
Identification of MicroRNA for Intermuscular Bone Development in Blunt Snout Bream (Megalobrama amblycephala)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
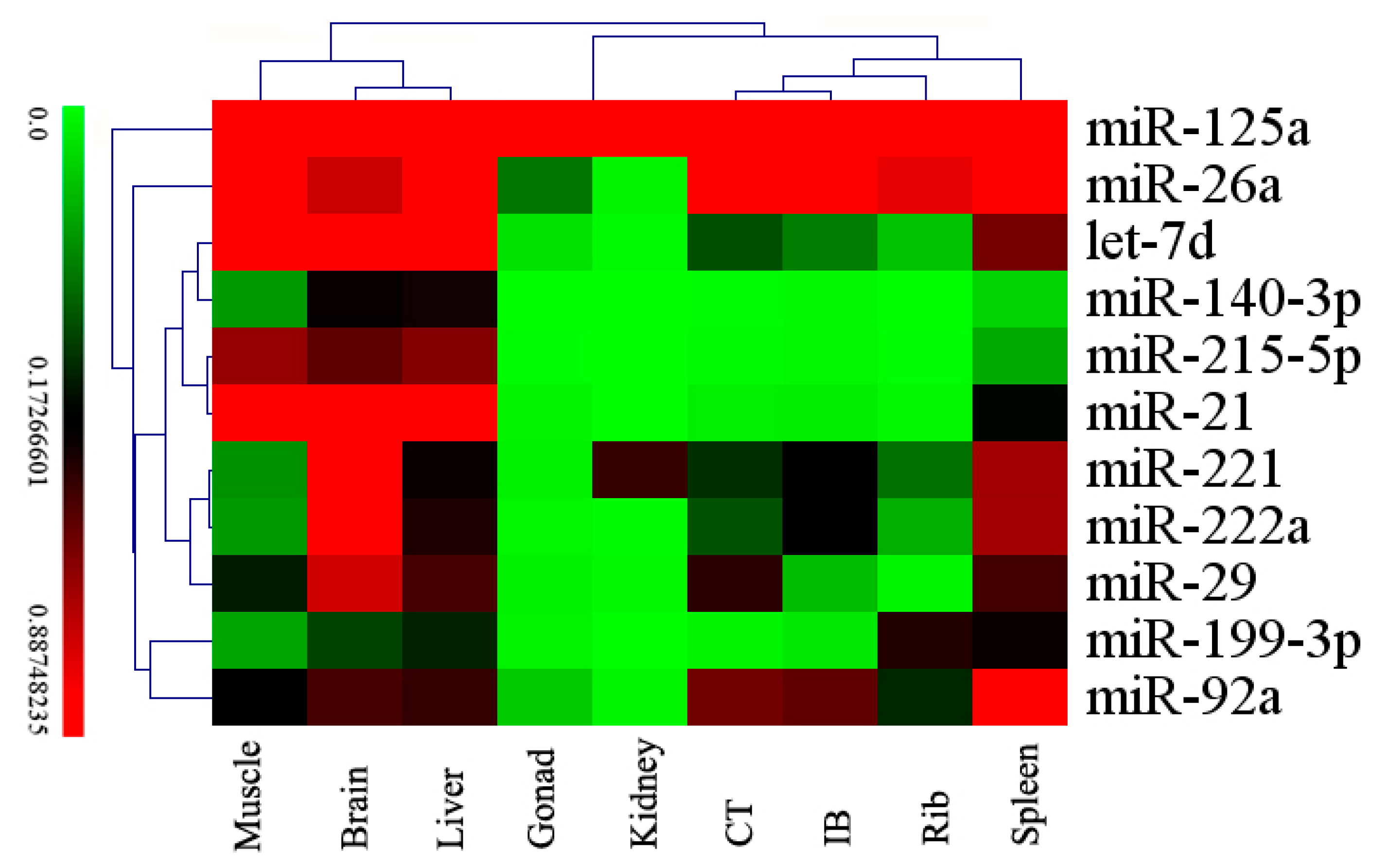
2. Results and Discussion
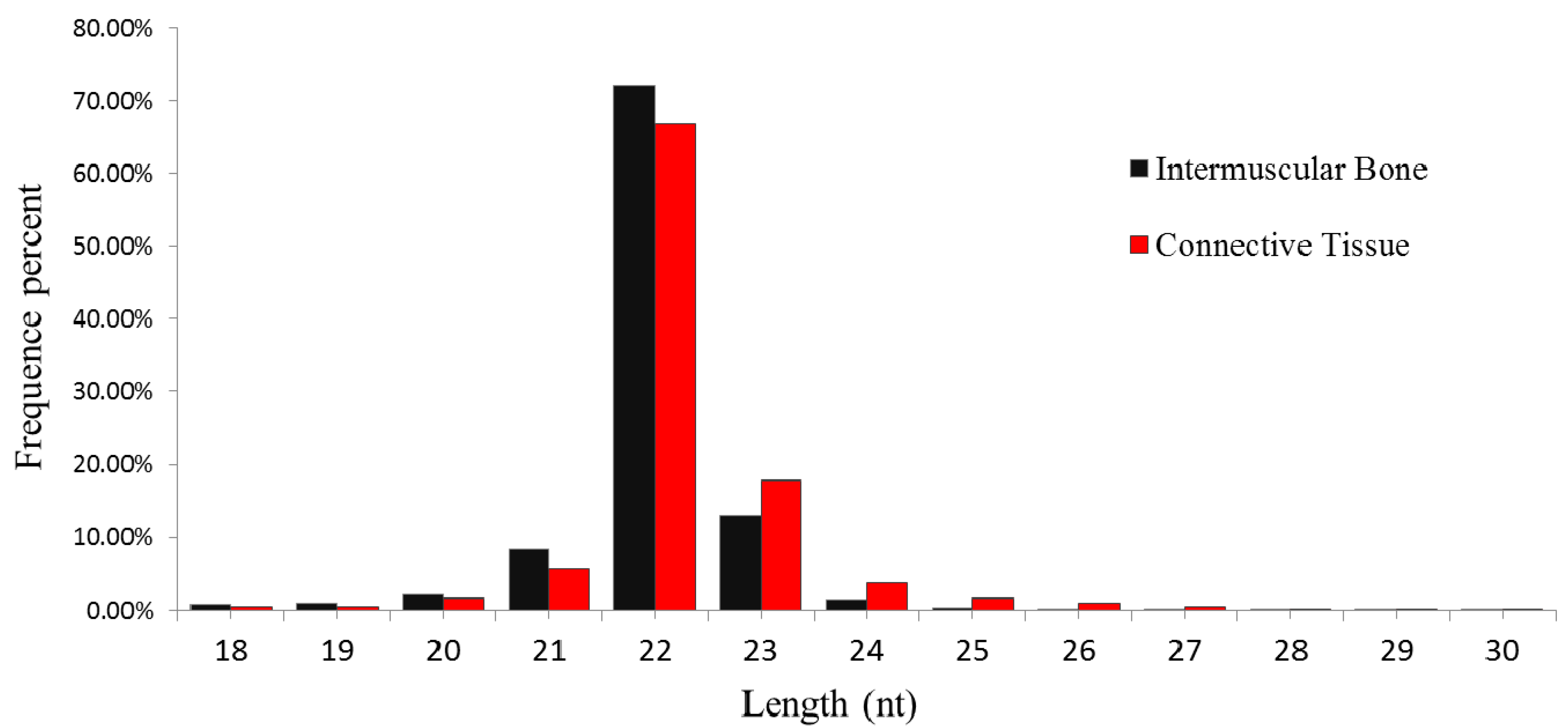
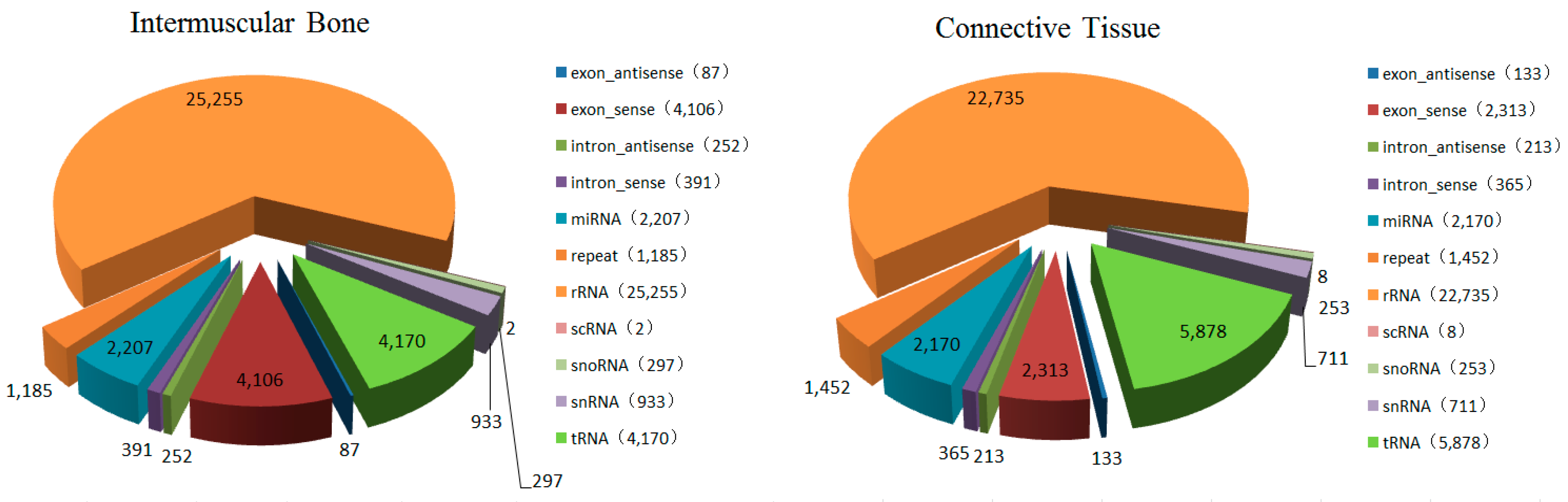
2.1. General Features of Small RNAs


2.2. Identification of Conserved miRNAs
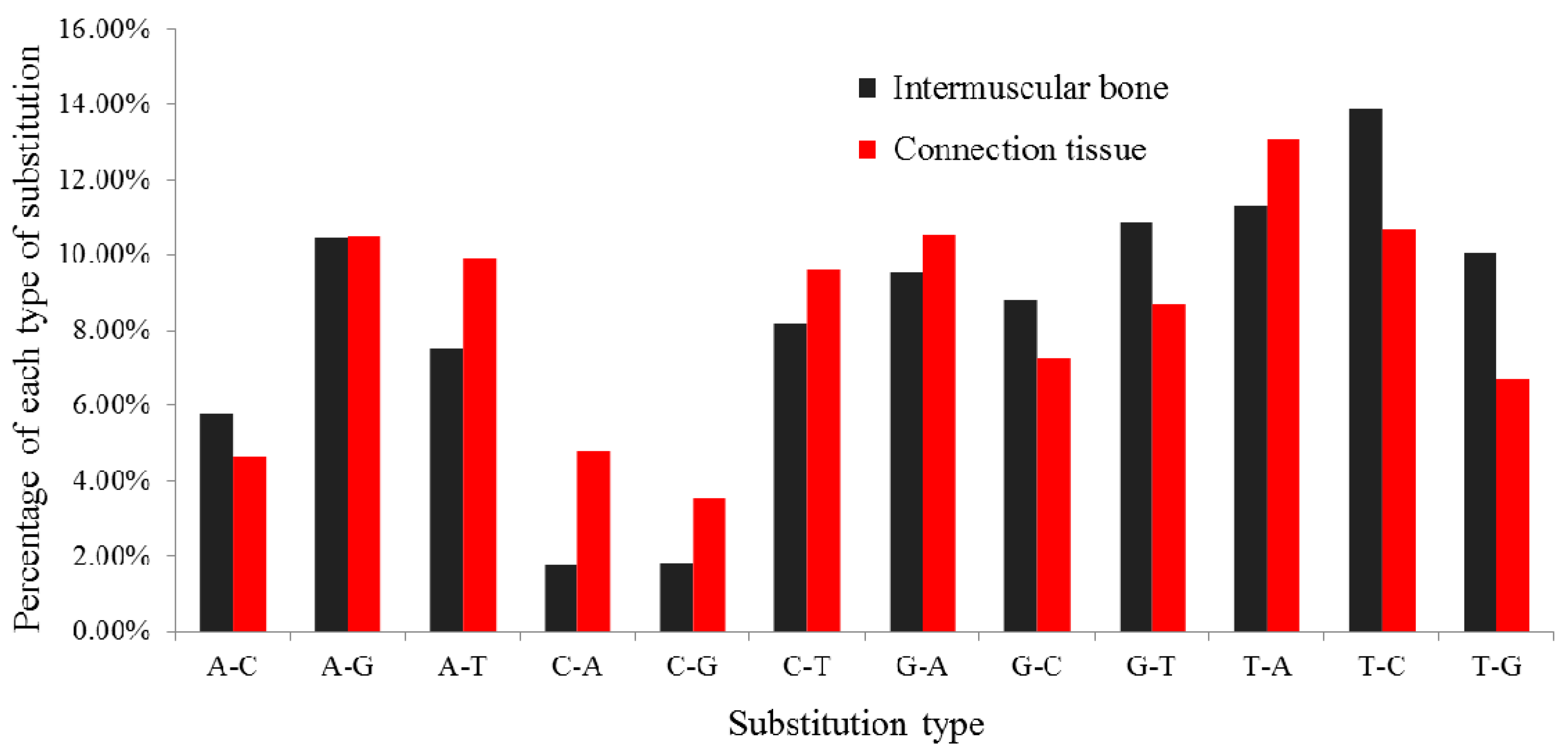
2.3. Statistics of Multiple IsomiRs in M. amblycephala


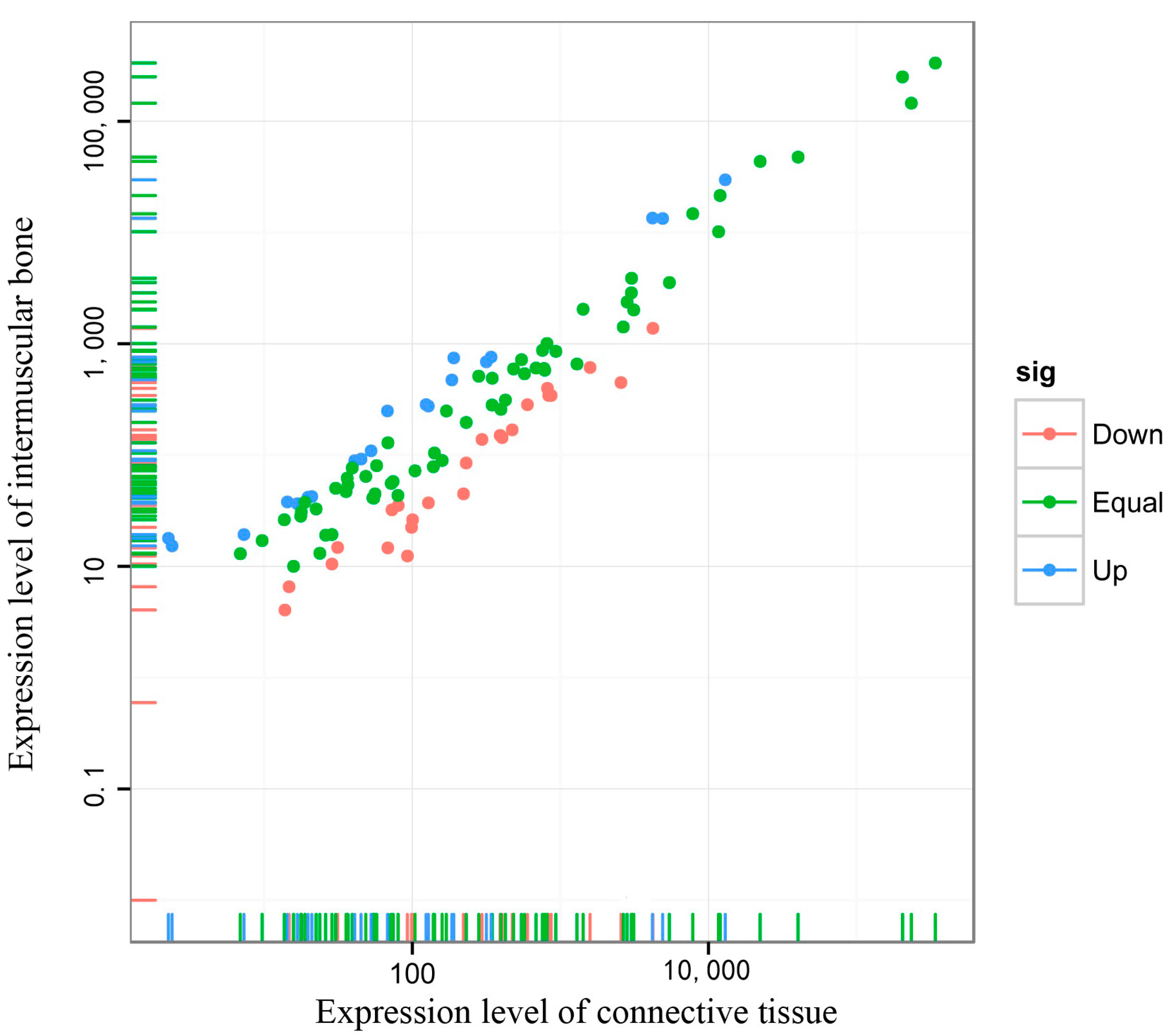
2.4. Differentially Expressed miRNAs


2.5. Prediction of Potential Targets of Differentially-Expressed miRNA
2.6. Function Analysis of Target Genes of Differentially-Expressed miRNAs
3. Experimental Section
3.1. Animals and Tissue Collection
3.2. Small RNA Isolation and cDNA Library Construction
3.3. Small RNA Sequence Analysis
3.4. Differential Expression Analysis of miRNAs
3.5. Prediction of miRNA Target Genes
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Patterson, C.; Johnson, G. Intermuscular bones and ligaments of teleostean fishes. Smithson Contrib. Zool. 1995, 559, 1–85. [Google Scholar] [CrossRef]
- Xie, C. Ichthyology (M); China Agricultural Press: Beijing, China, 2010; pp. 56–57. [Google Scholar]
- Bird, N.; Mabee, P. Developmental morphology of the axial skeleton of the zebrafish, Danio rerio (Ostariophysi, Cyprinidae). Dev. Dyn. 2003, 228, 337–357. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Dong, Z.; Su, S. The research progress on intermuscular bones of teleosts. Jiangsu Agric. Sci. 2012, 40, 234–235. [Google Scholar]
- Lü, Y.; Bao, B.; Jiang, Y. Comparative analysis of intermuscular bones in lower teleosts. J. Fish. China 2007, 31, 661–668. [Google Scholar]
- Bing, Z. On the myoseptal spines of the carp (Cyprinus carpio L.). Acta Zool. Sin. 1962, 14, 175–178. [Google Scholar]
