
BRCA1 and p53 Tumor Suppressor Molecules in Alzheimer’s Disease

{kind=link}
{kind=link}
{kind=link}
Abstract
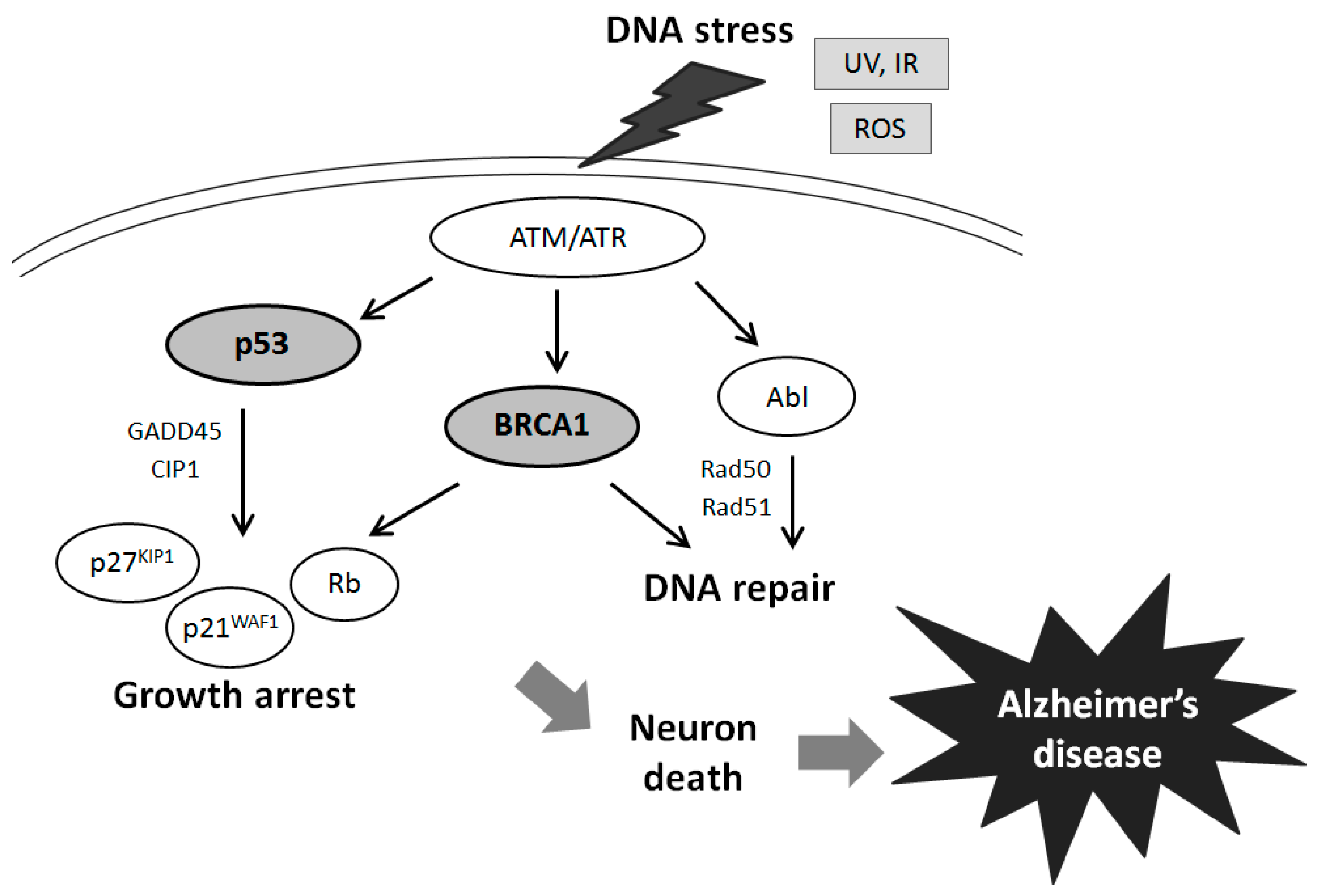
:1. Introduction
2. Relationship between AD and BRCA1 in the DNA Repair Pathway


3. Activation and Inactivation of the p53 Tumor Suppressor Involved in DNA Repair and AD

4. Some Diets Involved in Tumor Suppressor Expression May Contribute to the Neuro-Protection in AD
5. Perspective
6. Conclusions
Acknowledgments
Abbreviation
| AD | Alzheimer’s disease |
| ATM | ataxia telangiectasia-mutated |
| BRCA1 | breast cancer gene 1 |
| CDK | cyclin-dependent kinase |
| CDK2 | cyclin-dependent kinase 2 |
| DSBs | DNA double strand breaks |
| ROS | reactive oxygen species |
Conflicts of Interest
References
- Shastry, B.S. Molecular genetics of familial Alzheimer disease. Am. J. Med. Sci. 1998, 315, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Mazza, M.; Marano, G.; Traversi, G.; Bria, P.; Mazza, S. Primary cerebral blood flow deficiency and Alzheimer’s disease: Shadows and lights. J. Alzheimers Dis. 2011, 23, 375–389. [Google Scholar] [PubMed]
- Bloom, G.S. Amyloid-β and Tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Amyloid beta-induced glycogen synthase kinase 3β phosphorylated VDAC1 in Alzheimer’s disease: Implications for synaptic dysfunction and neuronal damage. Biochim. Biophys. Acta 2013, 1832, 1913–1921. [Google Scholar] [CrossRef] [PubMed]
- Herholz, K.; Ebmeier, K. Clinical amyloid imaging in Alzheimer’s disease. Lancet Neurol. 2011, 10, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Driver, J.A.; Lu, K.P. Pin1: A new genetic link between Alzheimer’s disease, cancer and aging. Curr. Aging Sci. 2010, 3, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic. Biol. Med. 2007, 43, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Heijink, A.M.; Krajewska, M.; van Vugt, M.A. The DNA damage response during mitosis. Mutat. Res. 2013, 750, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, W.K. Initiating the uninitiated: Replication of damaged DNA and carcinogenesis. Cell Cycle 2007, 6, 1460–1467. [Google Scholar] [CrossRef] [PubMed]
- Neganova, I.; Vilella, F.; Atkinson, S.P.; Lloret, M.; Passos, J.F.; von Zglinicki, T.; O’Connor, J.E.; Burks, D.; Jones, R.; Armstrong, L.; et al. An important role for CDK2 in G1 to S checkpoint activation and DNA damage response in human embryonic stem cells. Stem Cells 2011, 29, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F.; Concannon, P.; Gatti, R.A. Eighth International Workshop on Ataxia-Telangiectasia (ATW8). Cancer Res. 1999, 59, 3845–3849. [Google Scholar] [PubMed]
- Zinn, P.O.; Sathyan, P.; Mahajan, B.; Bruyere, J.; Hegi, M.; Majumder, S.; Colen, R.R. A novel volume-age-KPS (VAK) glioblastoma classification identifies a prognostic cognate microRNA-gene signature. PLoS One 2012, 7, e41522. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Mehmi, I.; Atlas, E.; Colomer, R.; Lupu, R. Novel signaling molecules implicated in tumor-associated fatty acid synthase-dependent breast cancer cell proliferation and survival: Role of exogenous dietary fatty acids, p53-p21WAF1/CIP1, ERK1/2 MAPK, p27KIP1, BRCA1, and NF-κB. Int. J. Oncol. 2004, 24, 591–608. [Google Scholar] [PubMed]
