
Repositioning of Thiourea-Containing Drugs as Tyrosinase Inhibitors

Abstract
:
1. Introduction
2. Results and Discussion
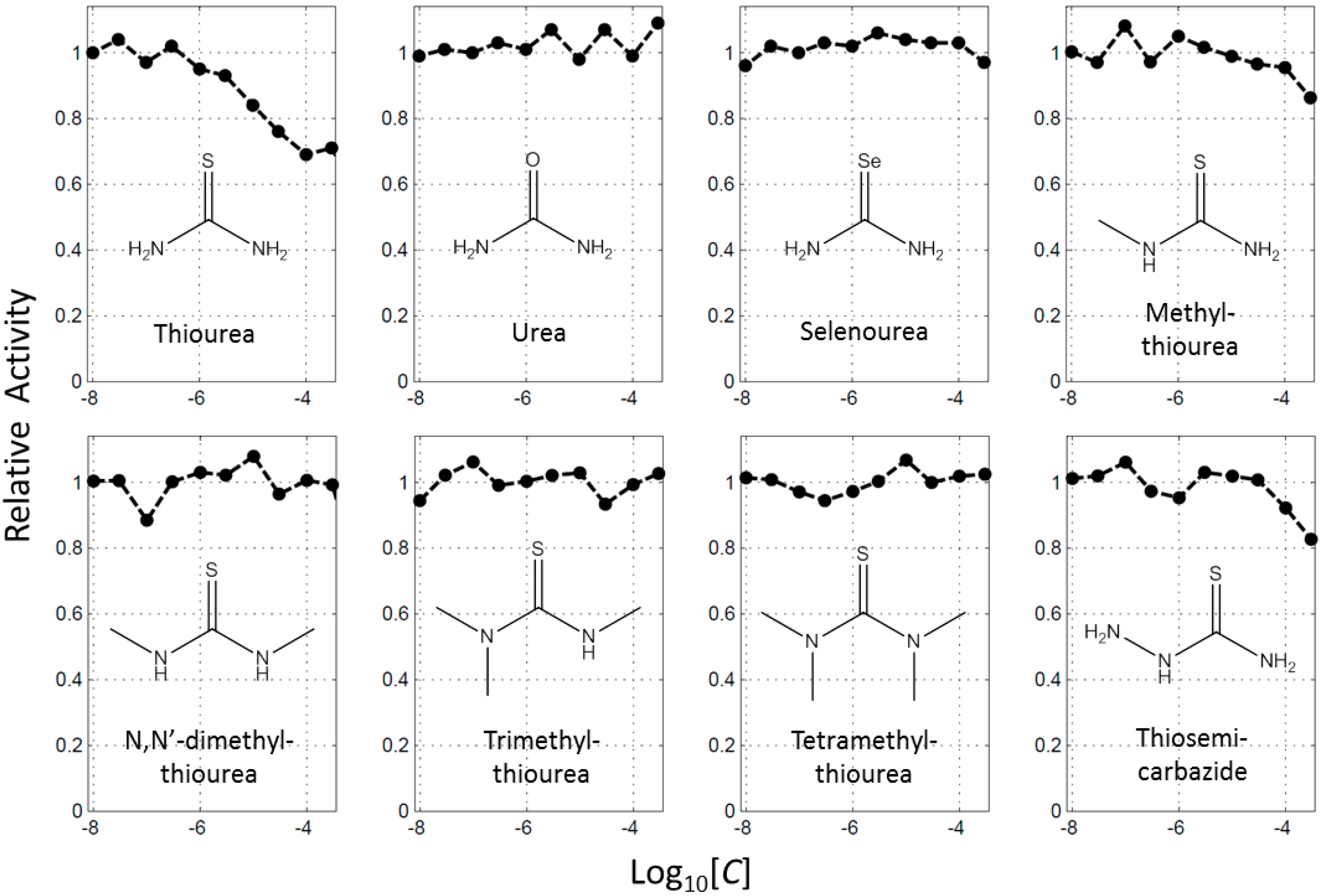
2.1. Thoiurea Itself Inhibits Tyrosinase
2.2. Thiourea-Containing Drugs Exhibit Inhibition of Tyrosinase


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID † | Chemical | 2D Structure | ZINC ID | Mw | IC50 (μM) | LE ‡ |
|---|---|---|---|---|---|---|
| 1 | Phenylthiourea (PTU) |  | ZINC03875720 | 152 | 1 | 0.84 |
| 2 | Kojic acid |  | ZINC13831818 | 142 | 29 | 0.64 |
| 3 | Thioacetazone |  | ZINC17970372 | 236 | 14 | 0.43 |
| 4 | Ambazone |  | ZINC18066619 | 237 | 15 | 0.42 |
| 5 | Methimazole |  | ZINC01187543 | 114 | 94 | 0.81 |
| 6 | Carbimazole |  | ZINC00001091 | 186 | >2000 | - |
| 7 | Thiouracil |  | ZINC05127810 | 128 | 215 | 0.64 |
| 8 | Methylthiouracil |  | ZINC05037820 | 142 | 266 | 0.56 |
| 9 | Propylthiouracil |  | ZINC04640636 | 170 | 375 | 0.44 |

2.3. Thiourea-Containing Drugs Exhibit Non-Competitive Inhibitory Kinetics
| ID | Mechanism | Ki or Kic (μM) † |
|---|---|---|
| 1 | Competitive | 0.2 |
| 2 | Competitive | 28 |
| 3 | Non-competitive | 18 |
| 4 | Non-competitive | 9 |
| 5 | Non-competitive | 73 |
| 7 | Non-competitive | 22 |
| 8 | Non-competitive | 170 |
| 9 | Non-competitive | 196 |

2.4. Ambazone Decreases Significantly Melanin Content in Mammalian Melanoma Cells
2.5. Thiourea-Containing Drugs Also Inhibit Mammalian Tyrosinase
| ID | MTT (%) | Melanin Content (%) ‡ | Cell Lysate (%) § |
|---|---|---|---|
| 20 µM | 20 µM | ||
| 1 | 113 ± 10 | 59 ± 7 | 5 ± 1 |
| 2 | 114 ± 10 | 108 ± 11 | 86 ± 8 |
| 3 | 95 ± 15 | 102 ± 10 | 74 ± 7 |
| 4 | 95 ± 12 | 80 ± 11 | 87 ± 9 |
| 5 | 100 ± 9 | 104 ± 12 | 93 ± 10 |
| 7 | 99 ± 10 | 106 ± 14 | 79 ± 9 |
| 8 | 108 ± 10 | 102 ± 14 | 78 ± 10 |
| 9 | 102 ± 12 | 102 ± 10 | 77 ± 9 |
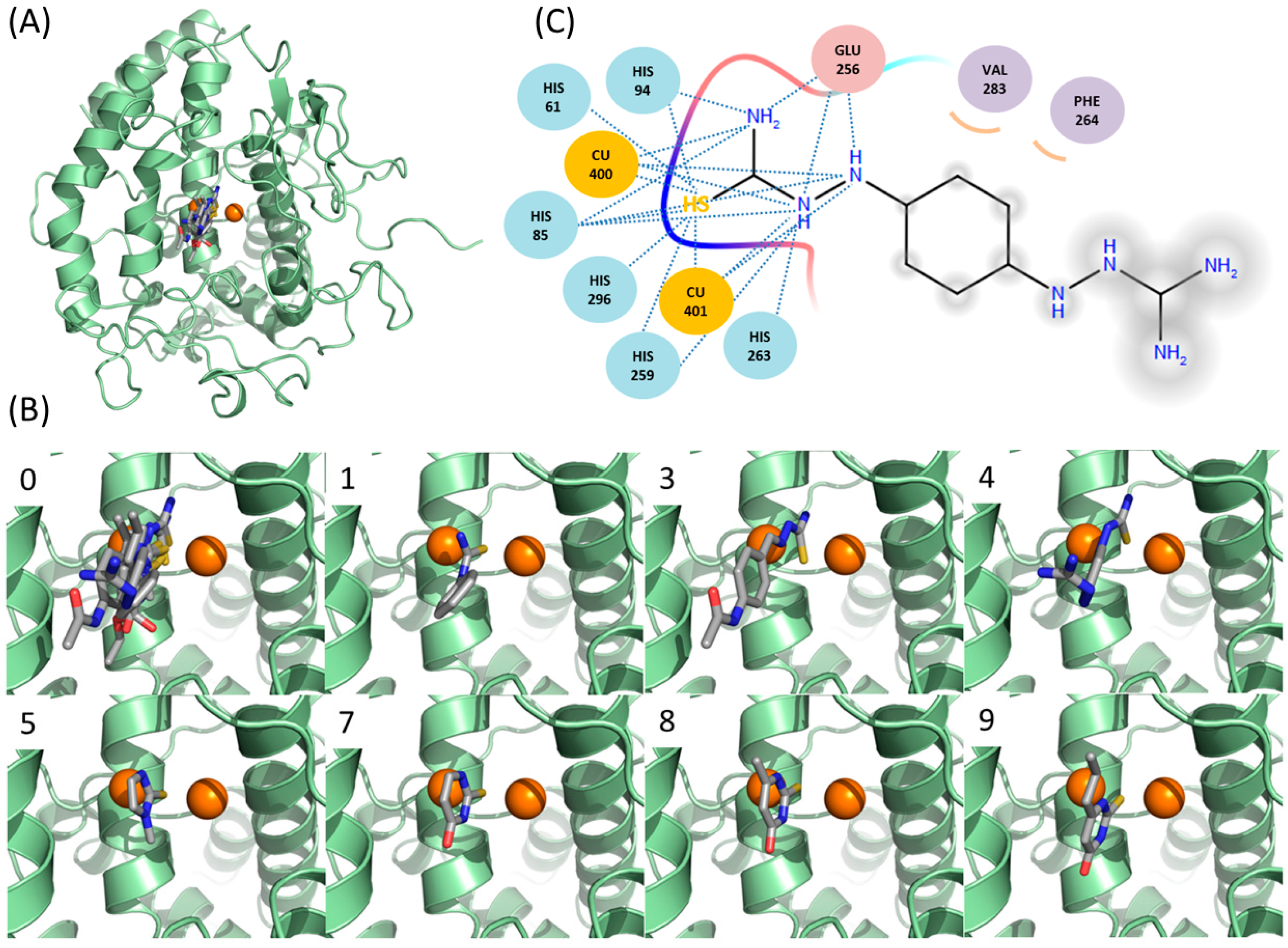
2.6. Docking Simulations Suggest that Thiourea Moieties of New Inhibitors Are Critical in Binding to Tyrosinase

2.7. Cheminformatics Identifies Antithyroid Drugs as a New Type of Inhibitors
| ID | ΣTc † | Max Tc ‡ | Closest Known Inhibitor § | IC50 (μM) || |
|---|---|---|---|---|
| 3 | 30.5 | 0.61 |  | 11 |
| 4 | 15.9 | 0.41 |  | 0.17 |
| 5 | 0.0 | 0.19 |  | 1 |
