
ADP-Ribosylation Factor 1 Regulates Proliferation, Migration, and Fusion in Early Stage of Osteoclast Differentiation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
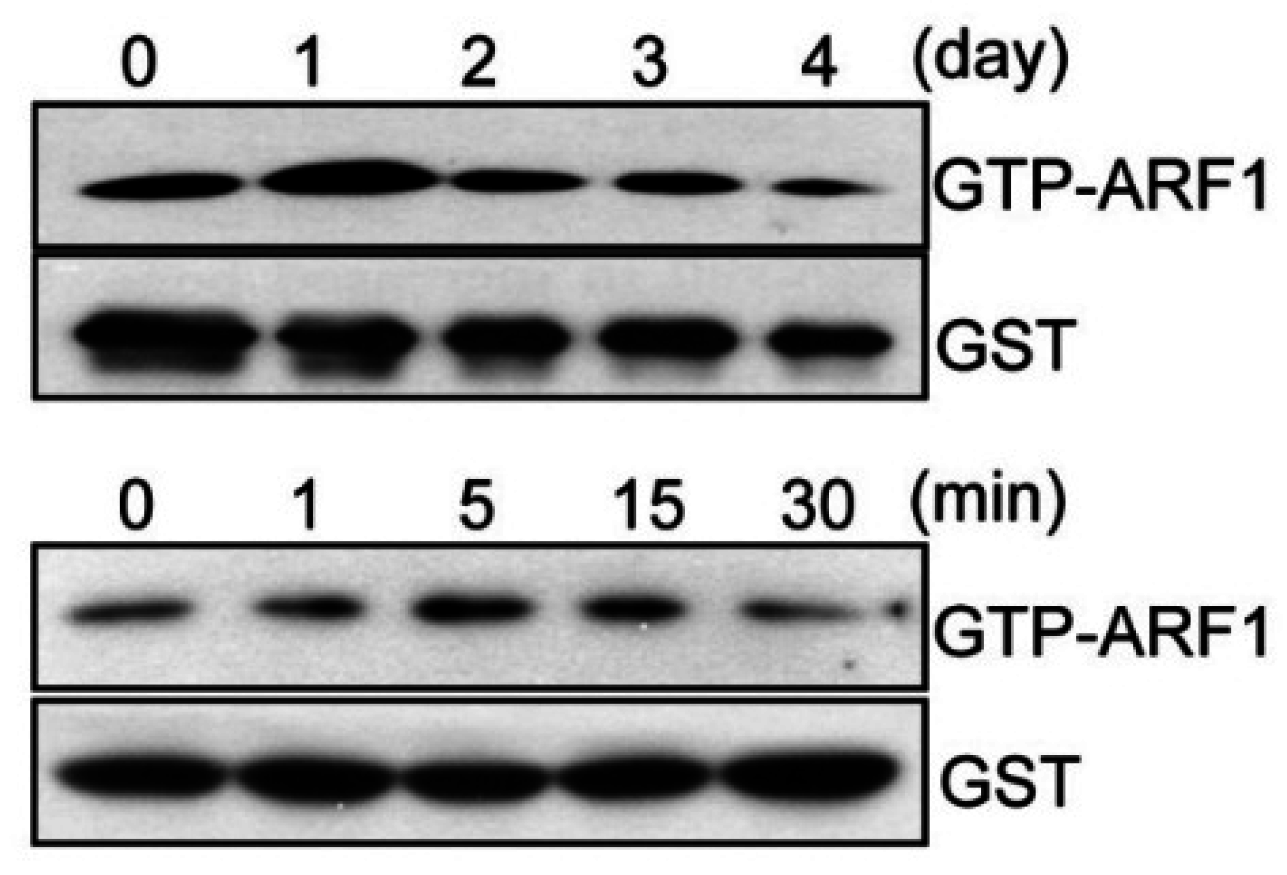
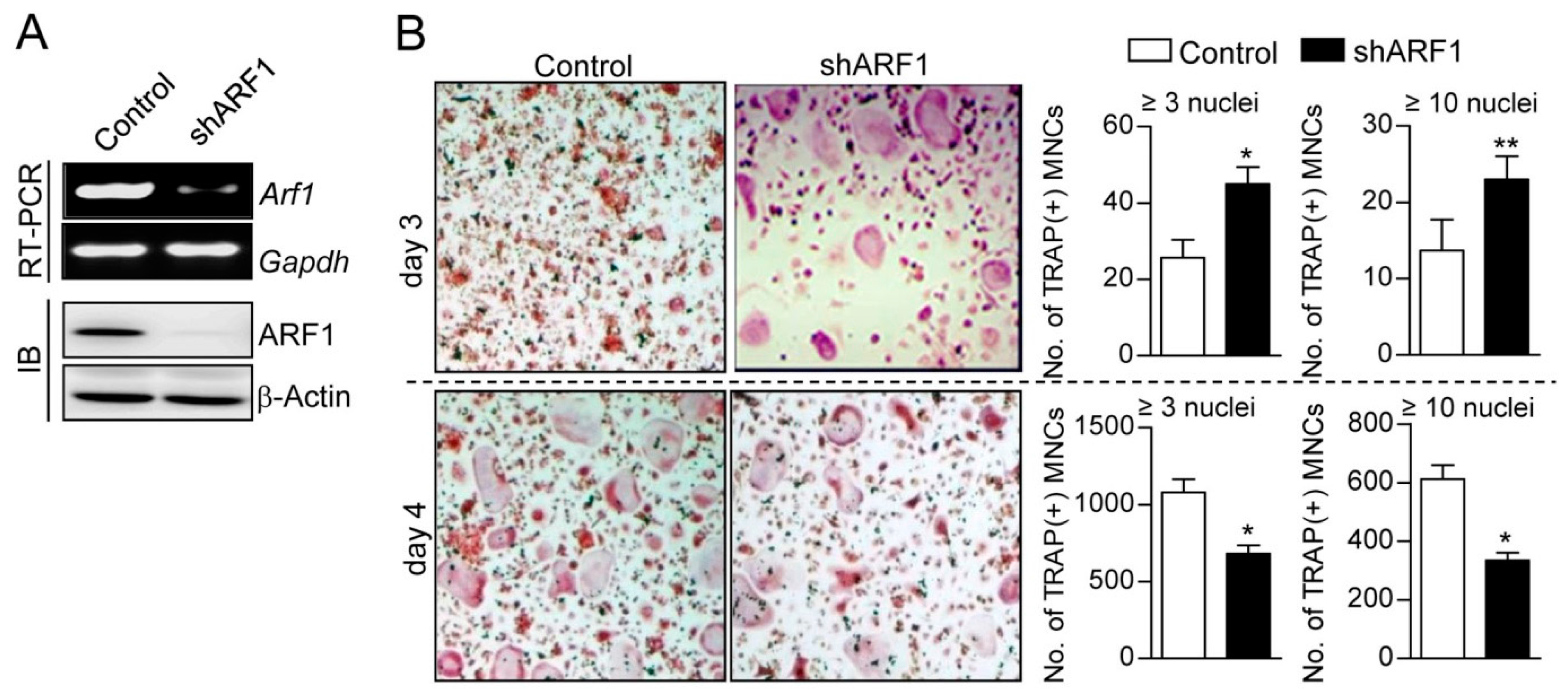
2.1. ARF1 Regulates Maturation Step of Osteoclast Differentiation


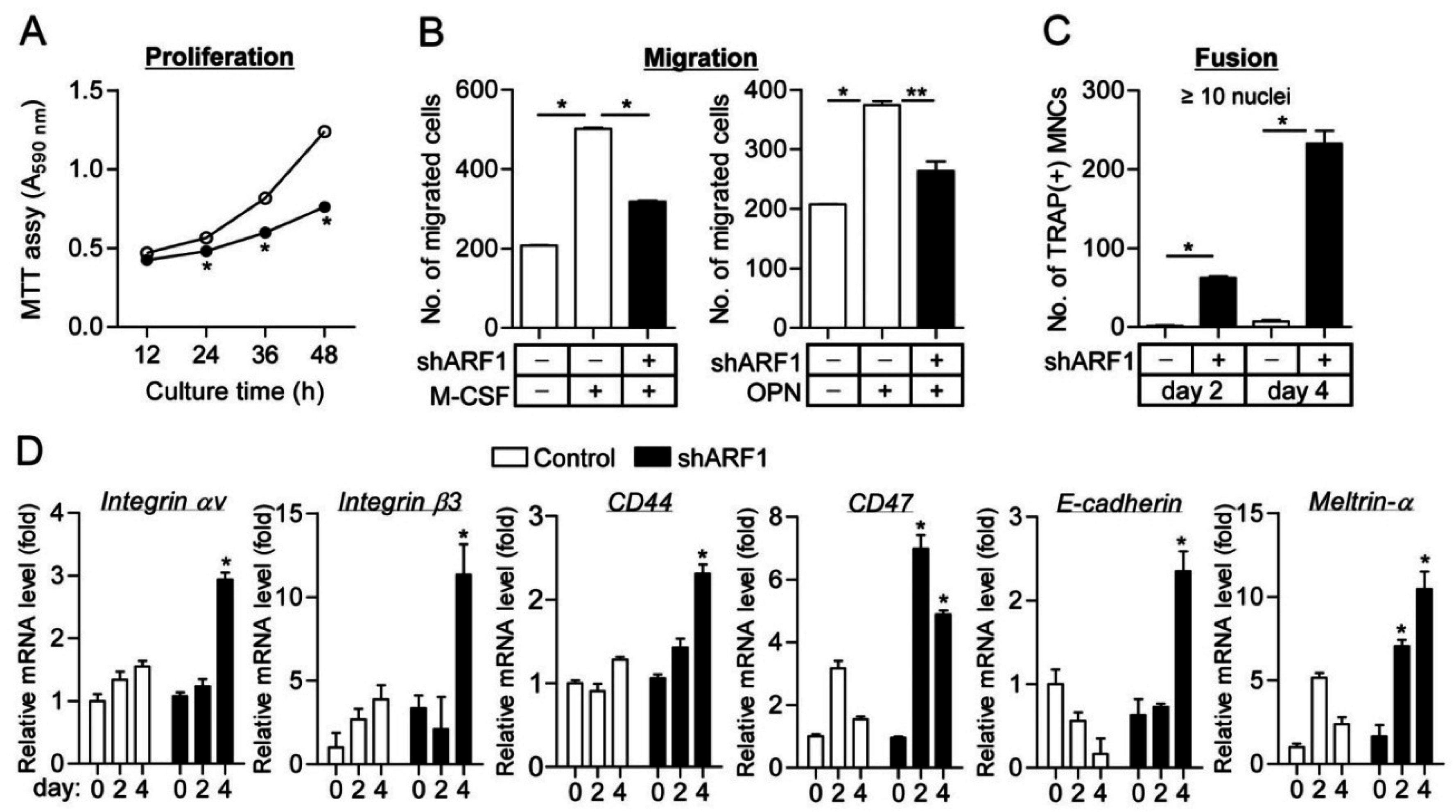
2.2. ARF1 Deficiency Suppresses Osteoclast Precursor Proliferation and Migration as Well as Potentiates Osteoclast Fusion

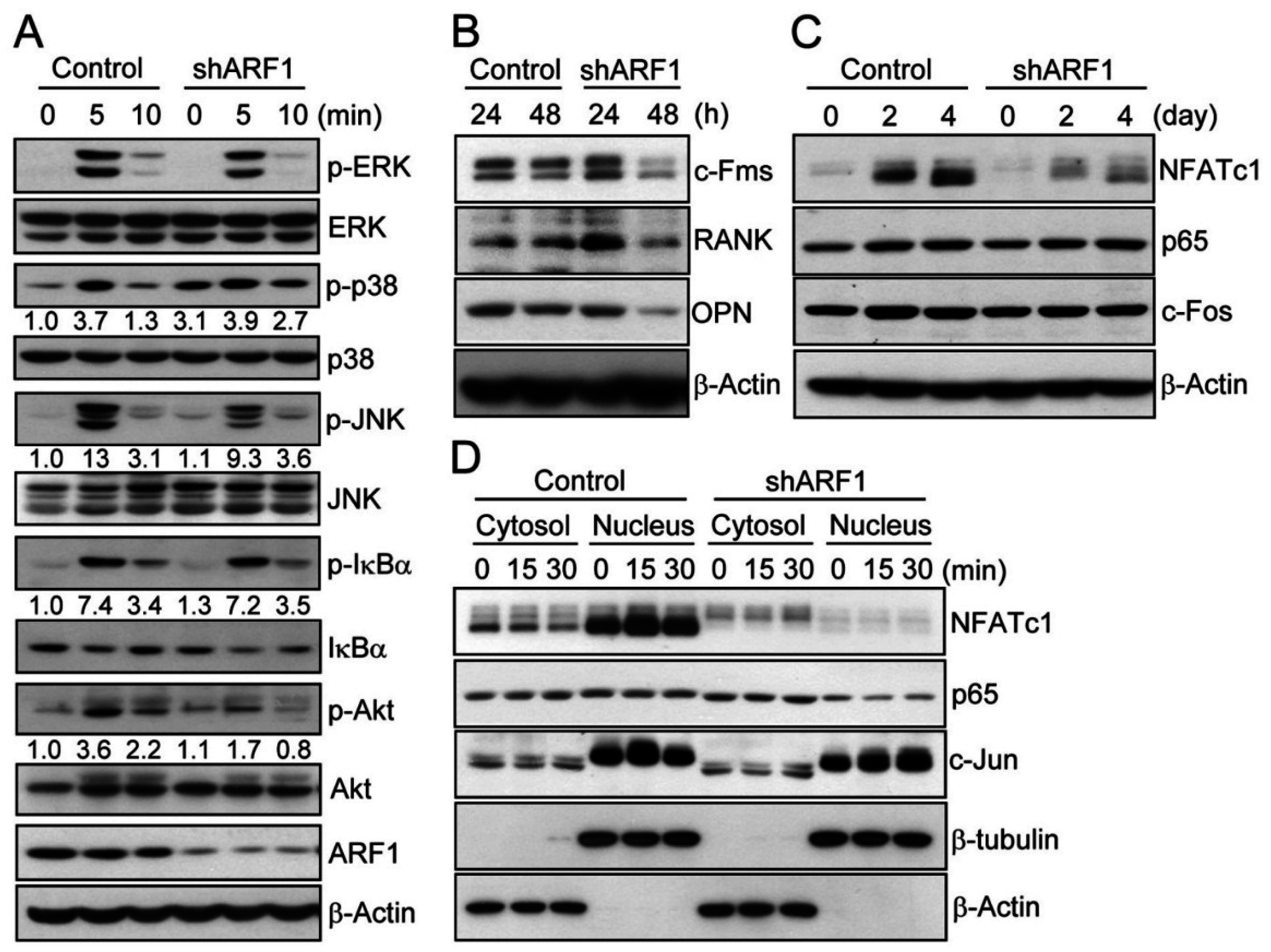
2.3. ARF1 Regulates Diverse Osteoclastogenic Signaling Pathways

3. Discussion
4. Materials and Methods
4.1. Osteoclast Precursor Preparation and Osteoclast Differentiation
4.2. Quantitative and Semi-Quantitative RT-PCR
4.3. ARF1 Activity Assay
4.4. Immunoblot Analysis
4.5. Knockdown of ARF1 by Short Hairpin RNA (shRNA)
4.6. Proliferation, Migration, and Fusion Assay
4.7. Subcellular Fractionation
4.8. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zaidi, M. Skeletal remodeling in health and disease. Nat. Med. 2007, 13, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.W.; Simon, M.C.; Anastasi, J.; Singh, H. Requirement of transcription factor pu.1 in the development of multiple hematopoietic lineages. Science 1994, 265, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- DeKoter, R.P.; Walsh, J.C.; Singh, H. Pu.1 regulates both cytokine-dependent proliferation and differentiation of granulocyte/macrophage progenitors. EMBO J. 1998, 17, 4456–4468. [Google Scholar] [CrossRef] [PubMed]
