
Vanadium Compounds as Pro-Inflammatory Agents: Effects on Cyclooxygenases


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
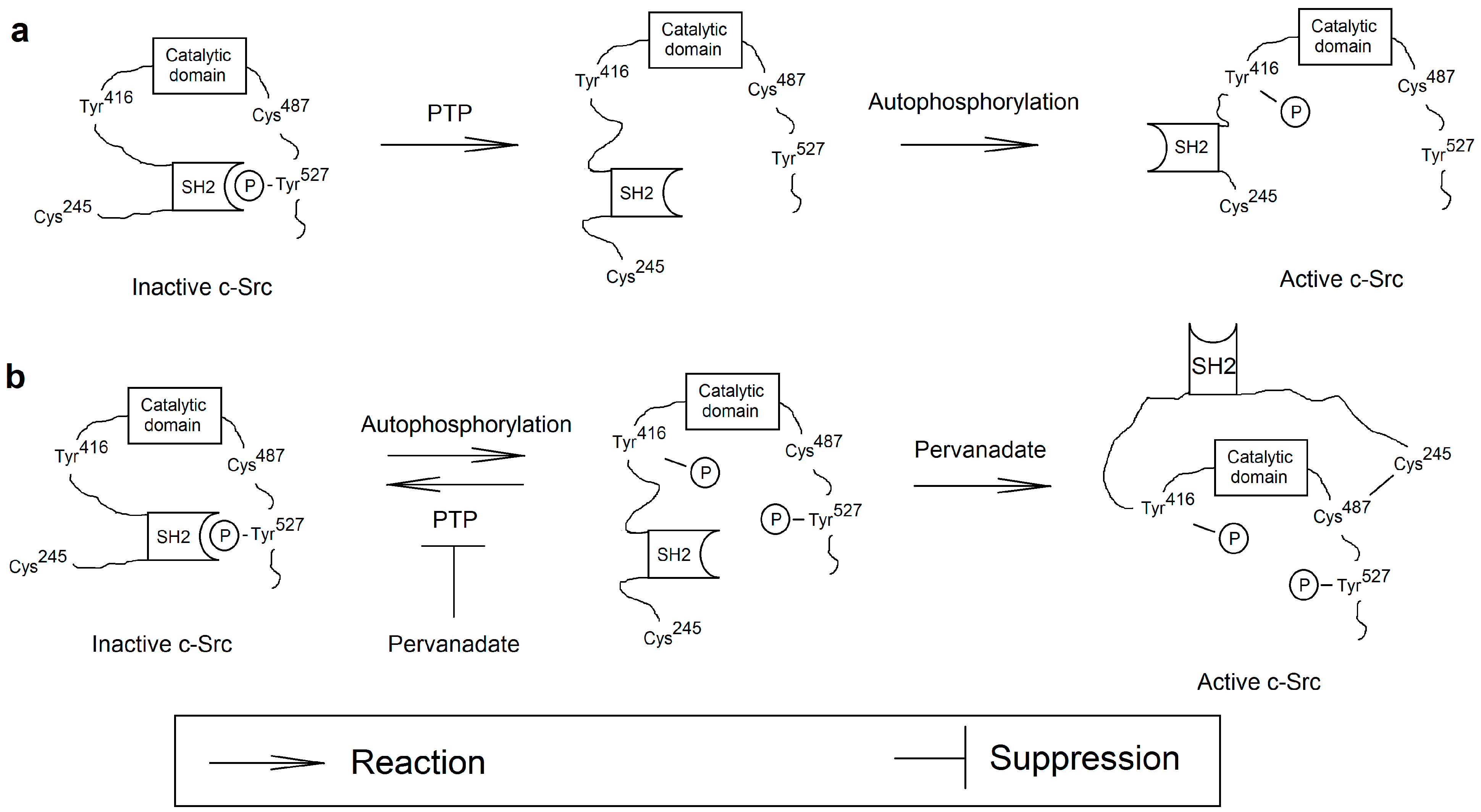{kind=link}
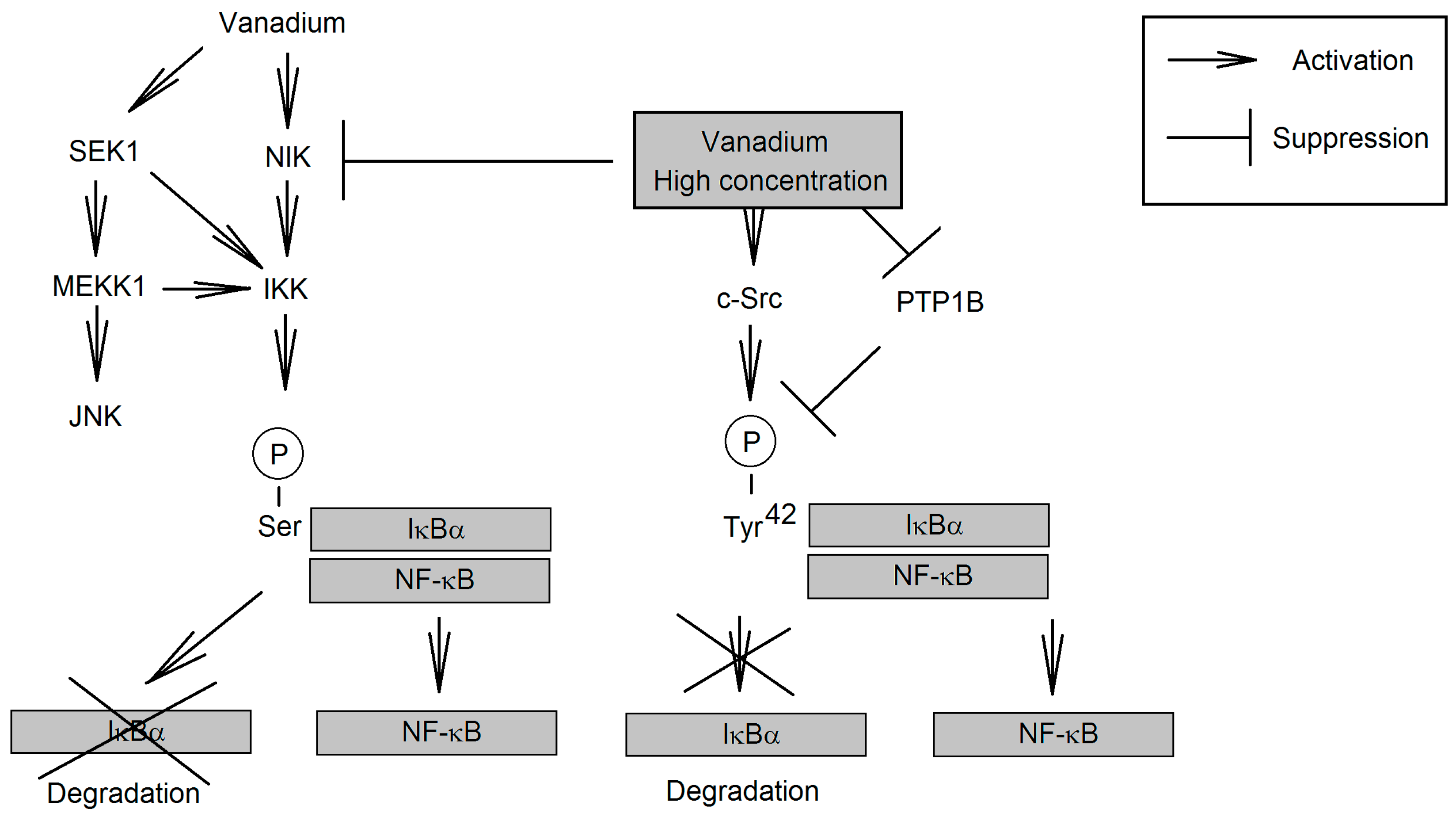{kind=link}
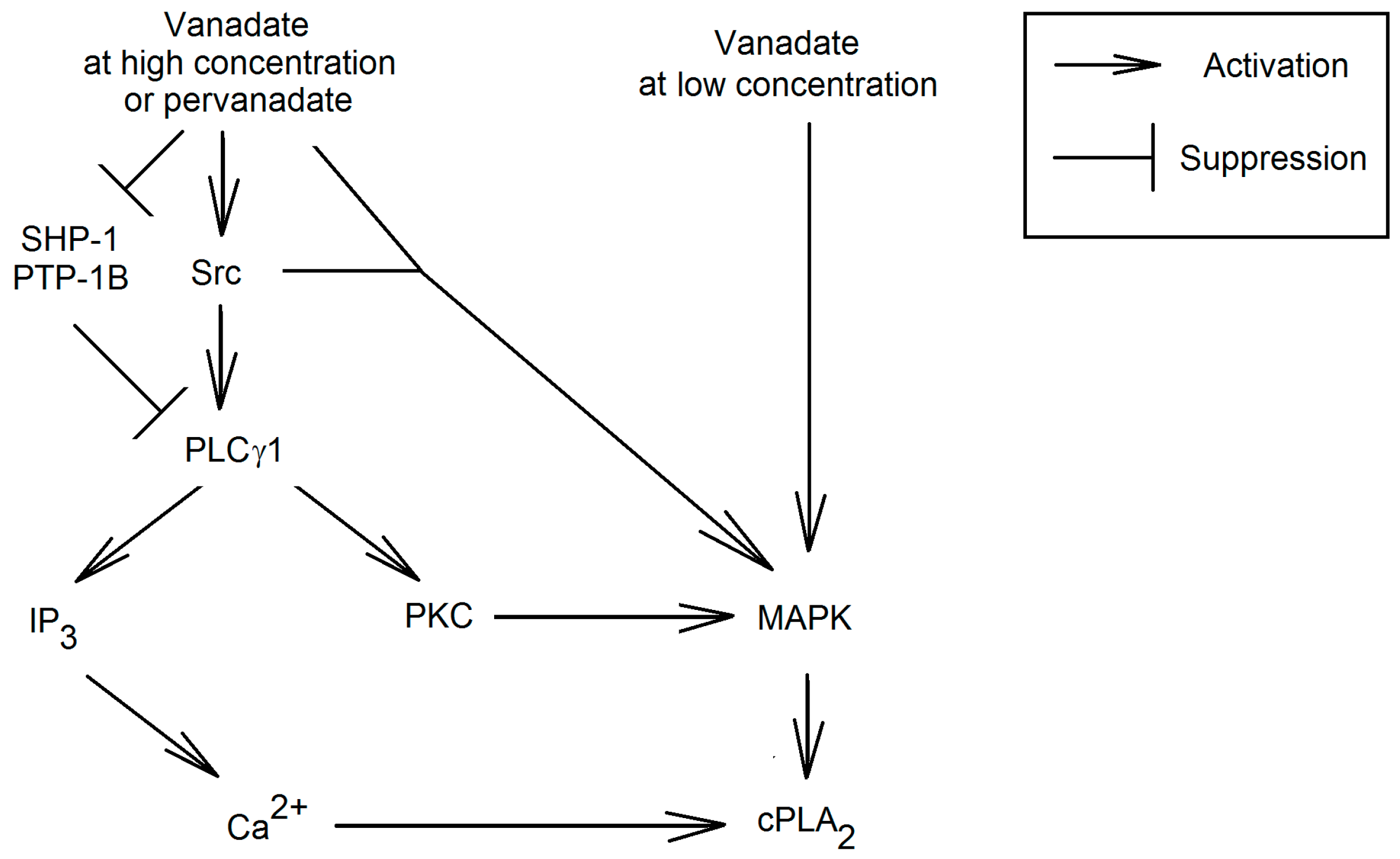{kind=link}
1. Introduction

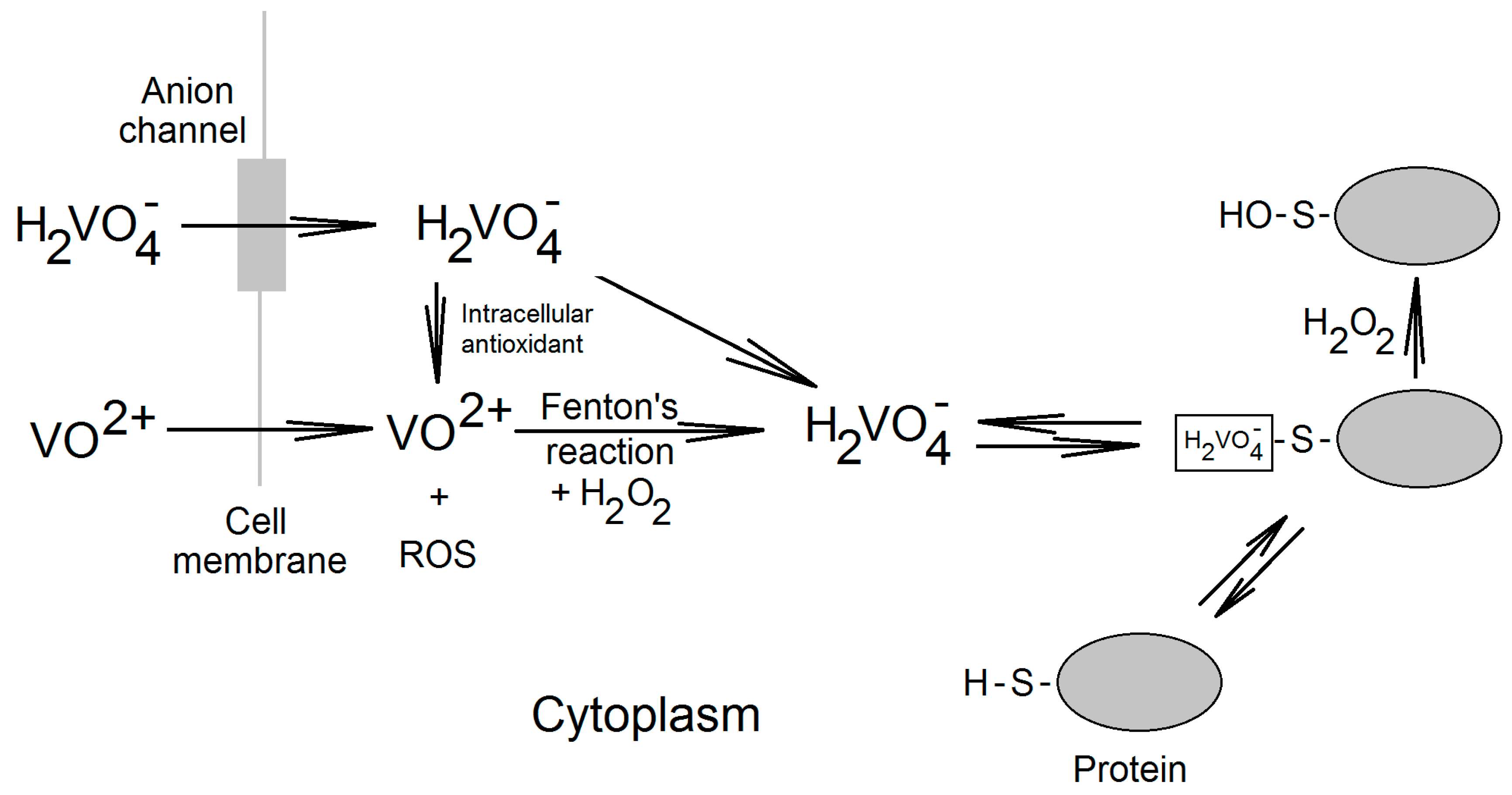
2. Vanadium Compounds in the Cell

3. Effect of Vanadium Compounds on the Expression of Cyclooxygenases
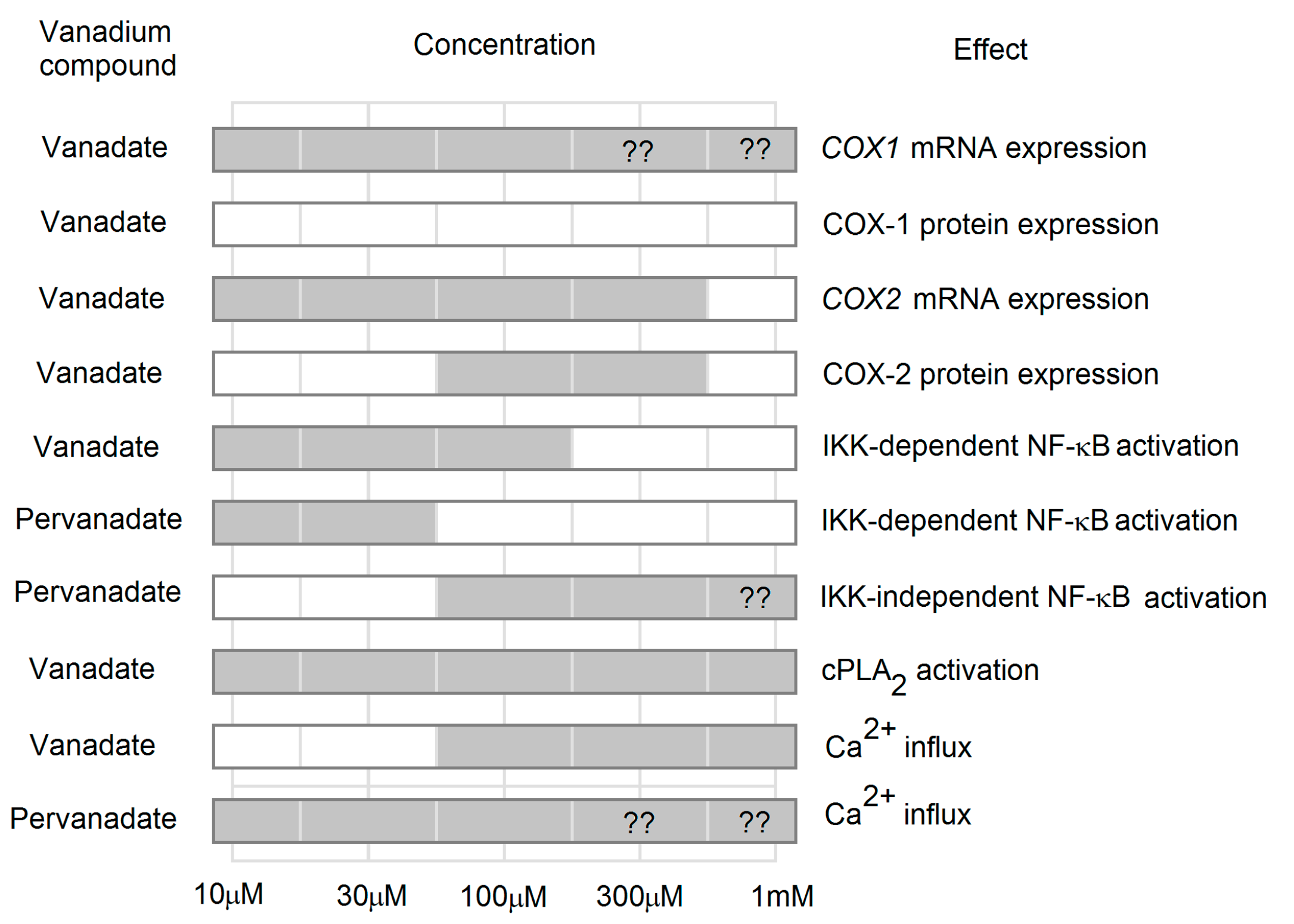
3.1. Expression of Cyclooxygenases
3.2. Effect on the Expression of Cyclooxygenase-1
3.3. Effect on the Expression of Cyclooxygenase-2
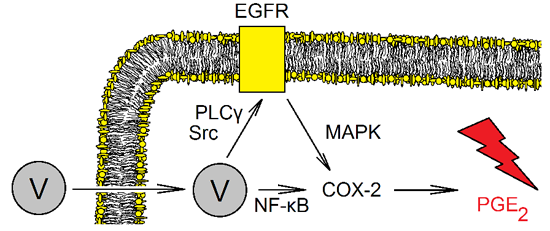
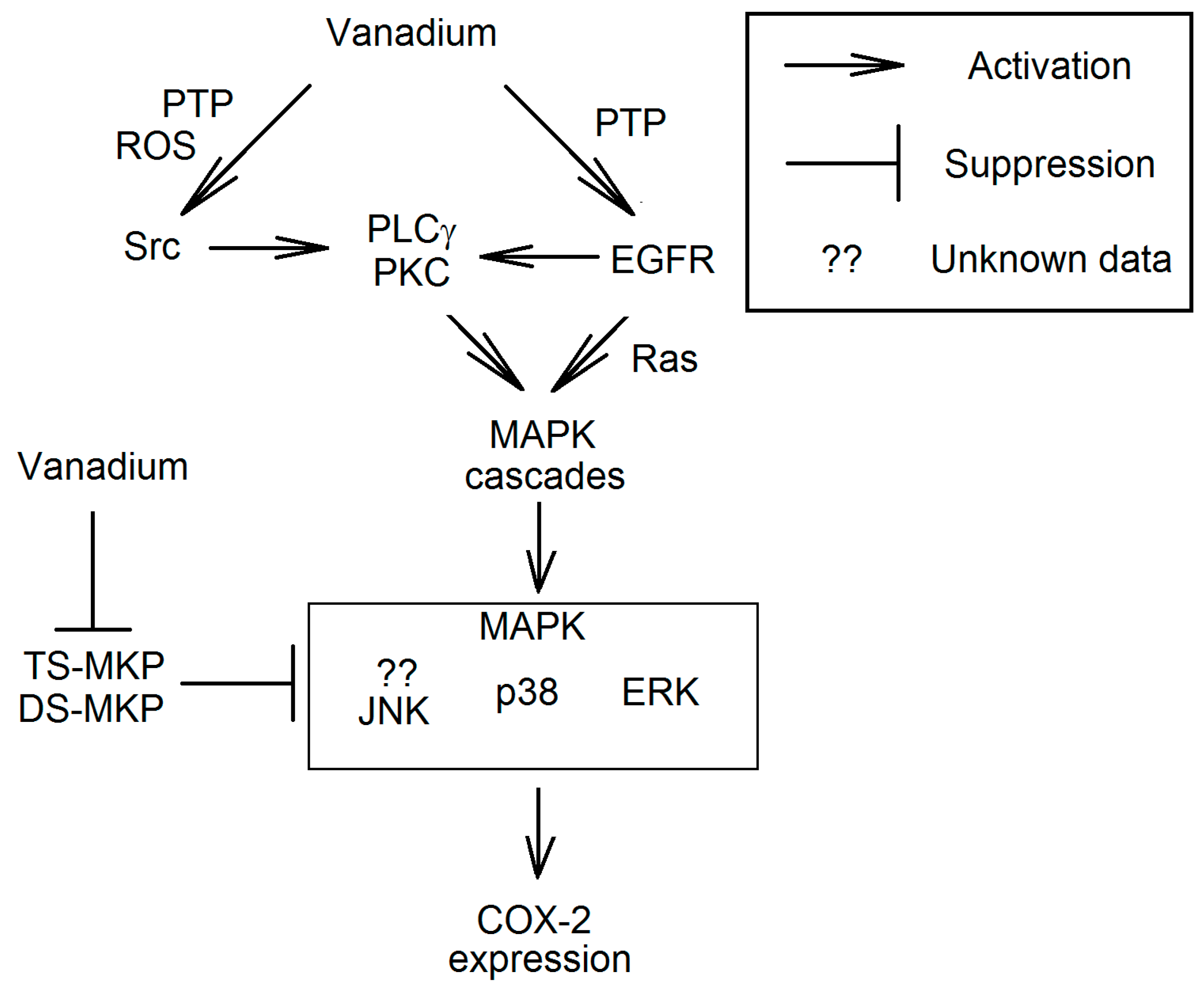
3.3.1. EGFR Signal Transduction Resulting in Increased Expression of Cyclooxygenase-2

3.3.2. Activation of MAPK Cascades Independent of EGFR
3.3.3. Activation of the Src

3.3.4. The Effect on MAPK Phosphatase
3.3.5. Effect on NF-κB

3.3.6. Effect on the Proteolysis of Cyclooxygenase-2
4. Effect on Cyclooxygenase Activity
5. Effect on the Supply of Substrate for Cyclooxygenases

6. Pro-Inflammatory Properties and Therapeutic Use of Vanadium Compounds
Conflicts of Interest
Abbreviations
| AA | arachidonic acid |
| AP-1 | activator protein-1 |
| ASK-1 | apoptosis signal-regulating kinase 1 |
| COX-1 | cyclooxygenase-1 |
| COX-2 | cyclooxygenase-2 |
| COX | cyclooxygenases |
| cPLA2 | cytoplasmic phospholipase A2 |
| DAG | diacylglycerol |
| DS.-MKP | dual specificity MKP |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| ERK | extracellular signal-regulated kinase |
| HUVEC | human umbilical vein in endothelial cell |
| IKK | IκB kinase |
| IP3 | inositol trisphosphate |
| IκB | inhibitor of NF-κB |
| JNK | c-Jun N-terminal kinase |
| MAPK | mitogen-activated protein kinase |
| MAPKK | mitogen-activated protein kinase kinase |
| MAPKKK | mitogen-activated protein kinase kinase kinase |
| MEKK1 | mitogen-activated protein kinase/ERK kinase kinase 1 |
| MKP | MAPK phosphatase |
| NF-κB | nuclear factor κB |
| NIK | NF-κB-inducing kinase |
| PDGFR | platelet-derived growth factor receptor |
| PGE2 | prostaglandin E2 |
| PKC | protein kinase C |
| PLCγ | phospholipase C-γ |
| PTP | protein tyrosine phosphatases |
| PTP-1B | protein tyrosine phosphatase-1B |
| ROS | reactive oxygen species |
| SEK1 | SAPK/ERK kinase 1 |
| SHP-1 | SH2 domain-containing protein tyrosine phosphatase 1 |
| SHP-2 | SH2 domain-containing protein tyrosine phosphatase 2 |
| Sp1 | specificity protein 1 |
| Src | non-receptor tyrosine kinases of the Src family |
| TS-MKP | tyrosine-specific MKP |
| TxA2 | thromboxane A2 |
References
- Thompson, K.H.; Orvig, C. Vanadium in diabetes: 100 years from Phase 0 to Phase I. J. Inorg. Biochem. 2006, 100, 1925–1935. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.H.; Lichter, J.; LeBel, C.; Scaife, M.C.; McNeill, J.H.; Orvig, C. Vanadium treatment of type 2 diabetes: A view to the future. J. Inorg. Biochem. 2009, 103, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Kurt, O.; Ozden, T.Y.; Ozsoy, N.; Tunali, S.; Can, A.; Akev, N.; Yanardag, R. Influence of vanadium supplementation on oxidative stress factors in the muscle of STZ-diabetic rats. Biometals 2011, 24, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Missaoui, S.; Ben Rhouma, K.; Yacoubi, M.T.; Sakly, M.; Tebourbi, O. Vanadyl sulfate treatment stimulates proliferation and regeneration of beta cells in pancreatic islets. J. Diabetes Res. 2014, 2014, 540242. [Google Scholar] [CrossRef] [PubMed]
- Pirmoradi, L.; Mohammadi, M.T.; Safaei, A.; Mesbah, F.; Dehghani, G.A. Does the relief of glucose toxicity act as a mediator in proliferative actions of vanadium on pancreatic islet beta cells in streptozocin diabetic rats? Iran. Biomed. J. 2014, 18, 173–180. [Google Scholar] [PubMed]