| 7 | 1.4 | 0.26 |  | 70 |
| 8 | 1.3 | 0.25 | ||
| 9 | 1.8 | 0.25 |  | 179 |

3. Materials and Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ramsden, C.A.; Riley, P.A. Tyrosinase: The four oxidation states of the active site and their relevance to enzymatic activation, oxidation and inactivation. Bioorg. Med. Chem. 2014, 22, 2388–2395. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, E.L.; Li, K.M.; Balu, N.; Saeed, M.; Devanesan, P.; Higginbotham, S.; Zhao, J.; Gross, M.L.; Rogan, E.G. Catechol ortho-quinones: The electrophilic compounds that form depurinating DNA adducts and could initiate cancer and other diseases. Carcinogenesis 2002, 23, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, M.; Miyazaki, I.; Ogawa, N. Dopamine- or L-DOPA-induced neurotoxicity: The role of dopamine quinone formation and tyrosinase in a model of Parkinson’s disease. Neurotox. Res. 2003, 5, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Li, X.; Jankovic, J. The association between Parkinson’s disease and melanoma. Int. J. Cancer 2011, 128, 2251–2260. [Google Scholar] [CrossRef] [PubMed]
- Sendoel, A.; Kohler, I.; Fellmann, C.; Lowe, S.W.; Hengartner, M.O. HIF-1 antagonizes p53-mediated apoptosis through a secreted neuronal tyrosinase. Nature 2010, 465, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S. An updated review of tyrosinase inhibitors. Int. J. Mol. Sci. 2009, 10, 2440–2475. [Google Scholar] [CrossRef] [PubMed]
- Loizzo, M.R.; Tundis, R.; Menichini, F. Natural and synthetic tyrosinase inhibitors as antibrowning agents: An update. Compr. Rev. Food Sci. Food Saf. 2012, 11, 378–398. [Google Scholar] [CrossRef]
- Khan, M.T. Novel tyrosinase inhibitors from natural resources—Their computational studies. Curr. Med. Chem. 2012, 19, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Uyama, H. Tyrosinase inhibitors from natural and synthetic sources: Structure, inhibition mechanism and perspective for the future. Cell. Mol. Life Sci. 2005, 62, 1707–1723. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.Y.; Sharma, V.K.; Sharma, N. Mushroom tyrosinase: Recent prospects. J. Agric. Food Chem. 2003, 51, 2837–2853. [Google Scholar] [CrossRef] [PubMed]