- Kelley, T.W.; Graham, M.M.; Doseff, A.I.; Pomerantz, R.W.; Lau, S.M.; Ostrowski, M.C.; Franke, T.F.; Marsh, C.B. Macrophage colony-stimulating factor promotes cell survival through akt/protein kinase b. J. Biol. Chem. 1999, 274, 26393–26398. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Kadono, Y.; Naito, A.; Matsumoto, K.; Yamamoto, T.; Tanaka, S.; Inoue, J. Segregation of traf6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J. 2001, 20, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Iotsova, V.; Caamano, J.; Loy, J.; Yang, Y.; Lewin, A.; Bravo, R. Osteopetrosis in mice lacking nf-kappaB1 and nf-kappaB2. Nat. Med. 1997, 3, 1285–1289. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F. Functions of ap1 (fos/jun) in bone development. Ann. Rheum. Dis. 2002, 61, 40–42. [Google Scholar] [CrossRef]
- Matsumoto, M.; Sudo, T.; Saito, T.; Osada, H.; Tsujimoto, M. Involvement of p38 mitogen-activated protein kinase signaling pathway in osteoclastogenesis mediated by receptor activator of nf-κB ligand (rankl). J. Biol. Chem. 2000, 275, 31155–31161. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and activation of the transcription factor nfatc1 (nfat2) integrate rankl signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef]
- Takayanagi, H. The role of nfat in osteoclast formation. Ann. N. Y. Acad. Sci. 2007, 1116, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Oursler, M.J. Recent advances in understanding the mechanisms of osteoclast precursor fusion. J. Cell. Biochem. 2010, 110, 1058–1062. [Google Scholar] [CrossRef] [PubMed]
- Bharti, A.C.; Takada, Y.; Shishodia, S.; Aggarwal, B.B. Evidence that receptor activator of nuclear factor (nf)-κB ligand can suppress cell proliferation and induce apoptosis through activation of a nf-kappab-independent and traf6-dependent mechanism. J. Biol. Chem. 2004, 279, 6065–6076. [Google Scholar] [CrossRef] [PubMed]
