Scholar] [CrossRef] [PubMed]
- Wickman, G.; Julian, L.; Olson, M.F. How apoptotic cells aid in the removal of their own cold dead bodies. Cell Death Differ. 2012, 19, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M. Ways of dying: Multiple pathways to apoptosis. Genes Dev. 2003, 17, 2481–2495. [Google Scholar] [CrossRef] [PubMed]
- Gill, C.; Mestril, R.; Samali, A. Losing heart: The role of apoptosis in heart disease - a novel therapeutic target? FASEB J. 2002, 16, 135–146. [Google Scholar] [CrossRef] [PubMed]
- James, T.N. The variable morphological coexistence of apoptosis and necrosis in human myocardial infarction: Significance for understanding its pathogenesis, clinical course, diagnosis and prognosis. Coron. Artery Dis. 1998, 9, 291–307. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Satyamoorthy, K.; Herlyn, M. N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res. 2001, 61, 3819–3825. [Google Scholar] [PubMed]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Burleson, K.O.; Kloner, R.A.; Babior, B.M.; Engler, R.L. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J. Clin. Investig. 1994, 94, 1621–1628. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.; Whelan, R.S.; Kitsis, R.N. Mechanisms of cell death in heart disease. Arterioscler. Thrombosis Vasc. Biol. 2012, 32, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Harvey, P.A.; Leinwand, L.A. Cellular mechanisms of cardiomyopathy. J. Cell Biol. 2011, 194, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.N. A decade of discoveries in cardiac biology. Nat. Med. 2004, 10, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Kajstura, J.; Cheng, W.; Reiss, K.; Clark, W.A.; Sonnenblick, E.H.; Krajewski, S.; Reed, J.C.; Olivetti, G.; Anversa, P. Apoptotic and necrotic myocyte cell deaths are independent contributing variables of infarct size in rats. Lab. Investg. 1996, 74, 86–107. [Google Scholar]
- Jeremias, I.; Kupatt, C.; Martin-Villalba, A.; Habazettl, H.; Schenkel, J.; Boekstegers, P.; Debatin, K.M. Involvement of CD95/Apo1/Fas in cell death after myocardial ischemia. Circulation 2000, 102, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Kurrelmeyer, K.M.; Michael, L.H.; Baumgarten, G.; Taffet, G.E.; Peschon, J.J.; Sivasubramanian, N.; Entman, M.L.; Mann, D.L. Endogenous tumor necrosis factor protects the adult cardiac myocyte against ischemic-induced apoptosis in a murine model of acute myocardial infarction. Proc. Nat. Acad. Sci. USA 2000, 97, 5456–5461. [Google Scholar] [CrossRef] [PubMed]
- Chiong, M.; Wang, Z.V.; Pedrozo, Z.; Cao, D.J.; Troncoso, R.; Ibacache, M.; Criollo, A.; Nemchenko, A.; Hill, J.A.; Lavandero, S. Cardiomyocyte death: Mechanisms and translational implications. Cell Death Dis. 2011, 2, e244. [Google Scholar] [CrossRef] [PubMed]
- Akyürek, Ő.; Akyürek, N.; Sayin, T.; Dinçer, I.; Berkalp, B.; Akyol, G.; Őzenci, M.; Oral, D. Association between the severity of heart failure and the susceptibility of myocytes to apoptosis in patients with idiopathic dilated cardiomyopathy. Int. J. Cardiol. 2001, 80, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Narula, J.; Pandey, P.; Arbustini, E.; Haider, N.; Narula, N.; Kolodgie, F.D.; Dal Bello, B.; Semigran, M.J.; Bielsa-Masdeu, A.; Dec, G.W.; et al. Apoptosis in heart failure: Release of cytochrome c from mitochondria and activation of caspase-3 in human cardiomyopathy. Proc. Nat. Acad. Sci. USA 1999, 96, 8144–8149. [Google Scholar] [CrossRef] [PubMed]
- Olivetti, G.; Abbi, R.; Quaini, F.; Kajstura, J.; Cheng, W.; Nitahara, J.A.; Quaini, E.; di Loreto, C.; Beltrami, C.A.; Krajewski, S.; et al. Apoptosis in the failing human heart. N. Engl. J. Med. 1997, 336, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Cirino, A.L.; Ho, C. Familial Hypertrophic Cardiomyopathy Overview; University of Washington: Seattle, WA, USA, 2011. [Google Scholar]