- Ubeid, A.A.; Do, S.; Nye, C.; Hantash, B.M. Potent low toxicity inhibition of human melanogenesis by novel indole-containing octapeptides. Biochim. Biophys. Acta 2012, 1820, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Solano, F.; Briganti, S.; Picardo, M.; Ghanem, G. Hypopigmenting agents: An updated review on biological, chemical and clinical aspects. Pigment Cell Res. 2006, 19, 550–571. [Google Scholar] [CrossRef] [PubMed]
- Halaouli, S.; Asther, M.; Sigoillot, J.C.; Hamdi, M.; Lomascolo, A. Fungal tyrosinases: New prospects in molecular characteristics, bioengineering and biotechnological applications. J. Appl. Microbiol. 2006, 100, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Klabunde, T.; Eicken, C.; Sacchettini, J.C.; Krebs, B. Crystal structure of a plant catechol oxidase containing a dicopper center. Nat. Struct. Biol. 1998, 5, 1084–1090. [Google Scholar] [CrossRef] [PubMed]
- Ismaya, W.T.; Rozeboom, H.J.; Weijn, A.; Mes, J.J.; Fusetti, F.; Wichers, H.J.; Dijkstra, B.W. Crystal structure of Agaricus bisporus mushroom tyrosinase: Identity of the tetramer subunits and interaction with tropolone. Biochemistry 2011, 50, 5477–5486. [Google Scholar] [CrossRef] [PubMed]
- Goldfeder, M.; Kanteev, M.; Isaschar-Ovdat, S.; Adir, N.; Fishman, A. Determination of tyrosinase substrate-binding modes reveals mechanistic differences between type-3 copper proteins. Nat. Commun. 2014, 5, 4505. [Google Scholar] [CrossRef] [PubMed]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Park, S.J.; Jee, J.G. Analogues of ethionamide, a drug used for multidrug-resistant tuberculosis, exhibit potent inhibition of tyrosinase. Eur. J. Med. Chem. 2015, 106, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.M.; Orlow, S.J. Degradation of tyrosinase induced by phenylthiourea occurs following Golgi maturation. Pigment Cell Res. 2005, 18, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Poma, A.; Bianchini, S.; Miranda, M. Inhibition of l-tyrosine-induced micronuclei production by phenylthiourea in human melanoma cells. Mutat. Res. 1999, 446, 143–148. [Google Scholar] [CrossRef]