- Pavard, S.; Metcalf, C.J. Negative selection on BRCA1 susceptibility alleles sheds light on the population genetics of late-onset diseases and aging theory. PLoS One 2007, 2, e1206. [Google Scholar] [CrossRef] [PubMed]
- Katsel, P.; Tan, W.; Fam, P.; Purohit, D.P.; Haroutunian, V. Correction: Cell cycle checkpoint abnormalities during dementia: A plausible association with the loss of protection against oxidative stress in Alzheimer’s disease. PLoS One 2013, 8, e68361. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Bohr, V.A.; Stevnsner, T. DNA repair deficiency in neurodegeneration. Prog. Neurobiol. 2011, 94, 166–200. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.B. Niacin requirements for genomic stability. Mutat. Res. 2012, 733, 14–20. [Google Scholar] [CrossRef] [PubMed]
- House, N.C.; Koch, M.R.; Freudenreich, C.H. Chromatin modifications and DNA repair: Beyond double-strand breaks. Front. Genet. 2014, 5, 296. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Hemström, T.H.; Joseph, B.; Schulte, G.; Lewensohn, R.; Zhivotovsky, B. PKC 412 sensitizes U1810 non-small cell lung cancer cells to DNA damage. Exp. Cell Res. 2005, 305, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Jia, H.; Li, L.; Zhang, G.; Zhao, M.; Cheng, Q.; Zheng, J.; Li, D. Inhibition of CK2 enhances UV-triggered apoptotic cell death in lung cancer cell lines. Oncol. Rep. 2013, 30, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Cantor, S.B.; Guillemette, S. Hereditary breast cancer and the BRCA1-associated FANCJ/BACH1/BRIP1. Future Oncol. 2011, 7, 253–261. [Google Scholar] [CrossRef]
- Konishi, H.; Mohseni, M.; Tamaki, A.; Garay, J.P.; Croessmann, S.; Karnan, S.; Ota, A.; Wong, H.Y.; Konishi, Y.; Karakas, B; et al. Mutation of a single allele of the cancer susceptibility gene BRCA1 leads to genomic instability in human breast epithelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17773–17778. [Google Scholar] [CrossRef]
- Salmena, L.; Narod, S. BRCA1 haploinsufficiency: Consequences for breast cancer. Womens Health 2012, 8, 127–129. [Google Scholar]
- Chai, Y.; Salmena, G.; Cui, J.; Liao, B.; Liu, S.; Aysola, K.; Yezdani, M.; Reddy, E.S.; Rao, V.N. c-Fos oncogene regulator Elk-1 interacts with BRCA1 splice variants BRCA1a/1b and enhances BRCA1a/1b-mediated growth suppression in breast cancer cells. Oncogene 2001, 20, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Le Page, F.; Randrianarison, V.; Marot, D.; Cabannes, J.; Perricaudet, M.; Feunteun, J.; Sarasin, A. BRCA1 and BRCA2 are necessary for the transcription-coupled repair of the oxidative 8-oxoguanine lesion in human cells. Cancer Res. 2000, 60, 5548–5552. [Google Scholar] [PubMed]
- Helmer, R.A.; Martínez-Zaguilán, R.; Dertien, J.S.; Fulford, C.; Foreman, O.; Peiris, V.; Chilton, B.S. Helicase-like transcription factor (Hltf) regulates G2/M transition, Wt1/Gata4/Hif-1a cardiac transcription networks, and collagen biogenesis. PLoS One 2013, 8, e80461. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Shukla, P.C.; Quan, A.; Al-Omran, M.; Lovren, F.; Pan, Y.; Brezden-Masley, C.; Ingram, A.J.; Stanford, W.L.; Teoh, H.; et al. BRCA1 is a novel target to improve endothelial dysfunction and retard atherosclerosis. J. Thorac. Cardiovasc. Surg. 2013, 146, 949–960.e4. [Google Scholar] [CrossRef] [PubMed]