- Johnson, G.D.; Patterson, C. The intermuscular system of acanthomorph fish, a commentary. Am. Mus. Novit. 2001, 1, 1–24. [Google Scholar]
- Ke, Z.; Zhang, W.; Jiang, Y. Developmental morphology of the intermuscular bone in Hypophthalmichthys molitrix. Chin. J. Zool. 2008, 43, 88–96. [Google Scholar]
- Jiang, Y.; Yang, L.; Bao, B. The epicentrals in several lower teleosts. J. Shanghai Fish. Univ. 2008, 17, 493–496. [Google Scholar]
- Fang, L.; Li, X. A review of research on intermuscular bone formation in lower teleosts. Fish. Sci. 2013, 32, 749–752. [Google Scholar]
- Li, L.; Zhong, Z.; Zeng, M. Comparative analysis of intermuscular bone in different ploidy fish. Sci. China Life Sci. 2013, 56, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Hobert, O. Gene regulation by transcription factors and microRNAs. Science 2008, 319, 1785–1786. [Google Scholar] [CrossRef] [PubMed]
- Bizuayehu, T.T.; Babiak, J.; Norberg, B.; Fernandes, J.; Johansen, S.D. Sex-biased miRNA expression in Atlantic halibut (Hippoglossus hippoglossus) brain and gonads. Sex. Dev. 2012, 6, 257–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Li, Q.; Wang, J. Identification and characterization of novel amphioxus microRNAs by Solexa sequencing. Genome Biol. 2009, 10. [Google Scholar] [CrossRef]
- Yan, X.; Ding, L.; Li, Y.; Zhang, X.; Liang, Y. Identification and profiling of microRNAs from skeletal muscle of the common carp. PLoS ONE 2012, 7, e30925. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.; Gao, Z.; Zhao, H.; Zeng, C.; Luo, W.; Chen, B.; Wang, W. Identification and characterization of microRNAs involved in growth of blunt snout bream (Megalobrama amblycephala) by Solexa sequencing. BMC Genomics 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Zhong, J.; Nie, C.; Gao, Z.; Zhang, X. The complete mitochondrial genome of the hybrid of Megalobrama amblycephala (♀) × Megalobrama terminalis (♂). Mitochondria DNA 2015, 2015. [Google Scholar] [CrossRef]