- Sun, L.; Shi, D.J.; Gao, X.C.; Mi, S.Y.; Yu, Y.; Han, Q. The protective effect of vanadium against diabetic cataracts in diabetic rat model. Biol. Trace Elem. Res. 2014, 158, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Soveid, M.; Dehghani, G.A.; Omrani, G.R. Long-term efficacy and safety of vanadium in the treatment of type 1 diabetes. Arch. Iran. Med. 2013, 16, 408–411. [Google Scholar] [PubMed]
- Evangelou, A.M. Vanadium in cancer treatment. Crit. Rev. Oncol. Hematol. 2002, 42, 249–265. [Google Scholar] [CrossRef]
- Bishayee, A.; Waghray, A.; Patel, M.A.; Chatterjee, M. Vanadium in the detection, prevention and treatment of cancer: the in vivo evidence. Cancer Lett. 2010, 294, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.T.; Liu, Y.J.; Wang, Q.; Yang, X.G.; Wang, K. Reactive-oxygen-species-mediated Cdc25C degradation results in differential antiproliferative activities of vanadate, tungstate, and molybdate in the PC-3 human prostate cancer cell line. J. Biol. Inorg. Chem. 2012, 17, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Suwalsky, M.; Fierro, P.; Villena, F.; Gallardo, M.J.; Jemiola-Rzeminska, M.; Strzalka, K.; Gul-Hinc, S.; Ronowska, A.; Zysk, M.; Szutowicz, A. Effects of sodium metavanadate on in vitro neuroblastoma and red blood cells. Arch. Biochem. Biophys. 2013, 535, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ma, Y.; Xu, Z.; Wang, D.; Zhao, B.; Pan, H.; Wang, J.; Xu, D.; Zhao, X.; Pan, S.; et al. Sodium orthovanadate inhibits growth of human hepatocellular carcinoma cells in vitro and in an orthotopic model in vivo. Cancer Lett. 2014, 351, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Kucera, J.; Byrne, A.R.; Mravcová, A.; Lener, J. Vanadium levels in hair and blood of normal and exposed persons. Sci. Total Environ. 1992, 115, 191–205. [Google Scholar] [CrossRef]
- Nadal, M.; Schuhmacher, M.; Domingo, J.L. Metal pollution of soils and vegetation in an area with petrochemical industry. Sci. Total Environ. 2004, 321, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Pourang, N.; Nikouyan, A.; Dennis, J.H. Trace element concentrations in fish, surficial sediments and water from northern part of the Persian Gulf. Environ. Monit. Assess. 2005, 109, 293–316. [Google Scholar] [CrossRef] [PubMed]
- Moreno, T.; Querol, X.; Alastuey, A.; de la Rosa, J.; Sánchez de la Campa, A.M.; Minguillón, M.; Pandolfi, M.; González-Castanedo, Y.; Monfort, E.; Gibbons, W. Variations in vanadium, nickel and lanthanoid element concentrations in urban air. Sci. Total Environ. 2010, 408, 4569–4579. [Google Scholar] [CrossRef] [PubMed]
- Guzmán-Morales, J.; Morton-Bermea, O.; Hernández-Álvarez, E.; Rodríguez-Salazar, M.T.; García-Arreola, M.E.; Tapia-Cruz, V. Assessment of atmospheric metal pollution in the urban area of Mexico City, using Ficus benjamina as biomonitor. Bull. Environ. Contam. Toxicol. 2011, 86, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Speight, J. Chemical composition. In The Chemistry and Technology of Petroleum, 3rd ed.; CRC Press: New York, NY, USA; Basel, Switzerland, 1999; pp. 215–243. [Google Scholar]
- Bednar, A.J.; Chappell, M.A.; Seiter, J.M.; Stanley, J.K.; Averett, D.E.; Jones, W.T.; Pettway, B.A.; Kennedy, A.J.; Hendrix, S.H.; Steevens, J.A. Geochemical investigations of metals release from submerged coal fly ash using extended elutriate tests. Chemosphere 2010, 81, 1393–1400. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.S.; Chang, C.L.; Shen, F.M. Whole blood vanadium in Taiwanese college students. Bull. Environ. Contam. Toxicol. 2004, 73, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Azay, J.; Brès, J.; Krosniak, M.; Teissedre, P.L.; Cabanis, J.C.; Serrano, J.J.; Cros, G. Vanadium pharmacokinetics and oral bioavailability upon single-dose administration of vanadyl sulfate to rats. Fundam. Clin. Pharmacol. 2001, 15, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.; Holko, P.; Ligeza, J.; Kordowiak, A.M. Sodium orthovanadate affects growth of some human epithelial cancer cells (A549, HTB44, DU145). Folia Biol. 2008, 56, 115–121. [Google Scholar] [CrossRef]