- Bradley-Whitman, M.A.; Timmons, M.D.; Beckett, T.L.; Murphy, M.P.; Lynn, B.C.; Lovell, M.A. Nucleic acid oxidation: An early feature of Alzheimer’s disease. J. Neurochem. 2014, 128, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Falik-Zaccai, T.C.; Keren, Z.; Slor, H. The versatile DNA nucleotide excision repair (NER) and its medical significance. Pediatr. Endocrinol. Rev. 2009, 7, 37–42. [Google Scholar] [PubMed]
- Keimpema, E.; Tortoriello, G.; Alpár, A.; Capsoni, S.; Arisi, I.; Calvigioni, D.; Hu, S.S.; Cattaneo, A.; Doherty, P.; Mackie, K.; et al. Nerve growth factor scales endocannabinoid signaling by regulating monoacylglycerol lipase turnover in developing cholinergic neurons. Proc. Natl. Acad. Sci. USA 2013, 110, 1935–1940. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.N.; August, A.; Hanafusa, H. Evidence for a transcriptional activation function of BRCA1 C-terminal region. Proc. Natl. Acad. Sci. USA 1996, 93, 13595–13599. [Google Scholar] [CrossRef] [PubMed]
- Ayi, T.C.; Tsan, J.T.; Hwang, L.Y.; Bowcock, A.M.; Baer, R. Conservation of function and primary structure in the BRCA1-associated RING domain (BARD1) protein. Oncogene 1998, 17, 2143–2148. [Google Scholar] [CrossRef] [PubMed]
- Reedy, M.B.; Hang, T.; Gallion, H.; Arnold, S.; Smith, S.A. Antisense inhibition of BRCA1 expression and molecular analysis of hereditary tumors indicate that functional inactivation of the p53 DNA damage response pathway is required for BRCA-associated tumorigenesis. Gynecol. Oncol. 2001, 81, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Jhanwar-Uniyal, M. BRCA1 in cancer, cell cycle and genomic stability. Front. Biosci. 2003, 8, s1107–s1117. [Google Scholar] [CrossRef] [PubMed]
- Cousineau, I.; Abaji, C.; Belmaaza, A. BRCA1 regulates RAD51 function in response to DNA damage and suppresses spontaneous sister chromatid replication slippage: Implications for sister chromatid cohesion, genome stability, and carcinogenesis. Cancer Res. 2005, 65, 11384–11391. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Kwon, Y.A.; Lee, Y.; Kim, H.; Yun, J.H.; Kim, S.; Kim, D.K. G1/S cell cycle checkpoint defect in lymphocytes from patients with Alzheimer’s disease. Psychiatry Investig. 2012, 9, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, E.; Beach, T.; Shen, Y.; Li, R.; Chang, Y. Deficiency of the Mre11 DNA repair complex in Alzheimer’s disease brains. Brain Res. Mol. Brain Res. 2004, 128, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Cao, L.; Li, C.; Xu, X.; Huber, L.J.; Chodosh, L.A.; Deng, C.X. Uterus hyperplasia and increased carcinogen-induced tumorigenesis in mice carrying a targeted mutation of the Chk2 phosphorylation site in Brca1. Mol. Cell Biol. 2004, 24, 9498–9507. [Google Scholar] [CrossRef] [PubMed]