- Luo, W.; Zeng, C.; Deng, W.; Robinson, N.; Wang, W.; Gao, Z. Genetic parameter estimates for growth-related traits of blunt snout bream (Megalobrama amblycephala) using microsatellite-based pedigree. Aquac. Res. 2014, 45, 1881–1888. [Google Scholar] [CrossRef]
- Li, X.; Liu, W.; Lu, K.; Xu, W.; Wang, Y. Dietary carbohydrate/lipid ratios affect stress, oxidative status and non-specific immune responses of fingerling blunt snout bream, Megalobrama amblycephala. Fish Shellfish Immunol. 2012, 33, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Luo, W.; Liu, H. Transcriptome analysis and SSR/SNP markers information of the blunt snout bream (Megalobrama amblycephala). PLoS ONE 2012, 7, e42637. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Yi, S.; Zhong, J.; Wang, W.; Jiang, E.; Chen, B.; Gao, Z. Development and morphological observation of intermuscular bones in Megalobrama amblycephala. Acta Hydrobiol. Sin. 2014, 38, 1143–1151. [Google Scholar]
- Zhu, Y.; Xue, W.; Wang, J.; Wan, Y.; Wang, S.; Xu, P.; Zhang, Y.; Li, J.; Sun, X. Identification of common carp (Cyprinus carpio) microRNAs and microRNA-related SNPs. BMC Genomics 2012, 13, 413. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Chen, J.; Li, X.; Ge, J.; Pan, J. Identification and characterization of MicroRNAs in channel catfish (Ictalurus punctatus) by using Solexa sequencing technology. PLoS ONE 2013, 8, e54174. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Shi, Z.; Wu, M.; Zhang, J.; Jia, L. Identification and differential expression of MicroRNAs during metamorphosis of the Japanese flounder (Paralichthys olivaceus). PLoS ONE 2011, 6, e22957. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Hansen, J.H.; Hedegaard, J.; Nielsen, R.O.; Panitz, F.; Bendixen, C.; Thomsen, B. MicroRNA identity and abundance in porcine skeletal muscles determined by deep sequencing. Anim. Genet. 