- Sankar, U.; Patel, K.; Rosol, T.J.; Ostrowski, M.C. Rankl coordinates cell cycle withdrawal and differentiation in osteoclasts through the cyclin-dependent kinase inhibitors p27kip1 and p21cip1. J. Bone Miner. Res. 2004, 19, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- D'Souza-Schorey, C.; Chavrier, P. Arf proteins: Roles in membrane traffic and beyond. Nat. Rev. Mol. Cell Biol. 2006, 7, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.R.; Casanova, J.E. Regulation of actin cytoskeleton dynamics by arf-family gtpases. Trends Cell Biol. 2008, 18, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, M.; Price, S.R.; Tsai, S.C.; Moss, J.; Vaughan, M. Molecular identification of adp-ribosylation factor mrnas and their expression in mammalian cells. J. Biol. Chem. 1991, 266, 2772–2777. [Google Scholar] [PubMed]
- Heckel, T.; Czupalla, C.; Expirto Santo, A.I.; Anitei, M.; Arantzazu Sanchez-Fernandez, M.; Mosch, K.; Krause, E.; Hoflack, B. Src-dependent repression of arf6 is required to maintain podosome-rich sealing zones in bone-digesting osteoclasts. Proc. Natl. Acad. Sci. USA 2009, 106, 1451–1456. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.H.; Pryce, B.A.; Tzeng, J.A.; Gonzalez, G.A.; Olson, E.N. Control of myoblast fusion by a guanine nucleotide exchange factor, loner, and its effector arf6. Cell 2003, 114, 751–762. [Google Scholar] [CrossRef]