- Coene, E.D.; Hollinshead, M.S.; Waeytens, A.A.; Schelfhout, V.R.; Eechaute, W.P.; Shaw, M.K.; van Oostveldt, P.M.; Vaux, D.J. Phosphorylated BRCA1 is predominantly located in the nucleus and mitochondria. Mol. Biol. Cell 2005, 16, 997–1010. [Google Scholar] [CrossRef] [PubMed]
- Rolyan, H.; Scheffold, A.; Heinrich, A.; Begus-Nahrmann, Y.; Langkopf, B.H.; Hölter, S.M.; Vogt-Weisenhorn, D.M.; Liss, B.; Wurst, W.; Lie, D.C.; et al. Telomere shortening reduces Alzheimer’s disease amyloid pathology in mice. Brain 2011, 134, 2044–2056. [Google Scholar] [CrossRef] [PubMed]
- Lanni, C.; Racchi, M.; Memo, M.; Govoni, S.; Uberti, D. p53 at the crossroads between cancer and neurodegeneration. Free Radic. Biol. Med. 2012, 52, 1727–1733. [Google Scholar] [CrossRef] [PubMed]
- Behrens, M.I.; Lendon, C.; Roe, C.M. A common biological mechanism in cancer and Alzheimer’s disease? Curr. Alzheimer Res. 2009, 6, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.L.; Sheu, L.F.; Yu, J.C.; Yang, T.L.; Chen, B.F.; Leu, F.J.; Shen, C.Y. Abnormality of the DNA double-strand-break checkpoint/repair genes, ATM, BRCA1 and TP53, in breast cancer is related to tumour grade. Br. J. Cancer 2004, 90, 1995–2001. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L. P21(WAF1/CIP1) may be a tumor suppressor after all. Cancer Biol. Ther. 2007, 6, 1171–1172. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Alquézar, C.; Bermejo-Pareja, F.; Bialopiotrowicz, E.; Wojda, U.; Martín-Requero, A. Downregulation of extracellular signal-regulated kinase 1/2 activity by calmodulin KII modulates p21Cip1 levels and survival of immortalized lymphocytes from Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 1090–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bialopiotrowicz, E.; Szybinska, A.; Kuzniewska, B.; Buizza, L.; Uberti, D.; Kuznicki, J.; Wojda, U. Highly pathogenic Alzheimer’s disease presenilin 1 P117R mutation causes a specific increase in p53 and p21 protein levels and cell cycle dysregulation in human lymphocytes. J. Alzheimers Dis. 2012, 32, 397–415. [Google Scholar] [PubMed]
- Muñoz-Fontela, C.; Macip, S.; Martínez-Sobrido, L.; Brown, L.; Ashour, J.; García-Sastre, A.; Lee, S.W.; Aaronson, S.A. Transcriptional role of p53 in interferon-mediated antiviral immunity. J. Exp. Med. 2008, 205, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Gehrau, R.C.; D’Astolfo, D.S.; Andreoli, V.; Bocco, J.L.; Koritschoner, N.P. Differential expression of the klf6 tumor suppressor gene upon cell damaging treatments in cancer cells. Mutat. Res. 2011, 707, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.; Hudson, J.W. p53-Dependent and cell specific epigenetic regulation of the polo-like kinases under oxidative stress. PLoS One 2014, 9, e87918. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Perez, V.A.; Strasberg-Rieber, M.; Rieber, M. Hypoxia and hypoxia mimetic cooperate to counteract tumor cell resistance to glucose starvation preferentially in tumor cells with mutant p53. Biochem. Biophys. Res. Commun. 2014, 443, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Wiech, M.; Olszewski, M.B.; Tracz-Gaszewska, Z.; Wawrzynow, B.; Zylicz, M.; Zylicz, A. Molecular mechanism of mutant p53 stabilization: the role of HSP70 and MDM2. PLoS One 2012, 7, e51426. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Achatz, M.I.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Pant, V.; Lozano, G. Limiting the power of p53 through the ubiquitin proteasome pathway. Genes Dev. 2014, 28, 1739–1751. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Hainaut, P. Massively regulated genes: The example of TP53. J. Pathol. 2010, 220, 164–173. [Google Scholar] [PubMed]