- Matsui, T.; Li, L.; Wu, J.C.; Cook, S.A.; Nagoshi, T.; Picard, M.H.; Liao, R.; Rosenzweig, A. Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J. Biol. Chem. 2002, 277, 22896–22901. [Google Scholar] [CrossRef] [PubMed]
- Santi Stacey, A.; Douglas Alison, C.; Lee, H. The Akt isoforms, their unique functions and potential as anticancer therapeutic targets. BioMol. Concepts 2011, 1, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Yamaji, K.; Fujimoto, S.; Ikeda, Y.; Masuda, K.; Nakamura, S.; Saito, Y.; Yutani, C. Apoptotic myocardial cell death in the setting of arrhythmogenic right ventricular cardiomyopathy. Acta Cardiol. 2005, 60, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M.; Xu, Y.; Hammond, H.L.; Willoughby, D.A.; Nathanson, L.; Rodriguez, M.M.; Vatta, M.; Lipshultz, S.E.; Lincoln, J. Myocardial alternative RNA splicing and gene expression profiling in early stage hypoplastic left heart syndrome. PLoS ONE 2012, 7, e29784. [Google Scholar] [CrossRef] [PubMed]
- Sedmera, D.; Hu, N.; Weiss, K.M.; Keller, B.B.; Denslow, S.; Thompson, R.P. Cellular changes in experimental left heart hypoplasia. Anat. Rec. 2002, 267, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.; d’Udekem, Y.; Ukoumunne, O.C.; Algar, E.M.; Newgreen, D.F.; Brizard, C.P. Differences in extra-cellular matrix and myocyte homeostasis between the neonatal right ventricle in hypoplastic left heart syndrome and truncus arteriosus. Eur. J. Cardio-Thorac. Surg. 2008, 34, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Wahbi, K.; Algalarrondo, V.; Bécane, H.M.; Fressart, V.R.; Beldjord, C.R.; Azibi, K.; Lazarus, A.; Berber, N.; Radvanyi-Hoffman, H.; Stojkovic, T.; et al. Brugada syndrome and abnormal splicing of SCN5A in myotonic dystrophy type 1. Arch. Cardiovasc. Dis. 2013, 106, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.W.; Hu, Y.W.; Ho, J.W.K.; Ikeda, S.; Polster, S.; John, R.; Hall, J.L.; Bisping, E.; Pieske, B.; dos Remedios, C.G.; et al. Heart failure-associated changes in RNA splicing of sarcomere genes. Circ. Cardiovasc. Genet. 2010, 3, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Arimura, T.; Ishikawa, T.; Nunoda, S.; Kawai, S.; Kimura, A. Dilated cardiomyopathy-associated BAG3 mutations impair Z-disc assembly and enhance sensitivity to apoptosis in cardiomyocytes. Hum. Mutat. 2011, 32, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Little, G.H.; Saw, A.; Bai, Y.; Dow, J.; Marjoram, P.; Simkhovich, B.; Leeka, J.; Kedes, L.; Kloner, R.A.; Poizat, C. Critical role of nuclear calcium/calmodulin-dependent protein kinase IIδb in cardiomyocyte survival in cardiomyopathy. J. Biol. Chem. 2009, 284, 24857–24868. [Google Scholar] [CrossRef] [PubMed]
- Vujic, A.; Robinson, E.L.; Ito, M.; Haider, S.; Ackers-Johnson, M.; See, K.; Methner, C.; Figg, N.; Brien, P.; Roderick, H.L.; et al. Experimental heart failure modelled by the cardiomyocyte-specific loss of an epigenome modifier, DNMT3B. J. Mol. Cell. Cardiol. 2015, 82, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Maatz, H.; Jens, M.; Liss, M.; Schafer, S.; Heinig, M.; Kirchner, M.; Adami, E.; Rintisch, C.; Dauksaite, V.; Radke, M.H.; et al. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J. Clin. Investig. 2014, 124, 3419–3430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannarelli, C.; Alique, M.; Rodriguez, D.T.; Yang, D.K.; Jeong, D.; Calcagno, C.; Hutter, R.; Millon, A.; Kovacic, J.C.; Weber, T.; et al. Alternatively spliced tissue factor promotes plaque angiogenesis through the activation of hypoxia-inducible factor-1α and vascular endothelial growth factor signaling. Circulation 2014, 130, 1274–1286. [Google Scholar] [CrossRef] [PubMed]