- McNicol, A.; Robertson, C.; Gerrard, J.M. Vanadate activates platelets by enhancing arachidonic acid release. Blood 1993, 81, 2329–2338. [Google Scholar] [PubMed]
- Tsujishita, Y.; Asaoka, Y.; Nishizuka, Y. Regulation of phospholipase A2 in human leukemia cell lines: Its implication for intracellular signaling. Proc. Natl. Acad. Sci. USA 1994, 91, 6274–6278. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.W.; Hsu, Y.W. Cycloheximide-induced cPLA(2) activation is via the MKP-1 down-regulation and ERK activation. Cell Signal. 2000, 12, 457–461. [Google Scholar] [CrossRef]
- Korbecki, J.; Baranowska-Bosiacka, I.; Gutowska, I.; Piotrowska, K.; Chlubek, D. Cyclooxygenase-1 as the main source of proinflammatory factors after sodium orthovanadate treatment. Biol. Trace Elem. Res. 2015, 163, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Greenhough, A.; Smartt, H.J.; Moore, A.E.; Roberts, H.R.; Williams, A.C.; Paraskeva, C.; Kaidi, A. The COX-2/PGE2 pathway: Key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 2009, 30, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Chien, P.S.; Mak, O.T.; Huang, H.J. Induction of COX-2 protein expression by vanadate in A549 human lung carcinoma cell line through EGF receptor and p38 MAPK-mediated pathway. Biochem. Biophys. Res. Commun. 2006, 339, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Boulassel, B.; Sadeg, N.; Roussel, O.; Perrin, M.; Belhadj-Tahar, H. Fatal poisoning by vanadium. Forensic Sci. Int. 2011, 206, e79–e81. [Google Scholar] [CrossRef] [PubMed]
- Crans, D.C.; Smee, J.J.; Gaidamauskas, E.; Yang, L. The chemistry and biochemistry of vanadium and the biological activities exerted by vanadium compounds. Chem. Rev. 2004, 104, 849–902. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, K.; Lu, J.; Crans, D.C. Membrane transport of vanadium compounds and the interaction with the erythrocyte membrane. Coord. Chem. Rev. 2003, 237, 103–111. [Google Scholar] [CrossRef]
- Bruech, M.; Quintanilla, M.E.; Legrum, W.; Koch, J.; Netter, K.J.; Fuhrmann, G.F. Effects of vanadate on intracellular reduction equivalents in mouse liver and the fate of vanadium in plasma, erythrocytes and liver. Toxicology 1984, 31, 283–295. [Google Scholar] [CrossRef]
- Ding, M.; Gannett, P.M.; Rojanasakul, Y.; Liu, K.; Shi, X. One-electron reduction of vanadate by ascorbate and related free radical generation at physiological pH. J. Inorg. Biochem. 1994, 55, 101–112. [Google Scholar] [CrossRef]
- Shi, X.L.; Dalal, N.S. Flavoenzymes reduce vanadium (V) and molecular oxygen and generate hydroxyl radical. Arch. Biochem. Biophys. 1991, 289, 355–361. [Google Scholar] [CrossRef]
- Shi, X.; Dalal, N.S. Hydroxyl radical generation in the NADH/microsomal reduction of vanadate. Free Radic. Res. Commun. 1992, 17, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Dalal, N.S. Vanadate-mediated hydroxyl radical generation from superoxide radical in the presence of NADH: Haber–Weiss vs. Fenton mechanism. Arch. Biochem. Biophys. 1993, 307, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Capella, L.S.; Gefé, M.R.; Silva, E.F.; Affonso-Mitidieri, O.; Lopes, A.G.; Rumjanek, V.M.; Capella, M.A. Mechanisms of vanadate-induced cellular toxicity: Role of cellular glutathione and NADPH. Arch. Biochem. Biophys. 2002, 406, 65–72. [Google Scholar] [CrossRef]
- Crans, D.C.; Zhang, B.; Gaidamauskas, E.; Keramidas, A.D.; Willsky, G.R.; Roberts, C.R. Is vanadate reduced by thiols under biological conditions? Changing the redox potential of V(V)/V(IV) by complexation in aqueous solution. Inorg. Chem. 2010, 49, 4245–4256. [Google Scholar] [CrossRef] [PubMed]