- Chalabi, N.; le Corre, L.; Maurizis, J.C.; Bignon, Y.J.; Bernard-Gallon, D.J. The effects of lycopene on the proliferation of human breast cells and BRCA1 and BRCA2 gene expression. Eur. J. Cancer 2004, 40, 1768–1775. [Google Scholar] [CrossRef] [PubMed]
- Chalabi, N.; Maurizis, J.C.; le Corre, L.; Delort, L.; Bignon, Y.J.; Bernard-Gallon, D.J. Quantification by affinity perfusion chromatography of phosphorylated BRCAl and BRCA2 proteins from tumor cells after lycopene treatment. J. Chromatogr. B 2005, 821, 188–193. [Google Scholar] [CrossRef]
- Lebda, M.A.; El-Neweshy, M.S.; El-Sayed, Y.S. Neurohepatic toxicity of subacute manganese chloride exposure and potential chemoprotective effects of lycopene. Neurotoxicology 2012, 33, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Bosviel, R.; Dumollard, E.; Déchelotte, P.; Bignon, Y.J.; Bernard-Gallon, D. Can soy phytoestrogens decrease DNA methylation in BRCA1 and BRCA2 oncosuppressor genes in breast cancer? OMICS 2012, 16, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Vissac-Sabatier, C.; Coxam, V.; Déchelotte, P.; Picherit, C.; Horcajada, M.N.; Davicco, M.J.; Lebecque, P.; Bignon, Y.J.; Bernard-Gallon, D. Phytoestrogen-rich diets modulate expression of Brca1 and Brca2 tumor suppressor genes in mammary glands of female Wistar rats. Cancer Res. 2003, 63, 6607–6612. [Google Scholar] [PubMed]
- Cabanes, A.; Wang, M.; Olivo, S.; DeAssis, S.; Gustafsson, J.A.; Khan, G.; Hilakivi-Clarke, L. Prepubertal estradiol and genistein exposures up-regulate BRCA1 mRNA and reduce mammary tumorigenesis. Carcinogenesis 2004, 25, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Meng, Q.; Auborn, K.; Carter, T.; Rosen, E.M. BRCA1 and BRCA2 as molecular targets for phytochemicals indole-3-carbinol and genistein in breast and prostate cancer cells. Br. J. Cancer 2006, 94, 407–426. [Google Scholar] [CrossRef] [PubMed]
- Soni, M.; Rahardjo, T.B.; Soekardi, R.; Sulistyowati, Y.; Lestariningsih; Yesufu-Udechuku, A.; Irsan, A.; Hogervorst, E. Phytoestrogens and cognitive function: A review. Maturitas 2014, 77, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.C.; Wu, J.M. Targeting CWR22Rv1 prostate cancer cell proliferation and gene expression by combinations of the phytochemicals EGCG, genistein and quercetin. Anticancer Res. 2009, 29, 4025–4032. [Google Scholar] [PubMed]