- Lee, S.Y.; Yang, J.S.; Hong, W.; Premont, R.T.; Hsu, V.W. Arfgap1 plays a central role in coupling copi cargo sorting with vesicle formation. J. Cell Biol. 2005, 168, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Boulay, P.L.; Cotton, M.; Melancon, P.; Claing, A. Adp-ribosylation factor 1 controls the activation of the phosphatidylinositol 3-kinase pathway to regulate epidermal growth factor-dependent growth and migration of breast cancer cells. J. Biol. Chem. 2008, 283, 36425–36434. [Google Scholar] [CrossRef] [PubMed]
- Puertollano, R.; Randazzo, P.A.; Presley, J.F.; Hartnell, L.M.; Bonifacino, J.S. The ggas promote arf-dependent recruitment of clathrin to the tgn. Cell 2001, 105, 93–102. [Google Scholar] [CrossRef]
- Stearns, T.; Willingham, M.C.; Botstein, D.; Kahn, R.A. Adp-ribosylation factor is functionally and physically associated with the golgi complex. Proc. Natl. Acad. Sci. USA 1990, 87, 1238–1242. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Hirose, M.; Hashimoto, A.; Morishige, M.; Yamada, A.; Hosaka, H.; Akagi, K.; Ogawa, E.; Oneyama, C.; Agatsuma, T.; et al. Targeting amap1 and cortactin binding bearing an atypical src homology 3/proline interface for prevention of breast cancer invasion and metastasis. Proc. Natl. Acad. Sci. USA 2006, 103, 7036–7041. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.B.; Kim, J.H.; Kim, K.; Youn, B.U.; Ko, A.; Lee, S.Y.; Kim, N. Akt induces osteoclast differentiation through regulating the gsk3beta/nfatc1 signaling cascade. J. Immunol. 2012, 188, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Li, C.; Wu, G. Regulation of α2B-adrenergic receptor-mediated extracellular signal-regulated kinase 1/2 (ERK1/2) activation by adp-ribosylation factor 1. J. Biol. Chem. 2011, 286, 43361–43369. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Frankel, P.; Jackson, D.; Rotunda, T.; Boshans, R.L.; D'Souza-Schorey, C.; Foster, D.A. Elevated phospholipase d activity in h-ras- but not k-ras-transformed cells by the synergistic action of rala and arf6. Mol. Cell. Biol. 2003, 23, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Rankovic, M.; Jacob, L.; Rankovic, V.; Brandenburg, L.O.; Schroder, H.; Hollt, V.; Koch, T. Adp-ribosylation factor 6 regulates mu-opioid receptor trafficking and signaling via activation of phospholipase d2. Cell Signal. 2009, 21, 1784–1793. [Google Scholar] [CrossRef] [PubMed]
- Knizhnik, A.V.; Kovaleva, O.V.; Komelkov, A.V.; Trukhanova, L.S.; Rybko, V.A.; Zborovskaya, I.B.; Tchevkina, E.M. Arf6 promotes cell proliferation via the pld-mtorc1 and p38mapk pathways. J. Cell Biol. 2012, 113, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Mbalaviele, G.; Chen, H.; Boyce, B.F.; Mundy, G.R.; Yoneda, T. The role of cadherin in the generation of multinucleated osteoclasts from mononuclear precursors in murine marrow. J. Clin. Investig. 1995, 95, 2757–2765. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Ke, J.Z.; Zhang, Q.; Ke, H.Z.; Chalouni, C.; Vignery, A. The intracellular domain of cd44 promotes the fusion of macrophages. Blood 2006, 107, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Sterling, H.; Chen, Y.; Saginario, C.; Brown, E.J.; Frazier, W.A.; Lindberg, F.P.; Vignery, A. CD47, a ligand for the macrophage fusion receptor, participates in macrophage multinucleation. J. Biol. Chem. 2000, 275, 37984–37992. [Google Scholar] [CrossRef] [PubMed]