2010, 41, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Novello, C.; Pazzaglia, L.; Cingolani, C.; Conti, A.; Quattrini, I.; Manara, M.C.; Tognon, M.; Picci, P.; Benassi, M.S. MiRNA expression profile in human osteosarcoma, role of miR-1 and miR-133b in proliferation and cell cycle control. Int. J. Oncol. 2013, 42, 667–675. [Google Scholar] [PubMed]
- Kim, H.K.; Lee, Y.S.; Sivaprasad, U.; Malhotra, A.; Dutta, A. Muscle-specific microRNA miR-206 promotes muscle differentiation. Cell Biol. 2006, 174, 677–687. [Google Scholar] [CrossRef]
- Sweetman, D.; Goljanek, K.; Rathjen, T.; Oustanina, S.; Braun, T.; Dalmay, T.; Münsterberg, A. Specific requirements of MRFs for the expression of muscle specific microRNAs, miR-1, miR-206, and miR-133. Dev. Biol. 2008, 321, 491–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inose, H.; Ochi, H.; Kimura, A. A microRNA regulatory mechanism of osteoblast differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 20794–20799. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Gross, C.; Fiedler, J. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signaling in fibroblasts. Nature 2008, 456, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Sugatani, T.; Hruska, K.A. Down-regulation of miR-21 biogenesis by estrogen action contributes to osteoclastic apoptosis. Cell Biochem. 2013, 114, 1217–1222. [Google Scholar] [CrossRef]
- Huang, S.; Wang, S.; Bian, C.; Yang, Z.; Zhou, H.; Zeng, Y.; Li, H.; Han, Q.; Zhao, R.C. Upregulation of miR-22 Promotes osteogenic differentiation and inhibits adipogenic differentiation of human adipose tissue-derived mesenchymal stem cells by repressing HDAC6 protein expression. Stem Cells Dev. 2012, 21, 2531–2540. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Choy, E.; Harmon, D.; Liu, X.; Susa, M.; Mankin, H.; Hornicek, F. MicroRNA-199a-3p is downregulated in human osteosarcoma and regulates cell proliferation and migration. Mol. Cancer Ther. 2011, 10, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Mondol, V.; Pasquinelli, A.E. Let’s make it happen, the role of let-7 microRNA in development. Curr. Top. Dev. Biol. 2012, 99, 1–30. [Google Scholar] [PubMed]
- Wang, X.; Cao, L.; Wang, Y.; Wang, X.; Liu, N.; You, Y. Regulation of let-7 and its target oncogenes (Review). Oncol. Lett. 2012, 3, 955–960. [Google Scholar] [PubMed]
- Johnson, C.D.; Esquela-Kerscher, A.; Stefani, G.; Byrom, M.; Kelnar, K.; Ovcharenko, D.; Wilson, M.; Wang, X.; Shelton, J.; Shingara, J.; et al. The let-7 microRNA represses cell Proliferation Pathways in Human Cells. Cancer Res. 2007, 67, 7713–7722. [Google Scholar] [CrossRef] [PubMed]
- Huleihel, L.; Ben-Yehudah, A. Let-7d microRNA affects mesenchymal phenotypic properties of lung fibroblasts. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L534–L542. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Deng, C.; Li, Y.P. TGF-β and BMP signaling in osteoblast Differentiation and Bone Formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, R. Discovery and characterization of miRNA genes in Atlantic salmon (Salmo salar) by use of a deep sequencing approach. BMC Genomics 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Ramachandra, R.K.; Salem, M.; Gahr, S.; Rexroad, C.; Yao, J. Cloning and characterization of microRNAs from rainbow trout (Oncorhynchus mykiss), their expression during early embryonic development. BMC Dev. Biol. 2008, 8. [Google Scholar] [CrossRef]
- Li, G.; Li, Y.; Li, X.; Ning, X.; Li, M.; Yang, G. MicroRNA identity and abundance in developing swine adipose tissue as determined by Solexa sequencing. Cell Biochem. 2011, 112, 1318–1328. [Google Scholar] [CrossRef]