- Schoenauer, R.; Emmert, M.; Felley, A.; Ehler, E.; Brokopp, C.; Weber, B.; Nemir, M.; Faggian, G.; Pedrazzini, T.; Falk, V.; et al. Eh-myomesin splice isoform is a novel marker for dilated cardiomyopathy. Basic Res. Cardiol. 2011, 106, 233–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, P.A.W.; Greig, A.; Mark, T.M.; Malouf, N.N.; Oakeley, A.E.; Ungerleider, R.M.; Allen, P.D.; Kay, B.K. Molecular basis of human cardiac troponin t isoforms expressed in the developing, adult, and failing heart. Circ. Res. 1995, 76, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Kim, J.O.; Oh, J.G.; Hong, S.E.; Kim do, H. Pressure-overload cardiac hypertrophy is associated with distinct alternative splicing due to altered expression of splicing factors. Mol. Cells 2014, 37, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Bachinski, L.L.; Roberts, R. Genomic organization and isoform-specific tissue expression of human NAPOR (CUGBP2) as a candidate gene for familial arrhythmogenic right ventricular dysplasia. Genomics 2001, 74, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Dalal, D.; Tichnell, C.; James, C.; Tucker, A.; Abraham, T.; Spevak, P.J.; Calkins, H.; Judge, D.P. Recessive arrhythmogenic right ventricular dysplasia due to novel cryptic splice mutation in PKP2. Hum. Mutat. 2006, 27, 1157. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, T.; Ladd, A.N. The importance of CELF control: Molecular and biological roles of the CUG-BP, Elav-like family of RNA-binding proteins. Wiley Interdiscip. Rev. RNA 2012, 3, 104–121. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yang, D.; Ding, J.-H.; Wang, W.; Chu, P.-H.; Dalton, N.D.; Wang, H.-Y.; Bermingham, J.R., Jr.; Ye, Z.; Liu, F.; et al. ASF/SF2-regulated CaMKIIδ alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell 2005, 120, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Patel, N.A.; Watson, J.E.; Apostolatos, H.; Kleiman, E.; Hanson, O.; Hagiwara, M.; Cooper, D.R. Akt2 regulation of CDC2-like kinases (CLK/STY), serine/arginine-rich (SR) protein phosphorylation, and insulin-induced alternative splicing of pkcbetaii messenger ribonucleic acid. Endocrinology 2009, 150, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.A.; Kaneko, S.; Apostolatos, H.S.; Bae, S.S.; Watson, J.E.; Davidowitz, K.; Chappell, D.S.; Birnbaum, M.J.; Cheng, J.Q.; Cooper, D.R. Molecular and genetic studies imply Akt-mediated signaling promotes protein kinase cβII alternative splicing via phosphorylation of serine/arginine-rich splicing factor SRp40. J. Biol. Chem. 2005, 280, 14302–14309. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.-H.; Zhang, W.-J.; Rao, Y.; Wu, J.Y. Regulation of Ich-1 pre-mRNA alternative splicing and apoptosis by mammalian splicing factors. Proc. Nat. Acad. Sci. USA 1998, 95, 9155–9160. [Google Scholar] [CrossRef] [PubMed]
- Droin, N.; Beauchemin, M.; Solary, E.; Bertrand, R. Identification of a caspase-2 isoform that behaves as an endogenous inhibitor of the caspase cascade. Cancer Res. 2000, 60, 7039–7047. [Google Scholar] [PubMed]
- Martinet, W.; Knaapen, M.W.M.; De Meyer, G.R.Y.; Herman, A.G.; Kockx, M.M. Overexpression of the anti-apoptotic caspase-2 short isoform in macrophage-derived foam cells of human atherosclerotic plaques. Am. J. Pathol. 2003, 162, 731–736. [Google Scholar] [CrossRef]
- Holly, T.A.; Drincic, A.; Byun, Y.; Nakamura, S.; Harris, K.; Klocke, F.J.; Cryns, V.L. Caspase inhibition reduces myocyte cell death induced by myocardial ischemia and reperfusion in vivo. J. Mol. Cell. Cardiol. 1999, 31, 1709–1715. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Vanden Hoek, T.L.; Wojcik, K.; Anderson, T.; Li, C.-Q.; Shao, Z.-H.; Becker, L.B.; Hamann, K.J. Caspase-dependent cytochrome c release and cell death in chick cardiomyocytes after simulated ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H2280–H2286. [Google Scholar] [CrossRef] [PubMed]