- Adjakly, M.; Bosviel, R.; Rabiau, N.; Boiteux, J.P.; Bignon, Y.J.; Guy, L.; Bernard-Gallon, D. DNA methylation and soy phytoestrogens: Quantitative study in DU-145 and PC-3 human prostate cancer cell lines. Epigenomics 2011, 3, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Dagdemir, A.; Durif, J.; Ngollo, M.; Bignon, Y.J.; Bernard-Gallon, D. Histone lysine trimethylation or acetylation can be modulated by phytoestrogen, estrogen or anti-HDAC in breast cancer cell lines. Epigenomics 2013, 5, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Roccisano, D.; Henneberg, M.; Saniotis, A. A possible cause of Alzheimer’s dementia—Industrial soy foods. Med. Hypotheses 2014, 82, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.X.; Li, Y.; Sun, C.; Jiang, D.; Lin, Y.J.; Jin, F.X.; Lee, S.K.; Jin, Y.H. p53-dependent Fas expression is critical for Ginsenoside Rh2 triggered caspase-8 activation in HeLa cells. Protein Cell 2014, 5, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, J.; Xing, Y.; Gong, L.; Li, H.; Wu, Z.; Li, Y.; Wang, J.; Wang, Y.; Dong, L.; Li, S. Effects of ginsenoside Rg1 or 17β-estradiol on a cognitively impaired, ovariectomized rat model of Alzheimer’s disease. Neuroscience 2012, 220, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yan, X.; Li, L.; Zhu, Y.; Qin, K.; Zhou, L.; Sun, D.; Zhang, X.; Ye, R.; Zhao, G. Ginsennoside rd attenuates cognitive dysfunction in a rat model of Alzheimer’s disease. Neurochem. Res. 2012, 37, 2738–2747. [Google Scholar] [CrossRef] [PubMed]
- Gali-Muhtasib, H.; Diab-Assaf, M.; Boltze, C.; Al-Hmaira, J.; Hartig, R.; Roessner, A.; Schneider-Stock, R. Thymoquinone extracted from black seed triggers apoptotic cell death in human colorectal cancer cells via a p53-dependent mechanism. Int. J. Oncol. 2004, 25, 857–866. [Google Scholar] [PubMed]
- Alhebshi, A.H.; Gotoh, M.; Suzuki, I. Thymoquinone protects cultured rat primary neurons against amyloid β-induced neurotoxicity. Biochem. Biophys. Res. Commun. 2013, 433, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Vaibhav, K.; Javed, H.; Khan, M.M.; Tabassum, R.; Ahmed, M.E.; Srivastava, P.; Khuwaja, G.; Islam, F.; Siddiqui, M.S.; et al. Attenuation of Aβ-induced neurotoxicity by thymoquinone via inhibition of mitochondrial dysfunction and oxidative stress. Mol. Cell. Biochem. 2012, 369, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, H.M.; Cho, Y.H.; Park, K.; Kim, E.J.; Jung, K.H.; Kim, C.H.; Kim, W.J.; Moon, S.K. Aqueous extract of Magnolia officinalis mediates proliferative capacity, p21WAF1 expression and TNF-α-induced NF-κB activity in human urinary bladder cancer 5637 cells; involvement of p38 MAP kinase. Oncol. Rep. 2007, 18, 729–736. [Google Scholar] [PubMed]
- Lee, Y.J.; Choi, D.Y.; Han, S.B.; Kim, Y.H.; Kim, K.H.; Hwang, B.Y.; Kang, J.K.; Lee, B.J.; Oh, K.W.; Hong, J.T. Inhibitory effect of ethanol extract of Magnolia officinalis on memory impairment and amyloidogenesis in a transgenic mouse model of Alzheimer’s disease via regulating β-secretase activity. Phytother. Res. 2012, 26, 1884–1892. [Google Scholar] [CrossRef] [PubMed]