- Ebhardt, H.A.; Tsang, H.H.; Dai, D.C.; Liu, Y.; Bostan, B.; Fahlman, R.P. Meta-analysis of small RNA-sequencing errors reveals ubiquitous post-transcriptional RNA modifications. Nucleic Acids Res. 2009, 37, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbauer, F.; Morin, R.D.; Argiropoulos, B.; Petriv, O.I.; Griffith, M. In-depth characterization of the microRNA transcriptome in a leukemia progression model. Genome Res. 2008, 18, 1787–1797. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Zhao, Q.; Zhu, D.; Yu, J. Characterization of microRNAs expression during maize seed development. BMC Genomics 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Ro, S.; Park, C.; Young, D.; Sanders, K.; Yan, W. Tissue-dependent paired expression of miRNAs. Nucleic Acids Res. 2007, 35, 5944–5953. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Valverde, S.L.; Taft, R.J.; Mattick, J.S. Dynamic isomiR regulation in Drosophila development. RNA 2010, 16, 1881–1888. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, A.M.; Ando, Y.; de Hoon, M.J.; Tomaru, Y.; Nishibu, T. A comprehensive survey of 3' animal miRNA modification events and a possible role for 3' adenylation in modulating miRNA targeting effectiveness. Genome Res. 2010, 20, 1398–1410. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Phillips, M.; Betel, D.; Mu, P.; Ventura, A.; Siepel, A.; Chen, K.; Lai, E. Widespread regulatory activity of vertebrate microRNA* species. RNA 2011, 17, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Laqtom, N.; Kelly, L.; Buck, A. Regulation of integrins and AKT signaling by miR-199-3p in HCMV-infected cells. BMC Genomics 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Herrera, B.M.; Lockstone, H.E.; Taylor, J.M. MicroRNA-125a is over-expressed in insulin target tissues in a spontaneous rat model of Type 2 Diabetes. BMC Med. Genomics 