- Huyer, G.; Liu, S.; Kelly, J.; Moffat, J.; Payette, P.; Kennedy, B.; Tsaprailis, G.; Gresser, M.J.; Ramachandran, C. Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J. Biol. Chem. 1997, 272, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.G.; Zhang, Z.Y. Redox regulation of protein tyrosine phosphatase activity by hydroxyl radical. Biochim. Biophys. Acta 2013, 1834, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Fantus, I.G.; Deragon, G.; Lai, R.; Tang, S. Modulation of insulin action by vanadate: Evidence of a role for phosphotyrosine phosphatase activity to alter cellular signaling. Mol. Cell. Biochem. 1995, 153, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Pugazhenthi, S.; Tanha, F.; Dahl, B.; Khandelwal, R.L. Decrease in protein tyrosine phosphatase activities in vanadate-treated obese Zucker (fa/fa) rat liver. Mol. Cell. Biochem. 1995, 153, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Pugazhenthi, S.; Tanha, F.; Dahl, B.; Khandelwal, R.L. Inhibition of a Src homology 2 domain containing protein tyrosine phosphatase by vanadate in the primary culture of hepatocytes. Arch. Biochem. Biophys. 1996, 335, 273–282. [Google Scholar] [CrossRef]
- Ostman, A.; Frijhoff, J.; Sandin, A.; Böhmer, F.D. Regulation of protein tyrosine phosphatases by reversible oxidation. J. Biochem. 2011, 150, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Tan, Z.; Diltz, C.D.; You, M.; Fischer, E.H. Activation of mitogen-activated protein (MAP) kinase pathway by pervanadate, a potent inhibitor of tyrosine phosphatases. J. Biol. Chem. 1996, 271, 22251–22255. [Google Scholar] [PubMed]
- Lee, K.; Esselman, W.J. Inhibition of PTPs by H2O2 regulates the activation of distinct MAPK pathways. Free Radic. Biol. Med. 2002, 33, 1121–1132. [Google Scholar] [CrossRef]
- Sturla, L.M.; Amorino, G.; Alexander, M.S.; Mikkelsen, R.B.; Valerie, K.; Schmidt-Ullrichr, R.K. Requirement of Tyr-992 and Tyr-1173 in phosphorylation of the epidermal growth factor receptor by ionizing radiation and modulation by SHP2. J. Biol. Chem. 2005, 280, 14597–145604. [Google Scholar] [CrossRef] [PubMed]
- Mbonye, U.R.; Song, I. Posttranscriptional and posttranslational determinants of cyclooxygenase expression. BMB Rep. 2009, 42, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N.; Yamashita, R.; Yamamoto, S.; Ishimura, K. Induction of cyclooxygenase-1 in a human megakaryoblastic cell line (CMK) differentiated by phorbol ester. Biochim. Biophys. Acta 1997, 1344, 103–110. [Google Scholar] [CrossRef]
- Okahara, K.; Sun, B.; Kambayashi, J. Upregulation of prostacyclin synthesis-related gene expression by shear stress in vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1922–1926. [Google Scholar] [CrossRef] [PubMed]
- Gibson, L.L.; Hahner, L.; Osborne-Lawrence, S.; German, Z.; Wu, K.K.; Chambliss, K.L.; Shaul, P.W. Molecular basis of estrogen-induced cyclooxygenase type 1 upregulation in endothelial cells. Circ. Res. 2005, 96, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, T.; Tohnai, N. Cyclooxygenase isozymes and their gene structures and expression. Prostaglandins Other Lipid Mediat. 2002, 68–69, 95–114. [Google Scholar] [CrossRef]
- Hwang, D.; Jang, B.C.; Yu, G.; Boudreau, M. Expression of mitogen-inducible cyclooxygenase induced by lipopolysaccharide: Mediation through both mitogen-activated protein kinase and NF-kappaB signaling pathways in macrophages. Biochem. Pharmacol. 1997, 54, 87–96. [Google Scholar] [CrossRef]
- Faour, W.H.; He, Y.; He, Q.W.; de Ladurantaye, M.; Quintero, M.; Mancini, A.; di Battista, J.A. Prostaglandin E(2) regulates the level and stability of cyclooxygenase-2 mRNA through activation of p38 mitogen-activated protein kinase in interleukin-1 beta-treated human synovial fibroblasts. J. Biol. Chem. 2001, 276, 31720–31731. [Google Scholar] [CrossRef] [PubMed]
- Parfenova, H.; Balabanova, L.; Leffler, C.W. Posttranslational regulation of cyclooxygenase by tyrosine phosphorylation in cerebral endothelial cells. Am. J. Physiol. 1998, 274, C72–C81. [Google Scholar] [PubMed]