- Heinke, M.Y.; Yao, M.; Chang, D.; Einstein, R.; dos Remedios, C.G. Apoptosis of ventricular and atrial myocytes from pacing-induced canine heart failure. Cardiovasc. Res. 2001, 49, 127–134. [Google Scholar] [CrossRef]
- Martin, L.J. Mitochondrial and cell death mechanisms in neurodegenerative diseases. Pharmaceuticals 2012, 3, 839–915. [Google Scholar] [CrossRef] [PubMed]
- Tzifi, F.; Economopoulou, C.; Gourgiotis, D.; Ardavanis, A.; Papageorgiou, S.; Scorilas, A. The role of BCL2 family of apoptosis regulator proteins in acute and chronic leukemias. Adv. Hematol. 2012, 2012, 15. [Google Scholar]
- Revil, T.; Toutant, J.; Shkreta, L.; Garneau, D.; Cloutier, P.; Chabot, B. Protein kinase C-dependent control of Bcl-x alternative splicing. Mol. Cell. Biol. 2007, 27, 8431–8441. [Google Scholar] [CrossRef] [PubMed]
- Bartling, B.; Milting, H.; Schumann, H.; Darmer, D.; Arusoglu, L.; Koerner, M.M.; El-Banayosy, A.; Koerfer, R.; Holtz, J.; Zerkowski, H.-R. Myocardial gene expression of regulators of myocyte apoptosis and myocyte calcium homeostasis during hemodynamic unloading by ventricular assist devices in patients with end-stage heart failure. Circulation 1999, 100, II216–II223. [Google Scholar] [CrossRef] [PubMed]
- Kunisada, K.; Tone, E.; Negoro, S.; Nakaoka, Y.; Oshima, Y.; Osugi, T.; Funamoto, M.; Izumi, M.; Fujio, Y.; Hirota, H.; et al. Bcl-xl reduces doxorubicin-induced myocardial damage but fails to control cardiac gene downregulation. Cardiovasc. Res. 2002, 53, 936–943. [Google Scholar] [CrossRef]
- Levanon, E.Y.; Sorek, R. The importance of alternative splicing in the drug discovery process. Targets 2003, 2, 109–114. [Google Scholar] [CrossRef]
- Pio, R.; Montuenga, L.M. Alternative splicing in lung cancer. J. Thorac. Oncol. 2009, 4, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.D.; Wang, G.; Luo, J.; Gu, Y.; Ping, P.; Chandrasekar, B. B-adrenergic receptor blockade modulates Bcl-xS expression and reduces apoptosis in failing myocardium. J. Mol. Cell. Cardiol. 2003, 35, 483–493. [Google Scholar] [CrossRef]
- Rohrbach, S.; Niemann, B.; Silber, R.E.; Holtz, J. Neuregulin receptors erbB2 and erbB4 in failing human myocardium. Basic Res. Cardiol. 2005, 100, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Montaigne, D.; Hurt, C.; Neviere, R. Mitochondria death/survival signaling pathways in cardiotoxicity induced by anthracyclines and anticancer-targeted therapies. Biochem. Res. Int. 2012, 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Hikisz, P.; Kilianska, Z. Puma, a critical mediator of cell death—One decade on from its discovery. Cell. Mol. Biol. Lett. 2012, 17, 646–669. [Google Scholar] [CrossRef] [PubMed]
- Kuribayashi, K.; Finnberg, N.K.; Jeffers, J.R.; Zambetti, G.P.; El-Deiry, W.S. The relative contribution of pro-apoptotic p53-target genes in the triggering of apoptosis following DNA damage in vitro and in vivo. Cell Cycle 2011, 10, 2380–2389. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.D.; Gallant-Behm, C.L.; Henry, R.E.; Fraikin, J.-L.; Espinosa, J.M. The p53 circuit board. Biochim. Biophys. Acta Rev. Cancer 2012, 1825, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, L. No PUMA, no death: Implications for p53-dependent apoptosis. Cancer Cell 2003, 4, 248–249. [Google Scholar] [CrossRef]