- Du, B.K.; Erway, W.F. Studies on the mechanism of action of thiourea and related compounds; inhibition of oxidative enzymes and oxidations catalyzed by copper. J. Biol. Chem. 1946, 165, 711–721. [Google Scholar]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Falzon, D.; Hill, G.; Pal, S.N.; Suwankesawong, W.; Jaramillo, E. Pharmacovigilance and tuberculosis: Applying the lessons of thioacetazone. Bull. World Health Organ. 2014, 92, 918–919. [Google Scholar] [CrossRef] [PubMed]
- Lober, G.; Hoffmann, H. Ambazone as a membrane active antitumor drug. Biophys. Chem. 1990, 35, 287–300. [Google Scholar] [CrossRef]
- Cooper, D.S. Antithyroid drugs. N. Engl. J. Med. 1984, 311, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.F.; Lai, S.T.; Guo, Y.C.; Chen, M.J. Inhibitory effects of novel synthetic methimazole derivatives on mushroom tyrosinase and melanogenesis. Bioorg. Med. Chem. 2014, 22, 2809–2815. [Google Scholar] [CrossRef] [PubMed]
- Neeley, E.; Fritch, G.; Fuller, A.; Wolfe, J.; Wright, J.; Flurkey, W. Variations in IC50 values with purity of mushroom tyrosinase. Int. J. Mol. Sci. 2009, 10, 3811–3823. [Google Scholar] [CrossRef] [PubMed]
- Flurkey, A.; Cooksey, J.; Reddy, A.; Spoonmore, K.; Rescigno, A.; Inlow, J.; Flurkey, W.H. Enzyme, protein, carbohydrate, and phenolic contaminants in commercial tyrosinase preparations: Potential problems affecting tyrosinase activity and inhibition studies. J. Agric. Food Chem. 2008, 56, 4760–4768. [Google Scholar] [CrossRef] [PubMed]
- Andrawis, A.; Kahn, V. Effect of methimazole on the activity of mushroom tyrosinase. Biochem. J. 1986, 235, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Thanigaimalai, P.; Lee, K.C.; Bang, S.C.; Lee, J.H.; Yun, C.Y.; Roh, E.; Hwang, B.Y.; Kim, Y.; Jung, S.H. Evaluation of 3,4-dihydroquinazoline-2(1H)-thiones as inhibitors of α-MSH-induced melanin production in melanoma B16 cells. Bioorg. Med. Chem. 2010, 18, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Keseru, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Ryazanova, A.D.; Alekseev, A.A.; Slepneva, I.A. The phenylthiourea is a competitive inhibitor of the enzymatic oxidation of DOPA by phenoloxidase. J. Enzym. Inhib. Med. Chem. 2012, 27, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.X.; Kubo, I. Kinetics of mushroom tyrosinase inhibition by quercetin. J. Agric. Food Chem. 2002, 50, 4108–4112. [Google Scholar] [CrossRef] [PubMed]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Kruger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef] [PubMed]