- Abe, E.; Mocharla, H.; Yamate, T.; Taguchi, Y.; Manolagas, S.C. Meltrin-α, a fusion protein involved in multinucleated giant cell and osteoclast formation. Calcif. Tissue Int. 1999, 64, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Helming, L.; Gordon, S. Molecular mediators of macrophage fusion. Trends Cell Biol. 2009, 19, 514–522. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.J.; Kim, H.; Lee, S.H.; Gu, D.R.; Lee, S.Y.; Lee, K.; Jeong, D. ADP-Ribosylation Factor 1 Regulates Proliferation, Migration, and Fusion in Early Stage of Osteoclast Differentiation. Int. J. Mol. Sci. 2015, 16, 29305-29314. https://doi.org/10.3390/ijms161226168
Kim MJ, Kim H, Lee SH, Gu DR, Lee SY, Lee K, Jeong D. ADP-Ribosylation Factor 1 Regulates Proliferation, Migration, and Fusion in Early Stage of Osteoclast Differentiation. International Journal of Molecular Sciences. 2015; 16(12):29305-29314. https://doi.org/10.3390/ijms161226168
Chicago/Turabian StyleKim, Min Jae, Hyunsoo Kim, Seoung Hoon Lee, Dong Ryun Gu, Soo Young Lee, Kyunghee Lee, and Daewon Jeong. 2015. "ADP-Ribosylation Factor 1 Regulates Proliferation, Migration, and Fusion in Early Stage of Osteoclast Differentiation" International Journal of Molecular Sciences 16, no. 12: 29305-29314. https://doi.org/10.3390/ijms161226168
APA StyleKim, M. J., Kim, H., Lee, S. H., Gu, D. R., Lee, S. Y., Lee, K., & Jeong, D. (2015). ADP-Ribosylation Factor 1 Regulates Proliferation, Migration, and Fusion in Early Stage of Osteoclast Differentiation. International Journal of Molecular Sciences, 16(12), 29305-29314. https://doi.org/10.3390/ijms161226168