- Alexanian, A.; Miller, B.; Chesnik, M.; Mirza, S.; Sorokin, A. Post-translational regulation of COX2 activity by FYN in prostate cancer cells. Oncotarget 2014, 5, 4232–4243. [Google Scholar] [PubMed]
- Hirai, K.; Takayama, H.; Tomo, K.; Okuma, M. Protein-tyrosine-kinase-dependent expression of cyclo-oxygenase-1 and -2 mRNAs in human endothelial cells. Biochem. J. 1997, 322, 373–377. [Google Scholar] [PubMed]
- Hirai, K.; Ezumi, Y.; Nishida, E.; Uchiyama, T.; Takayama, H. Comparative study of vanadate- and phorbol ester-induced cyclo-oxygenase-2 expression in human endothelial cells. Thromb. Haemost. 1999, 82, 1545–1552. [Google Scholar] [PubMed]
- DeLong, C.J.; Smith, W.L. An intronic enhancer regulates cyclooxygenase-1 gene expression. Biochem. Biophys. Res. Commun. 2005, 338, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Li, J.J.; Leonard, S.S.; Ye, J.P.; Shi, X.; Colburn, N.H.; Castranova, V.; Vallyathan, V. Vanadate-induced activation of activator protein-1: role of reactive oxygen species. Carcinogenesis 1999, 20, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.Y.; Wang, Y.T.; Yeh, S.H.; Liu, Y.W.; Chang, W.C.; Hung, J.J. Phosphorylation by c-Jun NH2-terminal kinase 1 regulates the stability of transcription factor Sp1 during mitosis. Mol. Biol. Cell 2008, 19, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Chu, S. Transcriptional regulation by post-transcriptional modification-role of phosphorylation in Sp1 transcriptional activity. Gene 2012, 508, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Demers, L.M.; Vallyathan, V.; Ding, M.; Lu, Y.; Castranova, V.; Shi, X. Vanadate induction of NF-kappaB involves IkappaB kinase beta and SAPK/ERK kinase 1 in macrophages. J. Biol. Chem. 1999, 274, 20307–20312. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Engelhardt, J.F. Interleukin-1beta induction of NFkappaB is partially regulated by H2O2-mediated activation of NFkappaB-inducing kinase. J. Biol. Chem. 2006, 281, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.J.; Lee, E.K.; Yu, B.P.; Chung, H.Y. Significance of protein tyrosine kinase/protein tyrosine phosphatase balance in the regulation of NF-kappaB signaling in the inflammatory process and aging. Free Radic. Biol. Med. 2009, 47, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Milarski, K.L.; Zhu, G.; Pearl, C.G.; McNamara, D.J.; Dobrusin, E.M.; MacLean, D.; Thieme-Sefler, A.; Zhang, Z.Y.; Sawyer, T.; Decker, S.J.; et al. Sequence specificity in recognition of the epidermal growth factor receptor by protein tyrosine phosphatase 1B. J. Biol. Chem. 1993, 268, 23634–23639. [Google Scholar] [PubMed]
- Agazie, Y.M.; Hayman, M.J. Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol. Cell. Biol. 2003, 23, 7875–7886. [Google Scholar] [CrossRef] [PubMed]
- Keilhack, H.; Tenev, T.; Nyakatura, E.; Godovac-Zimmermann, J.; Nielsen, L.; Seedorf, K.; Böhmer, F.D. Phosphotyrosine 1173 mediates binding of the protein-tyrosine phosphatase SHP-1 to the epidermal growth factor receptor and attenuation of receptor signaling. J. Biol. Chem. 1998, 273, 24839–24846. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signaling. Exp. Cell Res. 2003, 284, 31–53. [Google Scholar] [CrossRef]
- Wu, W.; Graves, L.M.; Jaspers, I.; Devlin, R.B.; Reed, W.; Samet, J.M. Activation of the EGF receptor signaling pathway in human airway epithelial cells exposed to metals. Am. J. Physiol. 1999, 277, L924–L931. [Google Scholar] [PubMed]