2009, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gañán-Gómez, I.; Wei, Y.; Yang, H.; Pierce, S.; Bueso-Ramos, C. Overexpression of miR-125a in myelodysplastic syndrome CD34+ cells Modulates NF-κB Activation and Enhances Erythroid Differentiation Arrest. PLoS ONE 2014, 9, e93404. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C. MicroRNA targets in Drosophila. Genome Biol. 2003, 5. [Google Scholar] [CrossRef]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Fell, D.A.; Dandekar, T. A general definition of metabolic pathways useful for systematic organization and analysis of complex metabolic networks. Nat. Biotechnol. 2000, 18, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Liu, H.; Mukherjee, R.; Yun, J.W. Downregulation of fetuin-B and zinc-α2-glycoprotein is linked to impaired fatty acid metabolism in liver cells. Cell. Physiol. Biochem. 2012, 30, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.A., II; Bialek, P.; Ahn, J.D.; Starbuck, M.; Patel, M.S.; Clevers, H.; Taketo, M.M.; Long, F.; McMahon, A.P.; Lang, R.A.; et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell 2005, 8, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease, from human mutations to treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; O’Keefe, R.; Chen, D. TGF-β signaling in chondrocytes. Front. Biosci. 2005, 10, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Keren, A.; Tamir, Y.; Bengal, E. The p38 MAPK signaling pathway, a major regulator of skeletal muscle development. Mol. Cell. Endocrinol. 2006, 252, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, M.B.; Shim, J.H.; Zou, W.; Sitara, D.; Schweitzer, M.; Hu, D.; Lotinun, S.; Sano, Y.; Baron, R.; Park, J.M.; et al. The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. Clin. Investig. 2010, 120, 2457–2473. [Google Scholar] [CrossRef] [Green Version]