- Hoi, C.P.; Ho, Y.P.; Baum, L.; Chow, A.H. Neuroprotective effect of honokiol and magnolol, compounds from Magnolia officinalis, on beta-amyloid-induced toxicity in PC12 cells. Phytother. Res. 2010, 24, 1538–1542. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.H.; Wen, J.K.; Miao, S.B.; Jia, Z.; Hu, H.J.; Sun, R.H.; Wu, Y.; Han, M. Baicalin inhibits PDGF-BB-stimulated vascular smooth muscle cell proliferation through suppressing PDGFRβ-ERK signaling and increase in p27 accumulation and prevents injury-induced neointimal hyperplasia. Cell Res. 2010, 20, 1252–1262. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, X.; Gao, P.; Tu, Y.; Zhao, M.; Li, J.; Zhang, S.; Liang, H. Baicalin attenuates Alzheimer-like pathological changes and memory deficits induced by amyloid β1–42 protein. Metab. Brain Dis. 2014. [Google Scholar] [CrossRef]
- Lee, S.J.; Park, K.; Ha, S.D.; Kim, W.J.; Moon, S.K. Gleditsia sinensis thorn extract inhibits human colon cancer cells: The role of ERK1/2, G2/M-phase cell cycle arrest and p53 expression. Phytother. Res. 2010, 24, 1870–1876. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Ryu, D.H.; Jang, L.C.; Cho, S.C.; Kim, W.J.; Moon, S.K. Suppressive effects of an ethanol extract of Gleditsia sinensis thorns on human SNU-5 gastric cancer cells. Oncol. Rep. 2013, 29, 1609–1616. [Google Scholar] [PubMed]
- Lu, Y.; Li, C.S.; Dong, Q. Chinese herb related molecules of cancer-cell-apoptosis: A minireview of progress between Kanglaite injection and related genes. J. Exp. Clin. Cancer Res. 2008, 27, 31. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Hung, C.M.; Tsai, J.C.; Lee, J.C.; Chen, Y.L.; Wei, C.W.; Kao, J.Y.; Way, T.D. Hispidulin potently inhibits human glioblastoma multiforme cells through activation of AMP-activated protein kinase (AMPK). J. Agric. Food Chem. 2010, 58, 9511–9517. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Merchant, J.L. ZBP-89 promotes growth arrest through stabilization of p53. Mol. Cell Biol. 2001, 21, 4670–4683. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; Maret, W. Imbalance between pro-oxidant and pro-antioxidant functions of zinc in disease. J. Alzheimers Dis. 2005, 8, 161–170. [Google Scholar] [PubMed]
- Hancock, S.M.; Finkelstein, D.I.; Adlard, P.A. Glia and zinc in ageing and Alzheimer’s disease: A mechanism for cognitive decline? Front. Aging Neurosci. 2014, 6. [Google Scholar] [CrossRef]
- Olcina, M.; Lecane, P.S.; Hammond, E.M. Targeting hypoxic cells through the DNA damage response. Clin. Cancer Res. 2010, 16, 5624–5629. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakanishi, A.; Minami, A.; Kitagishi, Y.; Ogura, Y.; Matsuda, S. BRCA1 and p53 Tumor Suppressor Molecules in Alzheimer’s Disease. Int. J. Mol. Sci. 2015, 16, 2879-2892. https://doi.org/10.3390/ijms16022879
Nakanishi A, Minami A, Kitagishi Y, Ogura Y, Matsuda S. BRCA1 and p53 Tumor Suppressor Molecules in Alzheimer’s Disease. International Journal of Molecular Sciences. 2015; 16(2):2879-2892. https://doi.org/10.3390/ijms16022879
Chicago/Turabian StyleNakanishi, Atsuko, Akari Minami, Yasuko Kitagishi, Yasunori Ogura, and Satoru Matsuda. 2015. "BRCA1 and p53 Tumor Suppressor Molecules in Alzheimer’s Disease" International Journal of Molecular Sciences 16, no. 2: 2879-2892. https://doi.org/10.3390/ijms16022879
APA StyleNakanishi, A., Minami, A., Kitagishi, Y., Ogura, Y., & Matsuda, S. (2015). BRCA1 and p53 Tumor Suppressor Molecules in Alzheimer’s Disease. International Journal of Molecular Sciences, 16(2), 2879-2892. https://doi.org/10.3390/ijms16022879