- Han, J.-W.; Flemington, C.; Houghton, A.B.; Gu, Z.; Zambetti, G.P.; Lutz, R.J.; Zhu, L.; Chittenden, T. Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc. Nat. Acad. Sci. USA 2001, 98, 11318–11323. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Tu, H.-C.; Ren, D.; Takeuchi, O.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.D.; Cheng, E.H.Y. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol. Cell 2009, 36, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Webster, K.A.; Graham, R.M.; Thompson, J.W.; Spiga, M.G.; Frazier, D.P.; Wilson, A.; Bishopric, N.H. Redox stress and the contributions of BH3-only proteins to infarction. Antioxid. Redox Signal. 2006, 8, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xing, D.; Liu, L. PUMA promotes bax translocation by both directly interacting with BAX and by competitive binding to Bcl-x l during UV-induced apoptosis. Mol. Biol. Cell. 2009, 20, 3077–3087. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, J.R.; Parganas, E.; Lee, Y.; Yang, C.; Wang, J.; Brennan, J.; MacLean, K.H.; Han, J.; Chittenden, T.; Ihle, J.N.; et al. PUMA is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 2003, 4, 321–328. [Google Scholar] [CrossRef]
- Li, Y.; Liu, X.; Rong, F. PUMA mediates the apoptotic signal of hypoxia/reoxygenation in cardiomyocytes through mitochondrial pathway. Shock 2011, 35, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Altin, S.E.; Schulze, P.C. P53-upregulated modulator of apoptosis (PUMA): A novel proapoptotic molecule in the failing heart. Circulation 2011, 124, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Toth, A.; Jeffers, J.R.; Nickson, P.; Min, J.-Y.; Morgan, J.P.; Zambetti, G.P.; Erhardt, P. Targeted deletion of Puma attenuates cardiomyocyte death and improves cardiac function during ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H52–H60. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.-Y.; Wang, N.-P.; Kerendi, F.; Halkos, M.; Kin, H.; Guyton, R.A.; Vinten-Johansen, J.; Zhao, Z.-Q. Hypoxic postconditioning reduces cardiomyocyte loss by inhibiting ros generation and intracellular Ca2+ overload. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1900–H1908. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lv, Z.; Liu, X.; Su, W.; Wang, C.; Li, N.; Song, D.; Tao, T. Hypoxic postconditioning inhibits endoplasmic reticulum stress-mediated cardiomyocyte apoptosis by targeting puma. Shock 2013, 39, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Mandl, A.; Huong Pham, L.; Toth, K.; Zambetti, G.; Erhardt, P. PUMA deletion delays cardiac dysfunction in murine heart failure models through attenuation of apoptosis. Circulation 2011, 124, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Nickson, P.; Toth, A.; Erhardt, P. PUMA is critical for neonatal cardiomyocyte apoptosis induced by endoplasmic reticulum stress. Cardiovasc. Res. 2007, 73, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.P.; Wu, G.J.; Wang, B.W.; Shyu, K.G. Regulation of puma induced by mechanical stress in rat cardiomyocytes. J. Biomed. Sci. 2012, 19, 72. [Google Scholar] [CrossRef] [PubMed]
- Yussman, M.G.; Toyokawa, T.; Odley, A.; Lynch, R.A.; Wu, G.; Colbert, M.C.; Aronow, B.J.; Lorenz, J.N.; Dorn, G.W. Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat. Med. 2002, 8, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Demaurex, N.; Distelhorst, C. Apoptosis—The calcium connection. Science 2003, 300, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Bruick, R.K. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Nat. Acad. Sci. USA 2000, 97, 9082–9087. [Google Scholar] [CrossRef] [PubMed]