- Fuentes, E.N.; Björnsson, B.T.; Valdés, J.A.; Einarsdottir, I.E.; Lorca, B.; Alvarez, M.; Molina, A. IGF-I/PI3K/Akt and IGF-I/MAPK/ERK pathways in vivo in skeletal muscle are regulated by nutrition and contribute to somatic growth in the fine flounder. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1532–R1542. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Cui, X.; Zhang, Y.; Yang, C.; Jiang, Y. Identification of miRNAs associated with sexual maturity in chicken ovary by Illumina small RNA deep sequencing. BMC Genomics 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [PubMed]
- Chen, C.; Ridzon, D.Z.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, S.-M.; Yi, S.-K.; Zhong, J.; Nie, C.-H.; Guan, N.-N.; Chen, B.-X.; Gao, Z.-X. Identification of MicroRNA for Intermuscular Bone Development in Blunt Snout Bream (Megalobrama amblycephala). Int. J. Mol. Sci. 2015, 16, 10686-10703. https://doi.org/10.3390/ijms160510686
Wan S-M, Yi S-K, Zhong J, Nie C-H, Guan N-N, Chen B-X, Gao Z-X. Identification of MicroRNA for Intermuscular Bone Development in Blunt Snout Bream (Megalobrama amblycephala). International Journal of Molecular Sciences. 2015; 16(5):10686-10703. https://doi.org/10.3390/ijms160510686
Chicago/Turabian StyleWan, Shi-Ming, Shao-Kui Yi, Jia Zhong, Chun-Hong Nie, Ning-Nan Guan, Bo-Xiang Chen, and Ze-Xia Gao. 2015. "Identification of MicroRNA for Intermuscular Bone Development in Blunt Snout Bream (Megalobrama amblycephala)" International Journal of Molecular Sciences 16, no. 5: 10686-10703. https://doi.org/10.3390/ijms160510686
APA StyleWan, S. -M., Yi, S. -K., Zhong, J., Nie, C. -H., Guan, N. -N., Chen, B. -X., & Gao, Z. -X. (2015). Identification of MicroRNA for Intermuscular Bone Development in Blunt Snout Bream (Megalobrama amblycephala). International Journal of Molecular Sciences, 16(5), 10686-10703. https://doi.org/10.3390/ijms160510686