- Wu, W.; Jaspers, I.; Zhang, W.; Graves, L.M.; Samet, J.M. Role of Ras in metal-induced EGF receptor signaling and NF-kappaB activation in human airway epithelial cells. Lung Cell. Mol. Physiol. 2002, 282, L1040–L1048. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.; Spring, S.C.; Terman, B.I. Comparison of the signaling mechanisms by which VEGF, H2O2, and phosphatase inhibitors activate endothelial cell ERK1/2 MAP-kinase. Microvasc. Res. 2005, 69, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Src kinase regulation by phosphorylation and dephosphorylation. Biochem. Biophys. Res. Commun. 2005, 331, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ingley, E. Src family kinases: Regulation of their activities, levels and identification of new pathways. Biochim. Biophys. Acta 2008, 1784, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure and regulation. Biochem. Biophys. Res. Commun. 2004, 324, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, P. Src redox regulation: There is more than meets the eye. Mol. Cells 2008, 26, 329–337. [Google Scholar] [PubMed]
- Giannoni, E.; Taddei, M.L.; Chiarugi, P. Src redox regulation: again in the front line. Free Radic. Biol. Med. 2010, 49, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Lluis, J.M.; Buricchi, F.; Chiarugi, P.; Morales, A.; Fernandez-Checa, J.C. Dual role of mitochondrial reactive oxygen species in hypoxia signaling: Activation of nuclear factor-κB via c-SRC and oxidant-dependent cell death. Cancer Res. 2007, 67, 7368–7377. [Google Scholar] [CrossRef] [PubMed]
- Boulven, I.; Robin, P.; Desmyter, C.; Harbon, S.; Leiber, D. Differential involvement of Src family kinases in pervanadate-mediated responses in rat myometrial cells. Cell Signal. 2002, 14, 341–349. [Google Scholar] [CrossRef]
- Fan, C.; Li, Q.; Ross, D.; Engelhardt, J.F. Tyrosine phosphorylation of I kappa B alpha activates NF kappa B through a redox-regulated and c-Src-dependent mechanism following hypoxia/reoxygenation. J. Biol. Chem. 2003, 278, 2072–2080. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, T.E.; Johnson, P.J.; Shalloway, D. Regulation by the autophosphorylation site in overexpressed pp60c-src. Mol. Cell. Biol. 1988, 8, 4541–4546. [Google Scholar] [PubMed]
- Irtegun, S.; Wood, R.J.; Ormsby, A.R.; Mulhern, T.D.; Hatters, D.M. Tyrosine 416 is phosphorylated in the closed, repressed conformation of c-Src. PLoS ONE 2013, 8, e71035. [Google Scholar] [CrossRef] [PubMed]
- Farooq, A.; Zhou, M.M. Structure and regulation of MAPK phosphatases. Cell Signal. 2004, 16, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, K.; Nishida, E. Regulation of MAP kinases by MAP kinase phosphatases. Biochim. Biophys. Acta 2007, 1773, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Turpeinen, T.; Nieminen, R.; Moilanen, E.; Korhonen, R. Mitogen-activated protein kinase phosphatase-1 negatively regulates the expression of interleukin-6, interleukin-8, and cyclooxygenase-2 in A549 human lung epithelial cells. J. Pharmacol. Exp. Ther. 2010, 333, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Misra-Press, A.; Rim, C.S.; Yao, H.; Roberson, M.S.; Stork, P.J. A novel mitogen-activated protein kinase phosphatase. Structure, expression, and regulation. J. Biol. Chem. 1995, 270, 14587–14596. [Google Scholar] [CrossRef] [PubMed]
- Muda, M.; Boschert, U.; Smith, A.; Antonsson, B.; Gillieron, C.; Chabert, C.; Camps, M.; Martinou, I.; Ashworth, A.; Arkinstall, S. Molecular cloning and functional characterization of a novel mitogen-activated protein kinase phosphatase, MKP-4. J. Biol. Chem. 1997, 272, 5141–5151. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Ybanez, M.D.; Ahmadi, S.; Yeh, K.; Kaplowitz, N. Redox regulation of tumor necrosis factor signaling. Antioxid. Redox Signal. 2009, 11, 2245–2263. [Google Scholar] [CrossRef] [PubMed]
- O’Dea, E.; Hoffmann, A. The regulatory logic of the NF-kappaB signaling system. Cold Spring Harb. Perspect. Biol. 2010, 2, a000216. [Google Scholar] [PubMed]