- Crow, M.T. Hypoxia, BNip3 proteins, and the mitochondrial death pathway in cardiomyocytes. Circ. Res. 2002, 91, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Gang, H.; Hai, Y.; Dhingra, R.; Gordon, J.W.; Yurkova, N.; Aviv, Y.; Li, H.; Aguilar, F.; Marshall, A.; Leygue, E.; et al. A novel hypoxia-inducible spliced variant of mitochondrial death gene BNip3 promotes survival of ventricular myocytes. Circ. Res. 2011, 108, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-D.; Kuo, W.-W.; Wu, C.-H.; Lin, Y.-M.; Lin, J.A.; Lu, M.-C.; Yang, A.-L.; Liu, J.-Y.; Wang, S.-G.P.; Liu, C.-J.; et al. Effects of short- and long-term hypobaric hypoxia on Bcl2 family in rat heart. Int. J. Cardiol. 2006, 108, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Quinsay, M.N.; Thomas, R.L.; Lee, Y.; Gustafsson, Ã.B. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 2011, 6, 855–862. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Gordon, R.E.; Kohlbrenner, E.; Benard, L.; Jeong, D.; Hajjar, R.J. Potential role of Bnip3 in cardiac remodeling, myocardial stiffness, and endoplasmic reticulum: Mitochondrial calcium homeostasis in diastolic and systolic heart failure. Circ. Heart Fail. 2013, 6, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Regula, K.M.; Ens, K.; Kirshenbaum, L.A. Inducible expression of Bnip3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ. Res. 2002, 91, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Diwan, A. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J. Clin. Investg. 2007, 117, 2825–2833. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, R.; Margulets, V.; Chowdhury, S.R.; Thliveris, J.; Jassal, D.; Fernyhough, P.; Dorn, G.W.; Kirshenbaum, L.A. Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc. Nat. Acad. Sci. USA 2014, 111, E5537–E5544. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Kirshenbaum, L.A. Molecular regulation of autophagy and apoptosis during ischemic and non-ischemic cardiomyopathy. Autophagy 2008, 4, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.L.; Kubli, D.A.; Gustafsson, Ã.B. Bnip3-mediated defects in oxidative phosphorylation promote mitophagy. Autophagy 2011, 7, 775–777. [Google Scholar] [CrossRef] [PubMed]
- Chaanine, A.H.; Jeong, D.; Liang, L.; Chemaly, E.R.; Fish, K.; Gordon, R.E.; Hajjar, R.J. JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker Bnip3 in pathological hypertrophy and in heart failure. Cell Death Dis. 2012, 3, 265. [Google Scholar] [CrossRef] [PubMed]
- Gálvez, A.S.; Brunskill, E.W.; Marreez, Y.; Benner, B.J.; Regula, K.M.; Kirschenbaum, L.A.; Dorn, G.W. Distinct pathways regulate proapoptotic Nix and Bnip3 in cardiac stress. J. Biol. Chem. 2006, 281, 1442–1448. [Google Scholar] [CrossRef] [PubMed]
- Feuerstein, G.Z.; Rozanski, D. G proteins and heart failure: Is Gαq a novel target for heart failure? Circ. Res. 2000, 87, 1085–1086. [Google Scholar] [CrossRef] [PubMed]
- Diwan, A.; Matkovich, S.J.; Yuan, Q.; Zhao, W.; Yatani, A.; Brown, J.H.; Molkentin, J.D.; Kranias, E.G.; Dorn, G.W., II. Endoplasmic reticulum-mitochondria crosstalk in Nix-mediated murine cell death. J. Clin. Investig. 2009, 119, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Decker, K.F.; Zheng, D.; Matkovich, S.J.; Jia, L.; Dorn, G.W. A nucleus-targeted alternately spliced Nix/Bnip3L protein isoform modifies nuclear factor kb (NF-κB)-mediated cardiac transcription. J. Biol. Chem. 2013, 288, 15455–15465. [Google Scholar] [CrossRef] [PubMed]
- Stoilov, P.; Lin, C.-H.; Damoiseaux, R.; Nikolic, J.; Black, D.L. A high-throughput screening strategy identifies cardiotonic steroids as alternative splicing modulators. Proc. Nat. Acad. Sci. USA 2008, 105, 11218–11223. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.G.; Wood, M.J. RNA splicing: Disease and therapy. Brief Funct. Genom. 2011, 10, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Bracco, L.; Kearsey, J. The relevance of alternative RNA splicing to pharmacogenomics. Trends Biotechnol. 2003, 21, 346–353. [Google Scholar] [CrossRef]