- Kruse, L.I.; Kaiser, C.; DeWolf, W.E., Jr.; Frazee, J.S.; Garvey, E.; Hilbert, E.L.; Faulkner, W.A.; Flaim, K.E.; Sawyer, J.L.; Berkowitz, B.A. Multisubstrate inhibitors of dopamine β-hydroxylase. 1. Some 1-phenyl and 1-phenyl-bridged derivatives of imidazole-2-thione. J. Med. Chem. 1986, 29, 2465–2472. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Roy, G.; Mugesh, G. Antithyroid drug carbimazole and its analogues: Synthesis and inhibition of peroxidase-catalyzed iodination of l-tyrosine. J. Med. Chem. 2008, 51, 7313–7317. [Google Scholar] [CrossRef] [PubMed]
- Elsalini, O.A.; Rohr, K.B. Phenylthiourea disrupts thyroid function in developing zebrafish. Dev. Genes Evol. 2003, 212, 593–598. [Google Scholar] [PubMed]
- Mysinger, M.M.; Shoichet, B.K. Rapid context-dependent ligand desolvation in molecular docking. J. Chem. Inf. Model. 2010, 50, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K.; Mysinger, M.M.; Huang, N.; Colizzi, F.; Wassam, P.; Cao, Y. Automated docking screens: A feasibility study. J. Med. Chem. 2009, 52, 5712–5720. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, L.L.C. The PyMOL Molecular Graphics System; Springer: New York, NY, USA, 2010. [Google Scholar]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cao, R.; Yi, W.; Ma, C.; Wan, Y.; Zhou, B.; Ma, L.; Song, H. A class of potent tyrosinase inhibitors: Alkylidenethiosemicarbazide compounds. Eur. J. Med. Chem. 2009, 44, 1773–1778. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Dubois, C.; Yahiaoui, S.; Haudecoeur, R.; Belle, C.; Song, H.; Hardre, R.; Reglier, M.; Boumendjel, A. Refinement of arylthiosemicarbazone pharmacophore in inhibition of mushroom tyrosinase. Eur. J. Med. Chem. 2011, 46, 4330–4335. [Google Scholar] [CrossRef] [PubMed]
- Ghani, U.; Ullah, N. New potent inhibitors of tyrosinase: Novel clues to binding of 1,3,4-thiadiazole-2(3H)-thiones, 1,3,4-oxadiazole-2(3H)-thiones, 4-amino-1,2,4-triazole-5(4H)-thiones, and substituted hydrazides to the dicopper active site. Bioorg. Med. Chem. 2010, 18, 4042–4048. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Cao, R.; Yi, W.; Chen, Z.; Wen, H.; Ma, L.; Song, H. Inhibitory effects of 5-benzylidene barbiturate derivatives on mushroom tyrosinase and their antibacterial activities. Eur. J. Med. Chem. 2009, 44, 4235–4243. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J. Using ZINC to acquire a virtual screening library. Curr. Protoc. Bioinform. 2008. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. Fundamentals of Enzyme Kinetics; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Dias, A.A.; Pinto, P.A.; Fraga, I.; Bezerra, R.M.F. Diagnosis of enzyme inhibition using excel solver: A combined dry and wet laboratory exercise. J. Chem. Educ. 2014, 91, 1017–1021. [Google Scholar] [CrossRef]
- Motulsky, H.J.; Ransnas, L.A. Fitting curves to data using nonlinear regression: A practical and nonmathematical review. FASEB J. 1987, 1, 365–374. [Google Scholar] [PubMed]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Lounkine, E.; Keiser, M.J.; Whitebread, S.; Mikhailov, D.; Hamon, J.; Jenkins, J.L.; Lavan, P.; Weber, E.; Doak, A.K.; Cote, S.; et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature 2012, 486, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Setola, V.; Irwin, J.J.; Laggner, C.; Abbas, A.I.; Hufeisen, S.J.; Jensen, N.H.; Kuijer, M.B.; Matos, R.C.; Tran, T.B.; et al. Predicting new molecular targets for known drugs. Nature 2009, 462, 175–181. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.; Jee, J.-G. Repositioning of Thiourea-Containing Drugs as Tyrosinase Inhibitors. Int. J. Mol. Sci. 2015, 16, 28534-28548. https://doi.org/10.3390/ijms161226114
Choi J, Jee J-G. Repositioning of Thiourea-Containing Drugs as Tyrosinase Inhibitors. International Journal of Molecular Sciences. 2015; 16(12):28534-28548. https://doi.org/10.3390/ijms161226114
Chicago/Turabian StyleChoi, Joonhyeok, and Jun-Goo Jee. 2015. "Repositioning of Thiourea-Containing Drugs as Tyrosinase Inhibitors" International Journal of Molecular Sciences 16, no. 12: 28534-28548. https://doi.org/10.3390/ijms161226114
APA StyleChoi, J., & Jee, J. -G. (2015). Repositioning of Thiourea-Containing Drugs as Tyrosinase Inhibitors. International Journal of Molecular Sciences, 16(12), 28534-28548. https://doi.org/10.3390/ijms161226114