- Barbeau, B.; Bernier, R.; Dumais, N.; Briand, G.; Olivier, M.; Faure, R.; Posner, B.I.; Tremblay, M. Activation of HIV-1 long terminal repeat transcription and virus replication via NF-kappaB-dependent and -independent pathways by potent phosphotyrosine phosphatase inhibitors, the peroxovanadium compounds. J. Biol. Chem. 1997, 272, 12968–12977. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.S.; Peters, R.T.; Dang, L.C.; Maniatis, T. MEKK1 activates both IkappaB kinase alpha and IkappaB kinase beta. Proc. Natl. Acad. Sci. USA 1998, 95, 9319–9324. [Google Scholar] [CrossRef] [PubMed]
- Herscovitch, M.; Comb, W.; Ennis, T.; Coleman, K.; Yong, S.; Armstead, B.; Kalaitzidis, D.; Chandani, S.; Gilmore, T.D. Intermolecular disulfide bond formation in the NEMO dimer requires Cys54 and Cys347. Biochem. Biophys. Res. Commun. 2008, 367, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Siomek, A. NF-κB signaling pathway and free radical impact. Acta Biochim. Pol. 2012, 59, 323–331. [Google Scholar] [PubMed]
- Imbert, V.; Rupec, R.A.; Livolsi, A.; Pahl, H.L.; Traenckner, E.B.; Mueller-Dieckmann, C.; Farahifar, D.; Rossi, B.; Auberger, P.; Baeuerle, P.A.; et al. Tyrosine phosphorylation of I kappa B-alpha activates NF-kappa B without proteolytic degradation of I kappa B-alpha. Cell 1996, 86, 787–798. [Google Scholar] [CrossRef]
- Singh, S.; Darnay, B.G.; Aggarwal, B.B. Site-specific tyrosine phosphorylation of IkappaBalpha negatively regulates its inducible phosphorylation and degradation. J. Biol. Chem. 1996, 271, 31049–31054. [Google Scholar] [CrossRef] [PubMed]
- Béraud, C.; Henzel, W.J.; Baeuerle, P.A. Involvement of regulatory and catalytic subunits of phosphoinositide 3-kinase in NF-kappaB activation. Proc. Natl. Acad. Sci. USA 1999, 96, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, A.; Manna, S.K.; Aggarwal, B.B. Pervanadate-induced nuclear factor-kappaB activation requires tyrosine phosphorylation and degradation of IkappaBalpha. Comparison with tumor necrosis factor-alpha. J. Biol. Chem. 2000, 275, 8549–8555. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Aggarwal, B.B. Protein-tyrosine phosphatase inhibitors block tumor necrosis factor-dependent activation of the nuclear transcription factor NF-kappa B. J. Biol. Chem. 1995, 270, 10631–10639. [Google Scholar] [PubMed]
- Kang, Y.J.; Mbonye, U.R.; DeLong, C.J.; Wada, M.; Smith, W.L. Regulation of intracellular cyclooxygenase levels by gene transcription and protein degradation. Prog. Lipid Res. 2007, 46, 108–125. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Waxman, L.; Goldberg, A.L. Vanadate inhibits the ATP-dependent degradation of proteins in reticulocytes without affecting ubiquitin conjugation. J. Biol. Chem. 1984, 259, 2803–2809. [Google Scholar] [PubMed]
- Kanayama, H.O.; Tamura, T.; Ugai, S.; Kagawa, S.; Tanahashi, N.; Yoshimura, T.; Tanaka, K.; Ichihara, A. Demonstration that a human 26S proteolytic complex consists of a proteasome and multiple associated protein components and hydrolyzes ATP and ubiquitin-ligated proteins by closely linked mechanisms. Eur. J. Biochem. 1992, 206, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Misra, A.; Srivastava, S.; Ankireddy, S.R.; Islam, N.S.; Chandra, T.; Kumar, A.; Barthwal, M.K.; Dikshit, M. Phospholipase C-γ2 via p38 and ERK1/2 MAP kinase mediates diperoxovanadate-asparagine induced human platelet aggregation and sCD40L release. Redox Rep. 2013, 18, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Kudo, I. Phospholipase A2. J. Biochem. 2002, 131, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Goldman, R.; Ferber, E.; Zor, U. Involvement of reactive oxygen species in phospholipase A2 activation: Inhibition of protein tyrosine phosphatases and activation of protein kinases. Adv. Exp. Med. Biol. 1997, 400A, 25–30. [Google Scholar]
- Helgadóttir, A.; Halldórsson, H.; Magnúsdóttir, K.; Kjeld, M.; Thorgeirsson, G. A role for tyrosine phosphorylation in generation of inositol phosphates and prostacyclin production in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Varecka, L.; Peterajová, E.; Sevcík, J. Vanadate changes Ca2+ influx pathway properties in human red blood cells. Gen. Physiol. Biophys. 1997, 16, 359–372. [Google Scholar] [PubMed]