- Pajares, M.J.; Ezponda, T.; Catena, R.; Calvo, A.; Pio, R.; Montuenga, L.M. Alternative splicing: An emerging topic in molecular and clinical oncology. Lancet Oncol. 2007, 8, 349–357. [Google Scholar] [CrossRef]
- Guo, J.; Fu, Y.-C.; Becerra, C.R. Dissecting role of regulatory factors in NF-κB pathway with siRNA. Acta Pharmacol. Sin. 2005, 26, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.H.P.; Lim, S.; Wong, W.S.F. Antisense oligonucleotides: From design to therapuetic application. Clin. Exp. Pharmacol. Physiol. 2006, 33, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Jabir, N.R.; Tabrez, S.; Ashraf, G.M.; Shakil, S.; Damanhouri, G.A.; Kamal, M.A. Nanotechnology-based approaches in anticancer research. Int. J. Nanomed. 2012, 7, 4391–4408. [Google Scholar]
- Schroeder, A.; Heller, D.A.; Winslow, M.M.; Dahlman, J.E.; Pratt, G.W.; Langer, R.; Jacks, T.; Anderson, D.G. Treating metastatic cancer with nanotechnology. Nat. Rev. Cancer 2012, 12, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Votruba, A.R.; Farokhzad, O.C.; Langer, R. Nanotechnology in drug delivery and tissue engineering: From discovery to applications. Nano Lett. 2010, 10, 3223–3230. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Pennesi, G.; Donatelli, F.; Cammarota, R.; de Flora, S.; Noonan, D.M. Cardiotoxicity of anticancer drugs: The need for cardio-oncology and cardio-oncological prevention. J. Nat. Cancer Inst. 2009, 102, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Zöller, B.; Ji, J.; Sundquist, J.; Sundquist, K. Risk of coronary heart disease in patients with cancer: A nationwide follow-up study from sweden. Eur. J. Cancer 2012, 48, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Monsuez, J.-J.; Charniot, J.-C.; Vignat, N.l.; Artigou, J.-Y. Cardiac side-effects of cancer chemotherapy. Int. J. Cardiol. 2012, 144, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Ravindra, T.; Lakshmi, N.K.; Chaitanya, K.; Surender, V.; Ahuja, Y.R. Clinical relevance of alternative splicing. Indian J. Hum. Genet. 2006, 12, 45–52. [Google Scholar] [CrossRef]
- Gellert, P.; Teranishi, M.; Jenniches, K.; de Gaspari, P.; John, D.; grosse Kreymborg, K.; Braun, T.; Uchida, S. Gene array analyzer: Alternative usage of gene arrays to study alternative splicing events. Nucleic Acids Res. 2012, 40, 2414–2425. [Google Scholar] [CrossRef] [PubMed]
- Manju, K.; Jat, R.K.; Gupta, A. A review on medicinal plants used as a source of anticancer agents. Int. J. Drug Res. 2012, 2, 177–183. [Google Scholar]
- Mishra, D.; Singh, R.K.; Srivastava, R.K. Ethno-medicinal plants used to cure different diseases by rural folks and tribes of north eastern tarai districts uttar pradesh. India. Res. J. Med. Plant 2012, 6, 286–299. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dlamini, Z.; Tshidino, S.C.; Hull, R. Abnormalities in Alternative Splicing of Apoptotic Genes and Cardiovascular Diseases. Int. J. Mol. Sci. 2015, 16, 27171-27190. https://doi.org/10.3390/ijms161126017
Dlamini Z, Tshidino SC, Hull R. Abnormalities in Alternative Splicing of Apoptotic Genes and Cardiovascular Diseases. International Journal of Molecular Sciences. 2015; 16(11):27171-27190. https://doi.org/10.3390/ijms161126017
Chicago/Turabian StyleDlamini, Zodwa, Shonisani C. Tshidino, and Rodney Hull. 2015. "Abnormalities in Alternative Splicing of Apoptotic Genes and Cardiovascular Diseases" International Journal of Molecular Sciences 16, no. 11: 27171-27190. https://doi.org/10.3390/ijms161126017
APA StyleDlamini, Z., Tshidino, S. C., & Hull, R. (2015). Abnormalities in Alternative Splicing of Apoptotic Genes and Cardiovascular Diseases. International Journal of Molecular Sciences, 16(11), 27171-27190. https://doi.org/10.3390/ijms161126017