- Törnquist, K.; Dugué, B.; Ekokoski, E. Protein tyrosine phosphorylation and calcium signaling in thyroid FRTL-5 cells. J. Cell. Physiol. 1998, 175, 211–219. [Google Scholar] [CrossRef]
- Randazzo, P.A.; Olshan, J.S.; Bijivi, A.A.; Jarett, L. The effect of orthovanadate on phosphoinositide metabolism in NIH 3T3 fibroblasts. Arch. Biochem. Biophys. 1992, 292, 258–265. [Google Scholar] [CrossRef]
- Bianchini, L.; Todderud, G.; Grinstein, S. Cytosolic [Ca2+] homeostasis and tyrosine phosphorylation of phospholipase C gamma 2 in HL60 granulocytes. J. Biol. Chem. 1993, 268, 3357–3363. [Google Scholar] [PubMed]
- Ohmori, T.; Yatomi, Y.; Wu, Y.; Osada, M.; Satoh, K.; Ozaki, Y. Wheat germ agglutinin-induced platelet activation via platelet endothelial cell adhesion molecule-1: Involvement of rapid phospholipase C gamma 2 activation by Src family kinases. Biochemistry 2001, 40, 12992–13001. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Ryu, S.H.; Suh, P.G. On/off-regulation of phospholipase C-gamma 1-mediated signal transduction. Adv. Enzym. Regul. 2007, 47, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Machide, M.; Kamitori, K.; Kohsaka, S. Hepatocyte growth factor-induced differential activation of phospholipase cgamma 1 and phosphatidylinositol 3-kinase is regulated by tyrosine phosphatase SHP-1 in astrocytes. J. Biol. Chem. 2000, 275, 31392–31398. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, N.; Shimohama, S.; Hayakawa, T.; Sumida, Y.; Fujimoto, S. Tyrosine phosphorylation and translocation of phospholipase C-gamma 2 in polymorphonuclear leukocytes treated with pervanadate. Biochim. Biophys. Acta 1996, 1314, 167–174. [Google Scholar] [CrossRef]
- Irani, K.; Pham, Y.; Coleman, L.D.; Roos, C.; Cooke, G.E.; Miodovnik, A.; Karim, N.; Wilhide, C.C.; Bray, P.F.; Goldschmidt-Clermont, P.J. Priming of platelet alphaIIbbeta3 by oxidants is associated with tyrosine phosphorylation of beta3. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1698–706. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, N.; Gaw Gonzalo, I.T.; Natarajan, R. Molecular mechanisms of high glucose-induced cyclooxygenase-2 expression in monocytes. Diabetes 2004, 53, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Kordowiak, A.M.; Klein, A.; Goc, A.; Dabroś, W. Comparison of the effect of VOSO4, Na3VO4 and NaVO3 on proliferation, viability and morphology of H35-19 rat hepatoma cell line. Pol. J. Pathol. 2007, 58, 51–57. [Google Scholar] [PubMed]
- Cuesta, S.; Francés, D.; García, G.B. ROS formation and antioxidant status in brain areas of rats exposed to sodium metavanadate. Neurotoxicol. Teratol. 2011, 33, 297–302. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korbecki, J.; Baranowska-Bosiacka, I.; Gutowska, I.; Chlubek, D. Vanadium Compounds as Pro-Inflammatory Agents: Effects on Cyclooxygenases. Int. J. Mol. Sci. 2015, 16, 12648-12668. https://doi.org/10.3390/ijms160612648
Korbecki J, Baranowska-Bosiacka I, Gutowska I, Chlubek D. Vanadium Compounds as Pro-Inflammatory Agents: Effects on Cyclooxygenases. International Journal of Molecular Sciences. 2015; 16(6):12648-12668. https://doi.org/10.3390/ijms160612648
Chicago/Turabian StyleKorbecki, Jan, Irena Baranowska-Bosiacka, Izabela Gutowska, and Dariusz Chlubek. 2015. "Vanadium Compounds as Pro-Inflammatory Agents: Effects on Cyclooxygenases" International Journal of Molecular Sciences 16, no. 6: 12648-12668. https://doi.org/10.3390/ijms160612648
APA StyleKorbecki, J., Baranowska-Bosiacka, I., Gutowska, I., & Chlubek, D. (2015). Vanadium Compounds as Pro-Inflammatory Agents: Effects on Cyclooxygenases. International Journal of Molecular Sciences, 16(6), 12648-12668. https://doi.org/10.3390/ijms160612648